
ABSTRACT
Methylenetetrahydrofolate reductase (MTHFR) is a key enzyme regulating the folate cycle and its genetic variations have been associated with various human diseases. Previously we identified that MTHFR is phosphorylated by cyclin-dependent kinase 1 (CDK1) at T34 and MTHFR underlies heterochromatin maintenance marked by H3K9me3 levels. Herein we demonstrate that pT34 creates a binding motif that docks MTHFR to the polo-binding domain (PBD) of polo-like kinase 1 (PLK1), a fundamental kinase that orchestrates many cell cycle events. We show that PLK1 phosphorylates MTHFR at T549 in vitro and in vivo. Further, we uncovered a role of MTHFR in replication. First, MTHFR depletion increased the fraction of cells in S phase. This defect could not be rescued by siRNA resistant plasmids harboring T549A, but could be restored by overproduction of Suv4–20H2, the H4K20 methyltransferase. Moreover, siMTHFR attenuated H4K20me3 levels, which could be rescued by Suv4–20H2 overproduction. More importantly, we also investigated MTHFR-E429A, the protein product of an MTHFR single nucleotide variant. MTHFR-E429A overexpression also increased S phase cells and decreased H4K20me3 levels, and it is linked to a poor glioma prognosis in the Chinese population. Collectively, we have unveiled a vital role of PLK1-dependent phosphorylation of MTHFR in replication via histone methylation, and implicate folate metabolism with glioma.
Introduction
One-carbon metabolism, comprising folate and methionine metabolism, sits at the heart of cancer biology. Into the one-carbon pathway enter glucose and amino acids, and out of it one expects a wealth of components used for processes encompassing all aspects of life: biosynthesis, genome integrity and epigenetics.Citation1 The folate cycle begins with folic acid, which, upon cell entry, is reduced to tetrahydrofolate (THF). THF, in turn, is modified to 5,10-methylene-THF (me-THF). This is when methylenetetrahydrofolate reductase (MTHFR) comes into play, and reduces me-THF to 5-methyltetrahydrofolate (mTHF). It is exactly mTHF that is at the hub of the folate cycle and the methionine cycle. mTHF is demethylated to release a carbon, which is taken up by homocysteine and thus generating methionine. Methionine, then, is adenylated to make S-adenosylmethionine (SAM), which is the ultimate methyl donor for methylation reactions in the cell. Thus, carbon units are transferred between the folate cycle and the methionine cycle. Its innate cyclic nature makes it ideal to be an integrator of nutrient status, cell growth and human diseases.Citation1
Among all the consequences of one-carbon metabolism, of particular interest, is epigenetics.Citation2 Epigenetics communicates nutrient status outside to cellular physiology inside, and dictates cellular homeostasis maintenance or chronicle disease conditions. Histone methylation, an indispensable part of epigenetics, involves transferring the methyl group from SAM to histones by histone methyltransferases (HMTs), resulting in mono-, di-, or trimethylated lysines or arginines. Recent years have witnessed a surge of interest in H4K20 methylation, whose monomethylated form is catalyzed by SET8 (also named PR-SET7 or SETD8) and di- and tri-methylated forms are catalyzed by SUV4–20H1 and SUV4–20H2.Citation3 Interestingly, H4K20me3, a hallmark of silenced heterochromatins, co-localizes with H3K9me3 at centromeres,Citation4 telomeres,Citation5 and pericentric heterochromatin regions.Citation6 In fact, it has been proposed that the heterochromatin marker H3K9me3 brings its reader heterochromatin protein 1 (HP1), which subsequently recruits SUV4–20H2, thus generating H4K20me3 at the heterochromatin regions.Citation7
There has been increasing awareness of the link between H4K20 methylation levels and cell cycle control, DNA damage response and DNA replication.Citation7 DNA replication is the intricate process of precise genome duplication. It needs to be carefully monitored, because replication failure could be a threat to genome integrity. Cells depleted of Set8, the HMT of H4K20me1, accumulate in S phase.Citation8-10 Set8 is normally degraded in S phase. When a non-degradable SET8 is introduced into cells, H4K20me3 aberrantly accumulates in S phase,Citation11 correlating with aberrant re-replication phenotypes.Citation12 These data suggest it is not methylation per se, but the precise timing of methylation that matters. Knockdown of SUV4–20H2, the essential enzyme that generates H4K20me3, also culminated in more S phase cell fractions.Citation13 Forced targeting of Set8 to genomic foci aberrantly recruits the origin of replication complex (ORC) in an H4K20me3 and SUV4–20H2-dependent fashion,Citation12 suggesting that H4K20me3 governs the replication pathway.
Perhaps most enlightening of all, is the finding that the replication protein machinery actually harbors readers of H4K20 methylation. During G1 phase of the cell cycle at the replication origins, a legion of the pre-replication complex (pre-RC) assembles, enlisting ORC, the minichromosome maintenance complex (MCM), Cdc6 and Cdt1.Citation14 The bromo-adjacent homology (BAH) domain of ORC1 reads H4K20me2,Citation15 while the WD repeat domain of ORCA/ORWD1 reads H4K20me3.Citation12,16 Indeed, knockout of both SUV4–20H1 and SUV4–20H2 in mice is perinatally lethal.Citation17 And ORCA associates with late-firing origins during G1 to regulate heterochromatin replication.Citation18 It is thus conceivable that H4K20me2/3 stabilizes the ORC complex, the disruption of which delays cell division and proliferation.
MTHFR has long been the culprit for a swathe of human diseases, for instance, occlusive vascular disease, neural tube defects and colon cancer.Citation19 Targeting MTHFR, methotrexate and pemetrexed have been approved for multiple cancers.Citation20 Two most intensively studied pathogenic single nucleotide variants (SNVs) are C667T and A1298C.Citation21 A role of MTHFR in cell cycle control has never been suspected, although its multitude of phosphorylation events caught attention a decade ago.Citation22 We reported before that T34 of MTHFR is phosphorylated by cyclin-dependent kinase 1 (CDK1), the crucial kinase that orchestrates cell cycle progression, and found that MTHFR maintains centromeric heterochromatin by modulating H3K9me3 levels.Citation19 Here we further demonstrate that pT34 actually creates a docking motif for the polo-binding domain (PBD) of polo-like kinase 1 (PLK1), another master kinase of cell division.Citation23 PLK1 in turn phosphorylates MTHFR at T549, thus affecting S phase progression via H4K20me3. Importantly, we show that E429A, encoded by A1286C (chr1:11854476:G:70 COSM3735923), is implicated in the prognosis of Chinese glioma patients, and might as well follow the same route by affecting the S phase and H4K20me3. We propose that the cell cycle role of MTHFR intersects histone methylation levels and tumorigenesis.
Results
Association between MTHFR and PLK1
Previously, we identified that Cdk1 phosphorylates MTHFR at T34 in vitro and in vivo.Citation19 A closer look surrounding T34 () reminded us of the motif (S-phospho S/T-P) that binds to the PBD of PLK1.Citation24 We also performed mass spectrometry (MS) analysis using HeLa cells extracts transiently expressing Flag-MTHFR, and PLK1 was identified in the anti-Flag immunoprecipitates (data not shown). Naturally, we tested the interaction between MTHFR and PLK1. HeLa cells extracts treated with Noc or mock treated were subject to IP assays using anti-MTHFR antibodies, and PLK1 was identified in the IP complexes (). Then Flag-MTHFR and HA-PLK1 were co-transfected into HeLa cells, and reciprocal coIP interaction was identified between these two proteins (). Moreover, recombinant GST-PLK1 efficiently pulled-down His-MTHFR (). Collectively, these data suggest that PLK1 directly interacts with MTHFR.
Figure 1. MTHFR interacts with PLK1. (A) A comparison of PLK1 polo-box domain (PBD) binding motif and various MTHFR homologues at T34. (B) MTHFR and PLK1 coIP. HeLa cells were synchronized in mitosis by thymidine-nocodazole block, then subject to IP by indicated antibodies. IgG immunoprecipitates were used as a negative control. (C-D) Epitope-tagged MTHFR and PLK1 were transfected into HeLa cells and subject to IP experiments with the antibodies indicated. (E-F) GST pull-down assays. Binding of GST-PLK1-FL with recombinant MTHFR was tested (E). The asterisk indicates a non-specific band. Binding between GST-PLK1-PBD and overexpressed MTHFR WT or T34A was examined in (F). (G) Domain structures of MTHFR. The boundaries of MTHFR-N or C fragments and mutations used in this study are indicated. (H) PLK1-kinase domain (KDN) was transfected into HeLa cells, together with MTHFR-FL, N or C constructs. Then coIP experiments were performed using antibodies indicated.
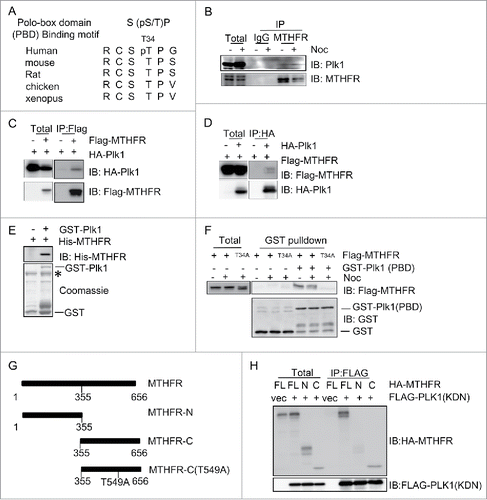
Then we investigated whether pT34 is essential for the interaction between MTHFR and PLK1. When we mutated T34 into phospho-deficient Ala and transfected wild-type (WT) and T34A of MTHFR into HeLa cells, PLK1-PBD failed to pull-down T34A (), suggesting that the phosphorylation of T34 is crucial for MTHFR to dock onto the PBD of PLK1. Since MTHFR consists of an N-terminal catalytic domain and a C-terminal regulatory domainCitation19 (), we examined which domain interacts with PLK1. Full-length (FL), N and C-termini of MTHFR were transfected into HeLa cells, together with the kinase domain (KDN) of PLK1. Only the C-terminus of MTHFR efficiently coIPed with PLK1-KDN (). Taken together, these data suggest that CDK1-induced T34 phosphorylation docks MTHFR onto PLK1 PBD, then PLK1-KDN binds with the MTHFR C-terminus.
Phosphorylation of MTHFR by PLK1
According to the established paradigm of PLK1 activation, PLK1 KDN will phosphorylate the substrate after it docks to the PBD domain.Citation25 Therefore, we tested whether PLK1 phosphorylates MTHFR by an in vitro kinase (IVK) assay. Since only the C-terminus binds with PLK1-KDN, we purified the C-terminus of MTHFR from E. coli, and incubated it with recombinant PLK1. PLK1 phosphorylated the MTHFR C-terminus efficiently (). We also tried to purify the N-terminal catalytic domain of MTHFR, but it does not express sufficiently, as previously reported.Citation26
Figure 2. PLK1 phosphorylates MTHFR at T549. (A) An in vitro kinase (IVK) assay using recombinant PLK1 and MTHFR-C in kinase buffers containing 32P-ATP. Coomassie blue (CBB) staining showed input MTHFR- C proteins. (B) T549 of MTHFR is phosphorylated by PLK1 in vitro. From this collision-induced dissociation spectrum, a phosphorylated peptide ENI(pT)NAPELQPNAVTWGIFPGR of MTHFR was identified. “b” and “y” ion series represent fragment ions containing the N- and C-termini of the peptide, respectively. (C) A comparison of T549 and the PLK1 phosphorylation consensus motif. The residues in red fit the consensus motif. (D)An IP-kinase assay with the MTHFR pT549 antibody. HeLa cells were transfected with Flag-PLK1 WT or kinase dead (KD), and then the anti-Flag immunoprecipitates were incubated with recombinant GST-MTHFR-WT or T549A. (E) MTHFR is phosphorylated at T549 in vivo. HeLa cells were transfected with HA-MTHFR-WT or T549A, treated with BI2536 (a PLK1 inhibitor) or without, then the total lysates were subject to IB with indicated antibodies.
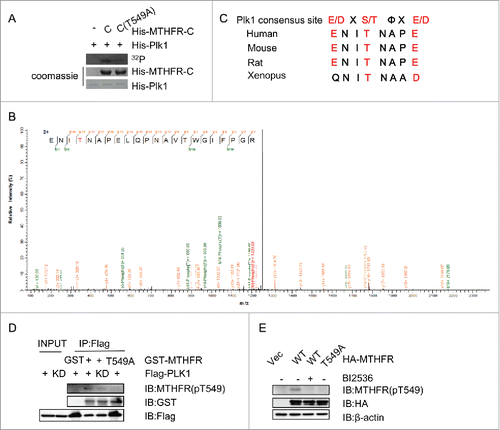
The IVK products were then analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS), and only one phosphorylation site, T549, was identified ().We then constructed the phospho-deficient T549A mutant, and recombinant MTHFR-T549A failed to be phosphorylated in the IVK assay (). Moreover, T549 also conforms to PLK1 consensus site and is conserved among various species (). The result indicated that T549 was the major phosphorylation site.
We proceeded to prepare the phospho-specific antibody targeting pT549. In an IP-kinase assay (), Flag-PLK1 immunoprecipitates efficiently phosphorylated recombinant MTHFR, but the PLK1-kinase dead (KD) did not, nor when MTHFR-T549A was used as the substrate, as detected by the pT549 antibodies. Then we examined whether the phosphorylation of T549 occurs in vivo. HeLa cells were transfected HA-MTHFR WT or T549A, treated or not treated with BI2536, an inhibitor of PLK1 (). Cell extracts were then subject to IB with pT549 antibodies, and a crisp band was only detected in the HA-MTHFR-WT lane without BI treatment, suggesting that PLK1-dependent phosphorylation of MTHFR T549 occurs in vivo.
MTHFR-depletion increases S phase cells
As PLK1 is a key kinase that orchestrates many mitotic event, including the recently identified process of replication,Citation25 we sought to examine whether MTHFR depletion is relevant to cell cycle control. MTHFR siRNA efficiently knocked down MTHFR proteins (). MTHFR-depleted and control HeLa cells were analyzed by flow cytometry using propidium iodide (PI) staining (). MTHFR depletion readily increased G1/S cells from ∼20% to ∼40% (), indicating that S phase cells accumulated in these cells.
Figure 3. MTHFR depleted cells accumulate in the S phase. HeLa cells were transfected with siMTHFR or control siRNA (A) and then analyzed by flow cytometry after PI staining (B). The percentage of cells in different cell cycle stages were shown in (B).The result is representative of 3 independent experiments. (C) Cells were pulsed with BrdU for 30 min, then stained with DAPI after BrdU staining, and examined by fluorescence microscopy. Scale bar, 10 µm. The percentage of BrdU positive cells were indicated in (D). More than 250 cells were counted in each experiment. Histograms represent mean ± SD of 3 independent experiments (p = 0.021< 0.05). Cells were pulse-labeled with BrdU as in (C), then measured for DNA synthesis rate and DNA content by flow cytometry. Quantitation was shown in (E) (p = 0.043 < 0.05).
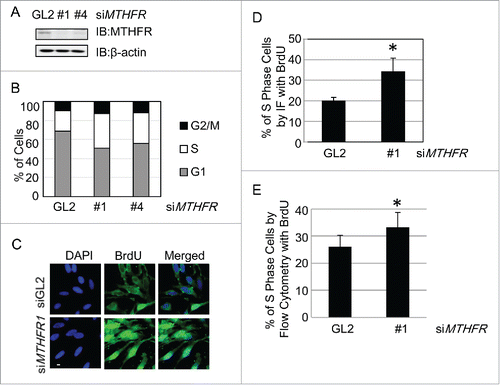
To further confirm that S phase cells did accumulate, we pulsed the cells with 5-bromo-2′-deoxyuridine (BrdU), then stained them with standard BrdU protocols (). As BrdU incorporation only occurs in the S-phase of the cell cycle, nuclear BrdU staining indicates S phase cells. As shown in , MTHFR-depleted cells increased ∼1.5-fold of BrdU positive cells as determined by immunofluorescence. We then used BrdU-labeled cells for flow cytometry analysis (). More siMTHFR cells were positive for BrdU staining, compared with WT (). These results indicated that more MTHFR-depleted cells were actively replicating their DNA compared with WT.
Then we sought after the possibility whether S phase accumulation was due to checkpoint activation. We examined Chk1 phosphorylation in MTHFR-depletion cells, and Chk1 was not activated (data not shown), suggesting that cell cycle progression did not arrest.
MTHFR(1286A > C) correlates with poor glioma survival rates in Han Chinese populations
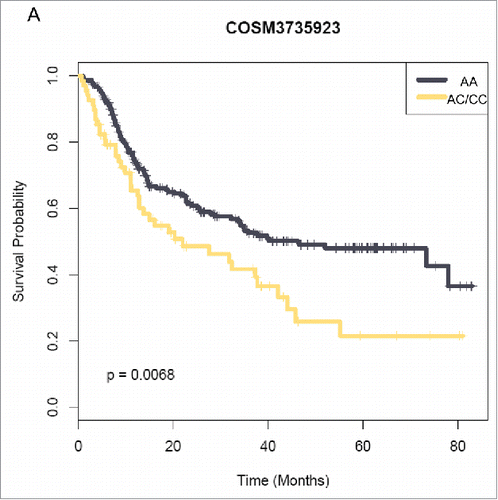
Recently, RNA-seq of 272 Han Chinese gliomas revealed a novel, recurrent fusion transcript involving the PTPRZ1 and MET gene (ZM) that was found in 15% of secondary glioblastoma (sGBMs).Citation27 The same analysis also unveiled a great many SNVs associated with both low grade gliomas and glioblastomas. Survival analysis revealed that the 7-year cumulative recurrence-free survival in sGBM patients carrying MTHFR(1286A>C)(chr1:11854476:G:70 COSM3735923) heterozygosity (AC:63 patients; CC: 8 patients) was significantly worse than those with WT MTHFR (p = 0.0068, ). As MTHFR(1286A>C) encodes MTHFR-E429A, we also incorporated it in our following studies.
Figure 4. MTHFR(1286A>C) correlates with a poor prognosis in Chinese glioma patients. Kaplan–Meier curves estimate the recurrence-free survival rates.
Cells harboring T549A and E429A failed to rescue the S phase accumulation defects
We then examined whether the phosphorylation of MTHFR by PLK1 contributes to S phase accumulation. We performed rescue assays using HA-MTHFR-res, T549A-res and E429A-res plasmids. These plasmids were transfected into HeLa cells and the resultant stable transfectants were treated with siMTHFR, as indicated in . Endogenous MTHFR was attenuated substantially (decreased ∼50%), while the exogenous HA-MTHFR-res, T549A-res and E429A-res were stably expressed, as shown by IB ().
Figure 5. T549A and E429A failed to rescue the S phase defects of MTHFR-depleted cells. (A) Western blots showing stable transfectants harboring rescue, rescue-T549A and rescue-E429A plasmids. (B) Cells in (A) were analyzed by BrdU staining and PI as described in materials and methods, and then analyzed by flow cytometry. (C) Quantitation of results in (B). Histograms represent mean ± SD of 3 independent experiments. Asterisks indicate significant differences from Lane 1 (p values are: p1–2 = 0.0003, p1–3 = 0.42, p1–4 = 0.024, p1–5 = 0.017). (D) Cells were transfected with control vectors, or MTHFR-WT, E429A plasmids. Then extracts were collected and analyzed by IB with the antibodies indicated.
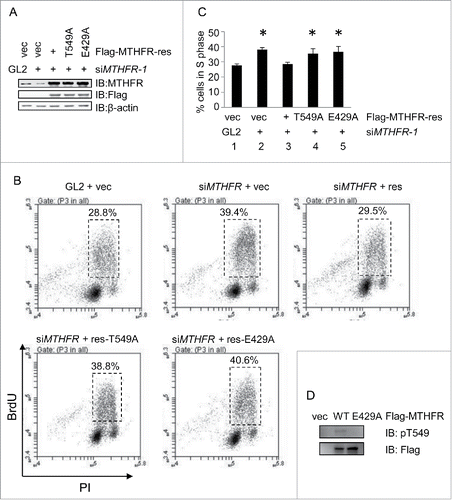
These cells were then pulsed with BrdU and subject to flow cytometry analysis (). MTHFR depletion increased BrdU incorporation. Re-expression of HA-MTHFR-res restored the defects (). The T549A-res and E429A plasmids, however, did not efficiently recover the S phase defects. The results were then subject to t-test (), and there is significant difference among WT, and T549A-res, E429A-res plasmids (). Thus, the rescue experiments indicate that pT549 and E429 are essential for MTHFR to participate in S phase progression.
Since MTHFR-T549A and E429A manifested similar cell cycle defects, we examined T549 phosphorylation levels in cells haboring E429A. Cells were transfected with MTHFR-WT and E429A plasmids, then extracts were collected and analyzed by Western analysis (). Indeed, pT549 was abolished in E429 cells, suggesting a common mechanism between these two mutants.
MTHFR-depletion dampens H4K20me3 levels
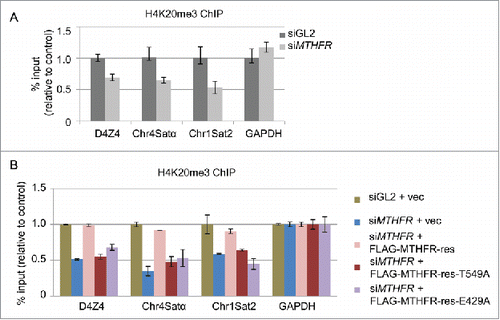
Previously we have demonstrated that MTHFR depletion results in relaxation of heterochromatin and a decrease of H3K9me3 levels.Citation19 Another marker of heterochromatin, H4K20me3, is mostly enriched in quiescent chromatin, and colocalizes with H3K9me37. Most importantly, knockdown of Suv4–20H1 and Suv4–20H2, the two enzymes that create H4K20me3, results in an increased fraction of cells in S phase.Citation13 Thus, we examined H4K20me3 levels by ChIP in MTHFR-depleted cells. And we found that siMTHFR decreased H4K20me3 levels across a panel of pericentric heterochromatin loci (). Moreover, re-expression of WT-res plasmids could efficiently rescue the H4K20me3 levels, but not expression of the T549A or E429A mutants (). Taken together, the results suggest that MTHFR might exert its impact on cell cycle progression via H4K20me3.
Figure 6. T549 phosphorylation of MTHFR influences H4K20me3 levels, a hallmark of heterochromatin. (A) HeLa cells depleted of MTHFR or control were analyzed by ChIP assays using H4K20me3 antibodies across a panel of pericentromeric heterochromatin regions. (B) HeLa cells were stably transfected with vector, rescue, rescue-T549A or E429A plasmids, and then analyzed by ChIP. qPCR data are presented as means ± SD of 3 independent experiments.
SUV4–20H2 suppresses the S phase accumulation and H4K20me3 defects
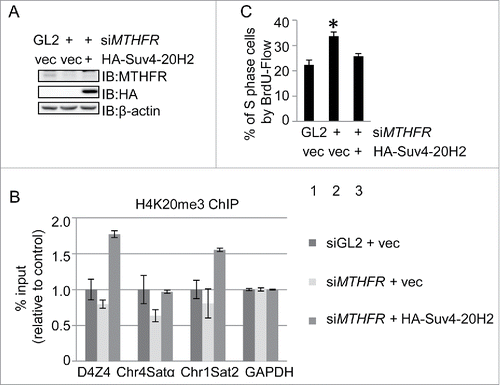
Genetically, if the S phase defects were caused by H4K20me3 defects, then overexpression of SUV4–20H2, the enzyme that converts H4K20me2 to H4K20me3, should rescue both defects. To test the hypothesis, we overexpressed HA-SUV4–20H2 in MTHFR-depleted cells (), and indeed H4K20me3 increased in MTHFR-depleted cells by ChIP analysis (). When the cell cycle profiles were measured by BrdU staining followed by flow cytometry, cells accumulating in S cells were significantly decreased (), thus unveiling the mechanistic link between MTHFR and cell cycle progression.
Figure 7. Overproduction of the H4K20 trimethyltransferase Suv4–20H2 rescues S phase and H4K20me3 levels in MTHFR-depleted cells. (A) IB indicated Suv4–20H2 overproduction in MTHFR-depleted cells. (B) ChIP analysis of the cells in A. qPCR data are presented as means ± SD of 3 independent experiments. (C) The cells in A were stained with BrdU and analyzed by flow cytometry, showing that Suv4–20H4 overproduction rescues the S phase accumulation defects in MTHFR-depleted cells. The asterisk indicates significant differences from Lane 1 (p values are: p1–2 = 0.002, p1–3 = 0.05).
Discussion
In a continued endeavor to study MTHFR, we identified Plk1 as its interacting partner. MTHFR is phosphorylated by CDK1 at T34, and thus targeted to the Plk1 PBD domain and in turn phosphorylated by Plk1 at T549. We show here that both MTHFR-T549A and E429A exhibit S phase accumulation as well as H4K20me3 level defects, both of which could be restored by SUV4–20H2 overproduction. As E429A glioma patients manifested poorer survival rates in Han Chinese and also decreased pT549 levels, we thus propose a model where MTHFR links the heterochromatin landscape, cell cycle profile and tumor progression.
Plk1's role in mitosis has long been appreciated, in centrosome duplication, mitotic Golgi dispersal, mitotic entry, chromosome segregation and cytokinesis.Citation28 Plk1 also governs replication, the inkling of which came as early as 1996, when Cdc5, the yeast homolog of human Plk1, was reported to phosphorylate Dbf4 in vitro, one of the DNA replication initiation proteins.Citation29 Then the coupling of Plk1 and replication became more obvious, as Plk1 is found to interact with and phosphorylate several components of the replication machinery: Mcm2 and 7,Citation30,31 Orc232 and the histone acetylase Hbo1.Citation33 And it came to no surprise that long-term Plk1 knockdown slowed S phase progression.Citation34 By showing that Plk1-phosphorylated MTHFR ensures a steady level of H4K20me3, our study adds to the already versatile role of Plk1 during the cell cycle.
The crosstalk between master cell cycle kinases and the epigenetics machinery is not few and far between. Suv39H1, the HMT specifically for H3K9me3, another heterochromatin marker, is phosphorylated by CDK2 during replication and then dissociates from chromatin, thus affecting heterochromatin replication timing.Citation35 Set8, the HMT for H4K20me1, is phosphorylated by CDK1 during mitosis, followed by chromatin dissociation, dephosphorylation and subsequent ubiquitination.Citation36 Plk1 has also been implicated in activating Haspin, the atypical mitotic kinase responsible for H3pT3 and subsequent targeting of Aurora B to centromeres.Citation37,38
Our results are in line with the report that knockdown SUV4–20H2, the HMT of H4K20me3, culminated in increase of cells in S phase.Citation13 It is conceivable that compromised H4K20me3 may destabilize the chromatin bound ORC complex and thus causing S phase defects.Citation12,39 Alternatively, H4K20me3 may execute its function via Rb. It has been known that H4K20me3 localization is dependent on Rb.Citation40 And recently it was shown that overexpression of SUV4–20H2 rescued the cohesin and H4K20me3 defects of Rb-depleted cells.Citation41 Since replication defects could very well lead to cohesin defects,Citation42 our data suggest that cohesin phenotypes in Rb-devoid cells might be secondary to replication.
As we reported before, MTHFR also affects H3K9me3 levels.Citation19 Intriguingly, KDM4D, the H3K9me3 demethylase, is recruited to replication origins by ORC and MCM complexes, which further enlist Cdc45, PCNA and polymerases.Citation43 It was postulated that H3K9me3 levels were attenuated by Kdm4d to facilitate formation of the pre-initiation complex. Concomitantly, SUMOylation of ORC2 is also involved in recruiting KDM5A, the demethylase to convert H3K4me3 to H3K4me2, the ablation of which caused re-replication of the heterochromatin.Citation44 Hence epigenetics may fine-tune replication through multiple channels.
It is possible that some methylation marks are more sensitive to SAM shortage than others. MTHFR depletion does not affect H3K9me1 or H3K9me219. The budding yeast homolog of MTHFR is Met13. Deletion of MET13 specifically reduced H3K4me3 and H3pT11, but not H3K4me1/2 or H3K36me3, suggesting that some types of chromatin modifications are more sensitive to serine derived from glucose.Citation45 H3K9 methylation was not examined due to lack of the catalytic enzyme in S. cerevisiae (Dr. Shanshan Li, personal communication).
A link between histone modification and metabolism could be conserved.Citation46 The yeast SESAME (Serine-responsive SAM-containing Metabolic Enzyme) Complex comprises Pyk1 (the yeast homolog of Pyruvate kinase M2 [PKM2]), Serine metabolic enzymes, SAM synthetases and an acetyl-CoA synthetase.Citation45 SESAME senses glycolysis and serine metabolism and thus modulates the crosstalk between H3K4me3 and H3pT11.Citation45 Threonine depletion in mouse embryonic stem cells decreased SAM and H3K4me3 levels, culminating in increased differentiation.Citation47 Congruently, methionine deprivation in human pluripotent stem cells decreases SAM levels, rapidly reduces H3K4me3 levels, modestly dampens DNA methylation levels and potentiates cellular differentiation.Citation48 Hence the cycle of glucose and one-carbon metabolism might be interconnected with cell cycle and cell fate.
Our results do not exclude the possibility that the cell cycle profile change might be contributed by DNA methylation alteration induced by MTHFR depletion. As reported previously, DNA methyltransferase 1 (DNMT1) knockdown triggered an intra-S-arrest of DNA replication.Citation49 siDNMT1 activates the genotoxic stress checkpoint kinase cascade, including Chk1 and Chk2.Citation50 In MTHFR-depleted cells, however, we did not observe Chk1 activation (data not shown), suggesting the mechanism might be different.
Glioma is particularly prone to metabolic disruptions, in particular, one-carbon metabolism.Citation51 Moreover, glioma cancer incidents are imbued with ample cases of histone alterations. In a tour de force study by Lewis et al.,Citation52 it was found that pediatric diffuse intrinsic pontine glioma (DIPG) samples containing the K27M mutation in histone H3 impaired H3K27me3 levels. Expression levels of enhancer of zeste homolog-2 (EZH2), the regulator of H3K27me3, can be used to stratify low- and high-grade astrocytic tumors.Citation53 Strikingly, small molecule EZH2 inhibitors could effectively abolish tumor growths in H3K27M mutant DIPG mouse models.Citation54 Hence, the possibility of targeting epigenetics for brain tumor therapeutics could be limitless.
H4K20me3 only constitutes less than 2% of cellular H4,Citation7 yet ripples across the metabolic network could cause undesired repercussions in cell cycle regulation, epigenetics and diseases states. We posit that Plk1 assists MTHFR in ascertaining H4K20me3 levels, so that a faithful and unerring replication will ensue.
Experimental procedures
Antibodies, plasmids, and cell culture
HeLa cells were grown in DMEM (Hyclone, UT, USA) containing 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C in a 5% CO2 incubator.
Rabbit polyclonal antibodies against MTHFR were purchased from Bethyl Laboratories Inc. (Montgomery, TX, USA, Cat. # A303–624A). Anti-PLK1 antibodies were from Santa Cruz (Cat. # sc17783). Antibodies against MTHFR phospho-T549 (pT549-Ab) were raised in rabbits using the sequence of CGENI(pT)NAPEL and manufactured by Beijing B&M Biotech Co., Ltd. MTHFR-T549A and T549D plasmids were generated using specific primers (sequences available upon request) following the manufacturer's instructions (QuickChange II, Stratagene). MTHFR siRNA sequences and plasmids were described previously.Citation19
In vitro kinase assays (IVK)
The PLK1 IVK assay was performed as described previously.Citation55 Briefly, recombinant PLK1 kinase (R & D systems) was incubated with purified GST-MTHFR with 10 mM HEPES (pH 7.5), 50 mM NaCl, 2 mM MgCl2, 1 mM Dithiothreitol, 1 mM EGTA, 1 mM ATP or 1 μCi of γ-[32P]ATP. After 20 min at 30°C, reactions were stopped by the sample buffer. Protein samples were separated by SDS-PAGE and phosphate incorporation was determined by PhosphorImager. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed as described.Citation56
Flow cytometry and cell cycle analysis
Flow cytometry analysis was performed as described.Citation57 Cells were collected by trypsinization and washed once with PBS, fixed with cold 75% ethanol and stored at 4°C overnight. Fixed cells were blocked with 2% BSA in PBST (PBS with 0.1% Tween 20) for 0.5 h at room temperature, and incubated with PBS containing propidium iodide (PI) (8 μg/ml) and RNAse A (1 μg/ml) at 37°C for 0.5 h, and then subjected to flow cytometry analysis.
For 5-bromo-2′-deoxyuridine (BrdU) incorporation, cells were fixed in 70% ethanol at 4°C for 1 h. BrdU incorporation was detected by using FITC-linked anti-BrdU (PharMingen) antibodies according to the manufacturer's protocols (HCl denaturation method). After washing, cells were resuspended in PI, then subject to flow cytometry analysis. Cells stained with BrdU were also examined with indirect immunofluorescence as described before .Citation57
Immunoprecipitation (IP) and immunoblotting (IB) assays
IP and IB experiments were performed as described before.Citation55 The following primary antibodies were used for IB: anti-β-actin (1:10000), anti-HA (1:1000), and anti-FLAG M2 (Sigma) (1:1000), anti-MTHFR (1:1000), anti-Plk1 (1:1000), anti-MTHFR-pT549 (1:100). Peroxidase-conjugated secondary antibodies were from JacksonImmuno Research. BI2356 (PLK1 inhibitor) was used at 1 μm for 2 hours. Nocodazole (Noc) was used at 100 ng/ml for 16 hours. Blotted proteins were visualized using the ECL detection system (Amersham). Signals were detected by a LAS-4000, and quantitatively analyzed by densitometry using the Multi Gauge software (Fujifilm). All western blots were repeated for at least 3 times.
Chromatin Immunoprecipitation (ChIP)
HeLa cells were cross-linked for 10 min with formaldehyde (final concentration 1%), and then stopped by glycine, followed by cell lysis and sonication. Cross-linked chromatin was incubated with corresponding antibodies at 4°C overnight. Cross-linking was reversed at 4°C for 2 h with the resultant purified DNA analyzed by quantitative polymerase chain reaction (qPCR) using primer sets for target heterochromatic loci. Primers sets were described before.Citation35
Abbreviations
| Plk1 | = | polo-like kinase 1 |
| MTHFR | = | Methylenetetrahydrofolate reductase |
| IP | = | Immunoprecipitation |
| IB | = | Immunoblotting |
| IVK | = | In vitro kinase assays |
| CDK1 | = | cyclin-dependent kinase 1 |
| PBD | = | the polo-binding domain |
| SAM | = | S-adenosylmethionine |
Disclosure of potential conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Author contributions
J. L. wrote the manuscript; J. L., X. X. and W-G. Z. designed the project and analyzed the data; Y. D. and M-Q. D. performed MS analysis; K.Y. and X-D. S analyzed the glioma data; X. L., S. N., Q. G. and B. Z. performed all the other experiments. All authors reviewed and approved the manuscript.
Acknowledgments
We thank Dr. Huiqiang Lou for support and members of the Xu laboratory for comments on the manuscript. This work was supported by the National Natural Science Foundation under Grant 31461143012 and 31130017 (to X.X.); the 973 projects under Grant 2013CB911002 and 2015CB910601 (to X. X.); the Beijing Nova Program Interdisciplinary Cooperation Project under Grant Z161100004916042 (to J. L.); and the CNU Interdisciplinary Project (to J. L.).
References
- Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572-83. doi:10.1038/nrc3557. PMID:23822983
- Mentch SJ, Locasale JW. One-carbon metabolism and epigenetics: understanding the specificity. Ann N Y Acad Sci. 2016;1363:91-8. doi:10.1111/nyas.12956. PMID:26647078
- Jorgensen S, Schotta G, Sorensen CS. Histone H4 lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013;41:2797-806. doi:10.1093/nar/gkt012. PMID:23345616
- Martens JH, O'Sullivan RJ, Braunschweig U, Opravil S, Radolf M, Steinlein P, Jenuwein T. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J. 2005;24:800-12. doi:10.1038/sj.emboj.7600545. PMID:15678104
- Benetti R, Garcia-Cao M, Blasco MA. Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat Genet. 2007;39:243-50. doi:10.1038/ng1952. PMID:17237781
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18:1251-62. doi:10.1101/gad.300704. PMID:15145825
- van Nuland R, Gozani O. Histone H4 Lysine 20 (H4K20) Methylation, Expanding the Signaling Potential of the Proteome One Methyl Moiety at a Time. Mol Cell Proteomics. 2016;15:755-64. doi:10.1074/mcp.R115.054742. PMID:26598646
- Houston SI, McManus KJ, Adams MM, Sims JK, Carpenter PB, Hendzel MJ, Rice JC. Catalytic function of the PR-Set7 histone H4 lysine 20 monomethyltransferase is essential for mitotic entry and genomic stability. J Biol Chem. 2008;283:19478-88. doi:10.1074/jbc.M710579200. PMID:18480059
- Tardat M, Murr R, Herceg Z, Sardet C, Julien E. PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J Cell Biol. 2007;179:1413-26. doi:10.1083/jcb.200706179. PMID:18158331
- Jorgensen S, Elvers I, Trelle MB, Menzel T, Eskildsen M, Jensen ON, Helleday T, Helin K, Sorensen CS. The histone methyltransferase SET8 is required for S-phase progression. J Cell Biol. 2007;179:1337-45. doi:10.1083/jcb.200706150. PMID:18166648
- Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, Jin J, Dyson NJ, Walter JC, Zou L. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol Cell. 2010;40:22-33. doi:10.1016/j.molcel.2010.09.015. PMID:20932472
- Beck DB, Burton A, Oda H, Ziegler-Birling C, Torres-Padilla ME, Reinberg D. The role of PR-Set7 in replication licensing depends on Suv4-20h. Genes Dev. 2012;26:2580-9. doi:10.1101/gad.195636.112. PMID:23152447
- Evertts AG, Manning AL, Wang X, Dyson NJ, Garcia BA, Coller HA. H4K20 methylation regulates quiescence and chromatin compaction. Mol Biol Cell. 2013;24:3025-37. doi:10.1091/mbc.E12-07-0529. PMID:23924899
- Arias EE, Walter JC. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev. 2007;21:497-518. doi:10.1101/gad.1508907. PMID:17344412
- Kuo AJ, Song J, Cheung P, Ishibe-Murakami S, Yamazoe S, Chen JK, Patel DJ, Gozani O. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature. 2012; 484:115-9. doi:10.1038/nature10956. PMID:22398447
- Shen Z, Sathyan KM, Geng Y, Zheng R, Chakraborty A, Freeman B, Wang F, Prasanth KV, Prasanth SG. A WD-repeat protein stabilizes ORC binding to chromatin. Mol Cell. 2010;40:99-111. doi:10.1016/j.molcel.2010.09.021. PMID:20932478
- Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callen E, Celeste A, Pagani M, Opravil S, De La Rosa-Velazquez IA, et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev 2008; 22:2048-61. doi:10.1101/gad.476008. PMID:18676810
- Wang Y, Khan A, Marks AB, Smith OK, Giri S, Lin YC, Creager R, MacAlpine DM, Prasanth KV, Aladjem MI, et al. Temporal association of ORCA/LRWD1 to late-firing origins during G1 dictates heterochromatin replication and organization. Nucleic Acids Res. 2017; 45:2490-502. doi:10.1093/nar/gkw1211. PMID:27924004
- Zhu B, Xiahou Z, Zhao H, Peng B, Xu X. MTHFR promotes heterochromatin maintenance. Biochem Biophys Res Commun. 2014;447:702-6. doi:10.1016/j.bbrc.2014.04.082. PMID:24769206
- Chabner BA, Roberts TG, Jr. Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5:65-72. doi:10.1038/nrc1529. PMID:15630416
- Yeh CC, Lai CY, Chang SN, Hsieh LL, Tang R, Sung FC, Lin YK. Polymorphisms of MTHFR C677T and A1298C associated with survival in patients with colorectal cancer treated with 5-fluorouracil-based chemotherapy. Int J Clin Oncol. 2017;22(3):484-493. doi:10.1007/s10147-016-1080-z. PMID:28044213.
- Yamada K, Strahler JR, Andrews PC, Matthews RG. Regulation of human methylenetetrahydrofolate reductase by phosphorylation. Proc Natl Acad Sci U S A. 2005;102:10454-9. doi:10.1073/pnas.0504786102. PMID:16024724
- Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol. 2009;10:265-75. doi:10.1038/nrm2653. PMID:19305416
- Elia AE, Cantley LC, Yaffe MB. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science. 2003;299:1228-31. doi:10.1126/science.1079079. PMID:12595692
- Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt-Dias M. Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol. 2014;15:433-52. doi:10.1038/nrm3819. PMID:24954208
- Guenther BD, Sheppard CA, Tran P, Rozen R, Matthews RG, Ludwig ML. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat Struct Biol. 1999;6:359-65. doi:10.1038/7594. PMID:10201405
- Bao ZS, Chen HM, Yang MY, Zhang CB, Yu K, Ye WL, Hu BQ, Yan W, Zhang W, Akers J, et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 2014;24:1765-73. doi:10.1101/gr.165126.113. PMID:25135958
- Archambault V, Lepine G, Kachaner D. Understanding the Polo Kinase machine. Oncogene. 2015;34:4799-807. doi:10.1038/onc.2014.451. PMID:25619835
- Hardy CF, Pautz A. A novel role for Cdc5p in DNA replication. Mol Cell Biol. 1996;16:6775-82. doi:10.1128/MCB.16.12.6775. PMID:8943332
- Tsvetkov L, Stern DF. Interaction of chromatin-associated Plk1 and Mcm7. J Biol Chem. 2005;280:11943-7. doi:10.1074/jbc.M413514200. PMID:15654075
- Stuermer A, Hoehn K, Faul T, Auth T, Brand N, Kneissl M, Putter V, Grummt F. Mouse pre-replicative complex proteins colocalise and interact with the centrosome. Eur J Cell Biol. 2007;86:37-50. doi:10.1016/j.ejcb.2006.09.002. PMID:17157410
- Song B, Liu XS, Davis K, Liu X. Plk1 phosphorylation of Orc2 promotes DNA replication under conditions of stress. Mol Cell Biol. 2011;31:4844-56. doi:10.1128/MCB.06110-11. PMID:21947279
- Wu ZQ, Liu X. Role for Plk1 phosphorylation of Hbo1 in regulation of replication licensing. Proc Natl Acad Sci U S A. 2008;105:1919-24. doi:10.1073/pnas.0712063105. PMID:18250300
- Lei M, Erikson RL. Plk1 depletion in nontransformed diploid cells activates the DNA-damage checkpoint. Oncogene. 2008;27:3935-43. doi:10.1038/onc.2008.36. PMID:18297112
- Park SH, Yu SE, Chai YG, Jang YK. CDK2-dependent phosphorylation of Suv39H1 is involved in control of heterochromatin replication during cell cycle progression. Nucleic Acids Res. 2014;42:6196-207. doi:10.1093/nar/gku263. PMID:24728993
- Wu S, Wang W, Kong X, Congdon LM, Yokomori K, Kirschner MW, Rice JC. Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev. 2010;24:2531-42. doi:10.1101/gad.1984210. PMID:20966048
- Zhou L, Tian X, Zhu C, Wang F, Higgins JM. Polo-like kinase-1 triggers histone phosphorylation by Haspin in mitosis. EMBO Rep. 2014;15:273-81. PMID:24413556
- Ghenoiu C, Wheelock MS, Funabiki H. Autoinhibition and Polo-dependent multisite phosphorylation restrict activity of the histone H3 kinase Haspin to mitosis. Mol Cell. 2013;52:734-45. doi:10.1016/j.molcel.2013.10.002. PMID:24184212
- Prioleau MN, MacAlpine DM. DNA replication origins-where do we begin? Genes Dev. 2016; 30:1683-97. doi:10.1101/gad.285114.116. PMID:27542827
- Gonzalo S, Garcia-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, Eguia R, Dean DC, Esteller M, Jenuwein T, et al. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat Cell Biol. 2005;7:420-8. doi:10.1038/ncb1235. PMID:15750587
- Manning AL, Yazinski SA, Nicolay B, Bryll A, Zou L, Dyson NJ. Suppression of genome instability in pRB-deficient cells by enhancement of chromosome cohesion. Mol Cell. 2014;53:993-1004. doi:10.1016/j.molcel.2014.01.032. PMID:24613344
- Zheng G, Yu H. Regulation of sister chromatid cohesion during the mitotic cell cycle. Sci China Life Sci. 2015;58:1089-98. doi:10.1007/s11427-015-4956-7. PMID:26511516
- Wu R, Wang Z, Zhang H, Gan H, Zhang Z. H3K9me3 demethylase Kdm4d facilitates the formation of pre-initiative complex and regulates DNA replication. Nucleic Acids Res. 2017;45:169-80. doi:10.1093/nar/gkw848. PMID:27679476
- Huang C, Cheng J, Bawa-Khalfe T, Yao X, Chin YE, Yeh ET. SUMOylated ORC2 Recruits a Histone Demethylase to Regulate Centromeric Histone Modification and Genomic Stability. Cell Rep. 2016;15:147-57. doi:10.1016/j.celrep.2016.02.091. PMID:27052177
- Li S, Swanson SK, Gogol M, Florens L, Washburn MP, Workman JL, Suganuma T. Serine and SAM Responsive Complex SESAME Regulates Histone Modification Crosstalk by Sensing Cellular Metabolism. Mol Cell. 2015;60:408-21. doi:10.1016/j.molcel.2015.09.024. PMID:26527276
- Suganuma T, Workman JL. Histone modification as a reflection of metabolism. Cell Cycle. 2016;15:481-2. doi:10.1080/15384101.2015.1128190. PMID:26726751
- Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science. 2013;339:222-6. doi:10.1126/science.1226603. PMID:23118012
- Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, Aburatani H, Kume K, Endo F, Kume S. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 2014;19:780-94. doi:10.1016/j.cmet.2014.03.017. PMID:24746804
- Milutinovic S, Zhuang Q, Niveleau A, Szyf M. Epigenomic stress response. Knockdown of DNA methyltransferase 1 triggers an intra-S-phase arrest of DNA replication and induction of stress response genes. J Biol Chem. 2003;278:14985-95.
- Unterberger A, Andrews SD, Weaver IC, Szyf M. DNA methyltransferase 1 knockdown activates a replication stress checkpoint. Mol Cell Biol. 2006; 26:7575-86. doi:10.1128/MCB.01887-05. PMID:17015478
- Strickland M, Stoll EA. Metabolic Reprogramming in Glioma. Front Cell Dev Biol. 2017;5:43. doi:10.3389/fcell.2017.00043. PMID:28491867
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013; 340:857-61. doi:10.1126/science.1232245. PMID:23539183
- Sharma V, Malgulwar PB, Purkait S, Patil V, Pathak P, Agrawal R, Kulshreshtha R, Mallick S, Julka PK, Suri A, et al. Genome-wide ChIP-seq analysis of EZH2-mediated H3K27me3 target gene profile highlights differences between low- and high-grade astrocytic tumors. Carcinogenesis 2017;38:152-61. PMID:27993893
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med. 2017;23:483-92. doi:10.1038/nm.4293. PMID:28263309
- Li J, Wang J, Hou W, Jing Z, Tian C, Han Y, Liao J, Dong MQ, Xu X. Phosphorylation of Ataxin-10 by polo-like kinase 1 is required for cytokinesis. Cell Cycle. 2011;10:2946-58. doi:10.4161/cc.10.17.15922. PMID:21857149
- Tian J, Tian C, Ding Y, Li Z, Geng Q, Xiahou Z, Wang J, Hou W, Liao J, Dong MQ, et al. Aurora B-dependent phosphorylation of Ataxin-10 promotes the interaction between Ataxin-10 and Plk1 in cytokinesis. Sci Rep. 2015;5:8360. doi:10.1038/srep08360. PMID:25666058
- Li J, Liu X, Liao J, Tian J, Wang J, Wang X, Zhang J, Xu X. MYPT1 sustains centromeric cohesion and the spindle-assembly checkpoint. J Genet Genomics. 2013;40:575-8. doi:10.1016/j.jgg.2013.08.005. PMID:24238611