
ABSTRACT
Our genetic information is organized into chromatin, which consists of histones and proteins involved in regulating DNA compaction, accessibility and function. Chromatin is decorated by histone modifications, which provide signals that coordinate DNA-based processes including transcription and DNA damage response (DDR) pathways. A major signal involved in these processes is acetylation, which when attached to lysines within proteins, including histones, can be recognized and read by bromodomain-containing proteins. We recently identified the bromodomain protein ZMYND8 (also known as RACK7 and PRKCBP1) as a critical DNA damage response factor involved in regulating transcriptional responses and DNA repair activities at DNA double-strand breaks. Other studies have further defined the molecular details for how ZMYND8 interacts with chromatin and other chromatin modifying proteins to exert its DNA damage response functions. ZMYND8 also plays essential roles in regulating transcription during normal cellular growth, perturbation of which promotes cellular processes involved in cancer initiation and progression. In addition to acetylation, histone methylation and demethylase enzymes have emerged as important regulators of ZMYND8. Here we discuss our current understanding of the molecular mechanisms that govern ZMYND8 function within chromatin, highlighting the importance of this protein for genome maintenance both during the DDR and in cancer.
Introduction
Genetic information is stored in DNA, access to which informs all cellular processes. The integrity of this information is vital for genome maintenance, which ensures cellular and organismal homeostasis. DNA is under constant threat of mutation as genotoxic agents from endogenous and exogenous sources react with DNA to cause errors including damaged bases and breaks in either one or two of the DNA strands. To identify and repair these DNA lesions, cells employ DNA damage response (DDR) pathways [Citation1,Citation2]. Numerous unique DDR pathways exist in cells, capable of recognizing various DNA lesions and mounting a proper response. As DNA is bound by histone proteins and organized into chromatin, chromatin-based DDR pathways are essential for orchestrating the repair of DNA lesions [Citation3–9]. The importance of DDR pathways are highlighted by the various diseases associated with DDR defects, including neurodegenerative disorders, immune deficiencies and cancer [Citation1–Citation10]. Thus, understanding the interplay between chromatin and the DDR is fundamental for obtaining mechanistic details of DDR activities in both normal and diseased contexts.
Histone proteins are highly modified by post-translational modifications (PTMs) including phosphorylation, methylation, and acetylation, which regulate chromatin structure and function [Citation11–15]. One mechanism whereby PTMs regulate chromatin structure and function is by promoting chromatin binding of “reader” proteins that contain PTM binding domains [Citation12,Citation16–18]. DNA damage results in dynamic changes to histone PTMs and the association of chromatin proteins at DNA damage sites [Citation19,Citation20], For example, the histone variant H2AX is phosphorylated on its C-terminus to form γH2AX at DNA double-strand breaks (DSBs) [Citation21]. γH2AX is recognized by the twin BRCT-domains of the mediator protein MDC1 [Citation22], which binds to damage sites through this interaction to promote various downstream DNA damage signaling and repair events. There are numerous other examples of how DNA damage factors bind to histone PTMs, including 53BP1 which acts as a bivalent histone PTM reader that recognizes both methylated and ubiquitylated histones [Citation23,Citation24], Given that PTMs can alter the structure and function of chromatin, DNA damage has the potential to not only alter DNA sequences but also disrupt the epigenome and processes reliant on histone PTMs, which include DNA-based events including replication, transcription and DNA repair. Interestingly, in addition to genome instability being a hallmark of cancer, epigenetic changes and mutations within chromatin regulatory proteins are also prevalent in this disease, suggesting a potential causal link between these processes in the initiation and progression of cancer [Citation25].
Emerging evidence has highlighted the importance of acetylation reader proteins in the DDR and cancer [Citation7,Citation26]. Acetylated lysine residues are bound by several different domains, including the bromodomain (BRD), which constitutes the major acetylation binding protein family in human cells. For example, there are 42 human proteins that contain one or more BRDs, the majority of which bind acetylated proteins [Citation27,Citation28], We identified one third of all human BRD proteins as being dynamically relocalized in response to DNA damage [Citation29], which together with other studies has established the importance of these chromatin proteins in the DDR [Citation7]. BRD proteins are mutated and/or aberrantly expressed in cancer, which has motivated targeting of BRD proteins as potential cancer therapies [Citation30–32]. These efforts are exemplified by the targeting of BRD4 by JQ1 and I-BET inhibitors [Citation33,Citation34], which have displayed remarkable effects on inhibiting cancer cell phenotypes in several pre-clinical studies [Citation35,Citation36], Thus, BRD proteins have the potential to function both in the DDR and in cancer. For example, mutations or inhibition of BRD proteins may alter cancer-related phenotypes while also creating DDR-deficient cells. This situation may be particularly relevant in scenarios where DNA damaging agents and epigenetic drugs, including as combination therapies, are employed as cancer treatments. Understanding the relationship between DDR and cancer functions of BRD proteins may help guide the use of therapies targeting BRD proteins and/or the DDR in cancer-relevant settings.
The Bromodomain protein ZMYND8
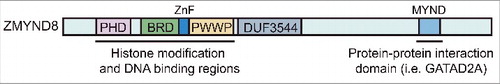
The bromodomain protein ZMYND8 (zinc finger MYND (Myeloid, Nervy and DEAF-1)-type containing 8) has recently been shown to act as an important chromatin factor in both the DDR and in cancer [Citation29,Citation37–Citation42]. ZMYND8 was first identified as an activated protein-kinase-C-binding protein (PRKCBP1) [Citation43]. Domain structure analyses identified a triple PHD-BRD-PWWP chromatin-binding module in its N-terminus and a C-terminal MYND domain (). Proteomic studies identified ZMYND8 as an interacting factor of several complexes involved in transcription regulation [Citation40,Citation41,Citation44–46]. More recent studies from our group and others have demonstrated ZMYND8 as an essential factor linking chromatin with DDR activities [Citation29,Citation40–42,Citation47]. Interestingly, several other studies have revealed a tumor suppressive function for ZMYND8 [Citation38,Citation39]. Here we provide a perspective on these recently identified functions of ZMYND8, which highlights its involvement in chromatin, DNA damage and cancer.
Figure 1. Bromodomain protein ZMYND8. Domain structure and functional regions of ZMYND8 are shown. PHD – Pleckstrin homology domain; BRD – Bromodomain; PWWP – Pro-Trp-Trp-Pro; ZnF – Zinc Finger domain; DUF – domain of unknown function; MYND – myeloid, Nervy, and DEAF-1 domain
ZMYND8 in the DNA damage response
Identification of ZMYND8 as a DNA damage response factor came from a screen we performed for human BRD proteins that relocalized upon laser damage in human cells [Citation29]. This study further revealed ZMYND8 as a crucial DDR factor that interacts with the NuRD chromatin remodeling complex to promote DNA damage-induced transcriptional silencing and repair of DNA double-strand breaks (DSBs) by homologous recombination (HR) [Citation29]. Several additional studies have since expanded our understanding of the molecular mechanisms utilized by ZMYND8 to function within the DDR [Citation40–Citation42,Citation47].
The first evidence that ZMYND8 participated in the DNA damage response was provided by its ability to rapidly accumulate at laser-induced DNA damage sites [Citation29]. The presence of a PHD-BRD-PWWP domain structure in ZMYND8 suggested that this protein has the potential to bind chromatin (). Analyses of ZMYND8, both in vitro and in cells, revealed that the BRD domain of ZMYND8 could recognize N-terminal histone H4 acetylations (H4ac) [Citation29,Citation37], which are marks mediated by the HAT TIP60 and are involved in the DDR [Citation48,Citation49]. A more comprehensive biochemical and structural study from the Filippakopoulos group and colleagues demonstrated that the N-terminal PHD-BRD-PWWP domains of ZMYND8 engaged chromatin through multivalent interactions [Citation40]. In addition to recognizing multiple histone PTMs including H3K14 acetylation, this triple module also participated in DNA binding. Deletion of any one of these chromatin reader domains resulted in a perturbation of damage-association of ZMYND8 [Citation40]. In addition, the HAT TIP60 is required for ZMYND8 recruitment to damage sites, which is consistent with the notion that acetylation signaling modulates ZMYND8 damage associations with chromatin [Citation29]. However, these requirements may be context specific as the human ZMYND8 gene encodes more than 20 isoforms and isoform 17 (NP_001268700), unlike isoform 1 (NP_898868), was shown to accumulate at damage sites independently of the BRD [Citation29,Citation41]. Although additional studies are needed to delineate the complex regulation of this gene, these studies established that ZMYND8 associates with chromatin using multivalent interactions, including histone PTMs and DNA, which are required to orchestrate the interactions between ZMYND8 and damaged chromatin.
To garner insights into the function of ZMYND8 at damage sites, affinity based mass spectrometry approaches have provided important information that has revealed mechanistically how ZMYND8 participates in DNA damage signaling and repair. We and others identified the NuRD chromatin-remodeling complex as a primary interactor of ZMYND8 [Citation29,Citation40,Citation41,Citation45,Citation46]. ZMYND8 also interacts with a complex of two or three other zinc finger proteins, depending on the study [Citation29,Citation40,Citation41, Citation44–46]. These zinc finger proteins ZNF532, ZNF592 and ZNF687 (referred to as the Z3 module with ZMYND8) [Citation44], participate in the regulation of transcription and multiple other cellular functions [Citation44,Citation45]. Structure function analyses identified the C-terminal MYND domain as the region of ZMYND8 responsible for interacting with NuRD () [Citation29]. Detailed biochemical analyses performed by Spruijt et al. revealed that the MYND domain of ZMYND8 specifically interacts with the PPPLΦ motif of NuRD complex factor GATAD2A and is also responsible for linking the Z3 module to the NuRD complex ((A)) [Citation41]. The NuRD complex contains both GATAD2A and a related factor GATAD2B, although the PPPLΦ motif is only found in GATAD2A. Interestingly, ZMYND8 was shown to only interact with the NuRD complex containing GATAD2A, even though both GATAD2A- and GATAD2B-containing NuRD complexes are recruited to DNA damage sites. ZNF592 and ZNF687 are also rapidly recruited to laser damage sites [Citation41,Citation42]. Taken together, these studies identified several ZMYND8-interacting factors, including the NuRD complex, which associate with DNA damage sites.
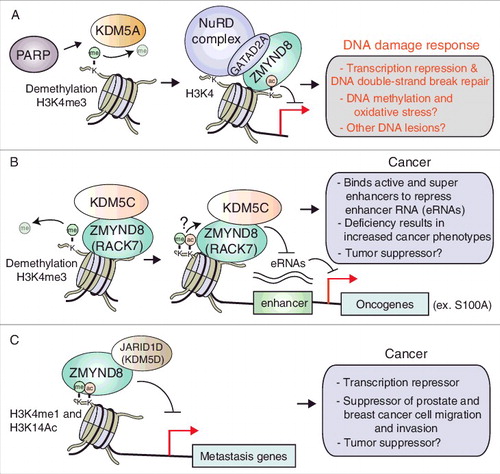
Figure 2. Involvement of ZMYND8 and demethylases in the DDR and cancer. (A) KDM5A-ZMYND8-NuRD pathway regulate transcription and promotes repair at DNA DSBs. (B) Enhancer RNA and oncogene transcriptional regulation by ZMYND8-KDM5C in breast cancer. (C) ZMYND8 cooperates with KDM5D to suppress transcription of metastasis genes in prostate cancer.
Numerous studies had previously established that the NuRD complex associates with DNA damage to facilitate the DDR [Citation50–55]. However, molecular insights were lacking for how the NuRD complex recognizes damaged DNA within chromatin. Given the ability of ZMYND8 to interact with chromatin at DNA lesions and its interactions with the NuRD complex, ZMYND8 represented a good candidate for linking the NuRD chromatin-remodeling complex to DNA damage sites. Indeed, cells deficient for ZMYND8 either by RNA interference or gene-editing by CRISPR-Cas9 displayed defective recruitment of the NuRD complex, including the components GATAD2A and CHD4, to DNA damage sites [Citation29,Citation41,Citation47], As has been demonstrated previously for the NuRD complex [Citation52–Citation54], recruitment of ZMYND8 requires poly(ADP-ribose) (PAR) as treatment of cells with a poly(ADP-ribose) polymerase (PARP) inhibitor or depletion of PARP1 by siRNA blocks ZMYND8 accumulation at damage sites [Citation41,Citation42]. Interestingly, ZNF687 appears to promote CHD4 recruitment but not ZMYND8 accumulation at DNA damage sites, which suggests that zinc finger proteins within the Z3 module may function downstream of ZMYND8 to promote NuRD accrual at damage sites [Citation41]. Thus, these studies establish that ZMYND8 is required for NuRD chromatin-remodeling complex recruitment to DNA damage.
The reliance on ZMYND8 for the recruitment of the NuRD chromatin-remodeling complex at DNA damage sites suggested that these factors shared functions within the DDR. Indeed, like the NuRD complex, ZMYND8 promotes the repair of DSBs by homologous recombination [Citation29,Citation41,Citation54]. The NuRD complex components CHD4 and ZMYND8 are recruited to DNA damage sites within chromatin that is being actively transcribed, as inhibition of transcription blocks their recruitment to damage sites [Citation29]. However, loss of CHD4 or ZMYND8 results in an inability to repress transcription that is proximal to DSBs [Citation29,Citation56]. Additionally, TIP60 is also required to repress transcription [Citation29]. Transcribing chromatin is acetylated but TIP60 may act to increase specific acetylations at damage sites to promote ZMYND8 recruitment. TIP60 could play additional roles in facilitating ZMYND8-NuRD DDR pathway including in DNA damage signaling and/or chromatin remodeling. Thus, the ability to regulate transcription by NuRD chromatin-remodeling complex and ZMYND8 appears to be engaged at DNA damage sites to repress transcription and facilitate DNA repair. We hypothesize that these activities are needed so that faithful repair by homologous recombination can occur without conflicts with other DNA-templated processes including transcription. Inhibiting transcription may be particularly important for HR as this repair pathway involves resecting DSBs to create long single-strand overhangs that can invade another DNA duplex during homology search, an essential process for HR. One could imagine that the presence of transcription associated factors and/or RNA:DNA hybrids in the vicinity of a DSB could interfere with HR by impeding DNA end-resection as well as homology search. However, R-loops have also been shown to promote HR repair either directly or by triggering DSB formation [Citation57,Citation58], although others have observed HR defects associated with R-loop formation [Citation59]. Given these diverse observations on the interplay between R-loops and HR, it is likely that these outcomes will be context specific. For example, R-loops are thought to induce DSBs from transcription-replication conflicts or by single-strand DNA (ssDNA) gaps at processed R-loops that are converted to DSBs during S-phase. DSBs caused by a DNA damaging agent independently from R-loops may promote different responses than R-loop dependent DSBs. More work is necessary to understand the complex relationship between transcription and DSB repair. Regardless, many observations point to the ZMYND8-NuRD axis as playing a prominent role in repairing DSBs associated with actively transcribing regions of the genome.
Actively transcribed chromatin is associated with specific histone modification patterns including the presence of trimethylated histone H3 at lysine 4 (H3K4me3) [Citation60]. Previous studies using modified histone peptides to identify chromatin-associated complexes have revealed that H3K4me3 perturbs the binding of NuRD and ZMYND8 to chromatin [Citation45,Citation61,Citation62]. Given that the ZMYND8-NuRD complex is recruited to transcriptionally active sites upon DNA damage, which are enriched with H3K4me3, these findings hinted at the potential need for H3K4me3 demethylation upon DNA damage to allow ZMYND8-NuRD to interact with H3K4me3-occupied chromatin. We provided answers to this question by our identification of KDM5A, a histone demethylase that acts on H3K4me3, as an important DNA damage factor that regulates the recruitment of the ZMYND8-NuRD complex to DSBs [Citation42]. KDM5A was a good candidate given that we identified it as a putative ZMYND8 interacting protein by proteomic analysis of ZMYND8 purified complexes [Citation29,Citation42]. Like ZMYND8-NuRD, KDM5A is recruited to laser-induced DNA damage sites in a PARP-dependent manner [Citation42]. KDM5A appears to act upstream of ZMYND8-NuRD, which is based on the findings that KDM5A is recruited to sites of damage before ZMYND8-NuRD and the association of KDM5A with damage sites is independent from ZMYND8-NuRD [Citation42]. ZMYND8 and KDM5A have been reported to interact, although the functional significance of these findings in the DDR is unclear. However, consistent with the requirement of H3K4me3 demethylation by KDM5A to allow chromatin association of ZMYND8-NuRD, a reduction of H3K4me3 is observed upon DNA damage [Citation42,Citation63]. Functionally, cells lacking KDM5A exhibit impaired transcriptional repression at DSBs and a deficiency in HR repair, which is similar to DDR defects associated with ZMYND8-NuRD impairment [Citation42]. Taken together, these studies demonstrate the necessity of remodeling chromatin by KDM5A by demethylating H3K4me3 to allow ZMYND8-NuRD to bind damaged chromatin to repress transcription and promote HR ((A)).
The NuRD chromatin-remodeling complex appears to play important roles at other damage sites that are independent from ZMYND8. For example, a GATAD2B-containing NuRD complex is recruited to DNA damage sites independently from ZMYND8 [Citation41]. Another study identified the zinc finger protein ZNF827 as a requisite factor involved in the recruitment of NuRD to telomeres to promote HR-mediated alternative lengthening of telomeres (ALT) in these cells [Citation64]. Upon oxidative damage, the excision repair protein OGG1 targets the NuRD complex to DNA damage sites to promote DNA methylation-mediated gene silencing at CpG islands of tumor suppressor genes (TSG) [Citation47]. Collectively, these studies highlight the ability of the NuRD chromatin-remodeling complex to multitask as a genome maintenance factor across diverse chromatin and genomic lesions through interactions with ZMYND8 and other NuRD co-factors. Important questions remain for how these interactions between the NuRD complex and these accessory factors, including ZMYND8, are controlled to allow normal gene expression regulation as well as various transcriptional and repair activities to occur at different DNA lesions across diverse chromatin environments.
ZMYND8 in cancer
In addition to its involvement in the DDR, ZMYND8 has also been found to play important roles in cancer. In line with this idea, aberrant expression or mutations of ZMYND8 have been identified in a wide range of human cancers, including breast, prostate, ovarian carcinoma, and acute erythroid leukemia [Citation38,Citation39,Citation65]. The Cancer Genome Atlas Research Network identified ZMYND8 within a recurrent somatic copy number alteration in high-grade serous ovarian cancer [Citation66]. Another study reported a translocation between ZMYND8 and RELA in a patient sample with acute erythroid leukemia, although contribution of this fusion protein in this cancer type is yet to be explored [Citation65]. Mechanistic studies of ZMYND8 in cancer have started to emerge, which have highlighted its vital function as a transcriptional co-repressor in cancer suppression [Citation38,Citation39].
In addition to collaborating with KDM5A to promote DDR processes, ZMYND8 also partners with other members of the H3K4me3-specific KDM5 family, including KDM5C and KDM5D, to regulate transcription and suppress cancer-associated phenotypes [Citation38,Citation39]. In breast cancer cells, ZMYND8 participates with KDM5C to “fine-tune” the expression of enhancer RNAs (eRNAs), thus regulating the activity of enhancers ((B)) [Citation38]. ZMYND8 occupies many active enhancers including super enhancers, which are enriched in the histone marks H3K4me1 and H3K27ac. ZMYND8 was shown to promote the recruitment of KDM5C to super enhancer regions, which is required to maintain low H3K4me3 levels resulting in the repression of enhancer activity. How ZMYND8 promotes the localization of KDM5C to these sites is unknown, although it is possible that ZMYND8 binds methyl and acetyl histone marks associated with these regions ((B)). Loss of ZMYND8 or KDM5C results in an increased level of H3K4me3, eRNA levels and a concomitant increase in gene expression of neighboring genes. Cells deficient for these genes also display enhanced tumor-promoting phenotypes including cell migration and invasion, as well as tumor growth in xenograft mouse tumor models [Citation38]. ZMYND8 and KDM5C regulate many genes, including the oncogenic family of S100A proteins ((B)), which were shown to be required, in part, for cancer-related phenotypes associated with loss of ZMYND8 or KDM5C [Citation38].
ZMYND8 has also been shown to suppress similar cancer-related phenotypes in prostate cancer cells. A recent study demonstrated that ZMYND8 and KDM5D function as transcriptional co-repressors involved in regulating metastasis-linked genes in prostate cancer ((C)) [Citation39]. ZMYND8 interacts with KDM5D using an undefined central region of the protein. This interaction is required to recruit KDM5D to chromatin regions highly enriched with H3K4me1 and H3K14ac marks ((C)). These dual marks can be recognized by the PHD-BRD domains of ZMYND8, suggesting that this signature is bound by ZMYND8. Genome-wide studies identified genes exhibiting this histone signature that were co-occupied by ZMYND8 and KDM5D. Loss of ZMYND8 or KDM5D resulted in expression of these genes, leading to elevated cell migration and high invasiveness of prostate cancer cells both in cell-based and mouse xenograft tumor models [Citation39].
ZMYND8 also appears to be a target gene for all-trans-retinoic-acid (ATRA), which is involved in ATRA-mediated inhibition of cancer cell proliferation and invasion [Citation37,Citation67]. Unlike other studies [Citation39,Citation40], ZMYND8 was shown to utilize its PHD-BRD-PWWP domains to specifically recognize di-methylation on K36 in histone H3.1, as well as H4K16ac, which is required for ZMYND8 to modulate the expression of other ATRA target genes [Citation37]. This group published additional studies identifying proliferation and cancer associated genes regulated by ZMYND8, including Ki67 and PCNA [Citation67], as well as genes mediating epithelial-mesenchymal transition (EMT), an important mechanism of cellular invasion [Citation68]. Consistent with ZMYND8 acting as a tumor suppressor, overexpression of ZMYND8 in an invasive breast cancer cell line reduced the tumor promoting ability of these cells in mice and human breast cancer tissues exhibiting reduced ZMYND8 expression [Citation68], which is consistent with other studies reporting a tumor suppressive function of ZMYND8 in different cancer types [Citation38,Citation39]. Taken together, these studies have revealed ZMYND8 as an important epigenetic regulator involved in tumor suppression in various cancers.
Concluding remarks
Here we have discussed the involvement of ZMYND8 in the DDR and in cancer. Researchers have made rapid progress in identifying the multiple mechanisms by which ZMYND8 interacts with chromatin and other proteins to regulate transcription and promote repair of damaged DNA as well as control transcription to suppress various cancers [Citation29,Citation37–Citation42]. Although these studies have revealed the importance of ZMYND8 in genetic and epigenetic regulation, many questions still remain. For example, PARP signaling has been shown to be required for the damage recruitment of the KDM5A-ZMYND8-NuRD axis [Citation41,Citation42,Citation52,Citation53]. However, mechanistic details for how PARP signaling promotes damage recognition and chromatin binding of these factors is lacking. Multiple studies have demonstrated a requirement for KDM5A-ZMYND8-NuRD in promoting HR [Citation29,Citation41,Citation42,Citation54], but molecular details for where in this multi-step process this pathway acts remain to be determined. It is tempting to speculate that although transcriptional repression is important in this pathway, additional functions that regulate other steps in the HR pathway are likely to also require ZMYND8 and NuRD. Mechanistically defining the individual steps of HR that require the activities of KDM5A-ZMYND8-NuRD, as well as delineating the chromatin environment and activities associated with this pathway, would provide critical insights into understanding how this pathway promotes genome integrity.
ZMYND8, as well as NuRD and KDM5A, are mutated or display aberrant expression in several cancers [Citation38,Citation39,Citation65,Citation69–73] In addition to transcriptional regulation, cells defective for KDM5A-ZMYND8-NuRD pathway may promote cancer initiation and progression through the loss of genome integrity function, a known hallmark of cancer. Targeting epigenetic and DNA repair pathways are strategies for cancer therapeutics [Citation7,Citation74–78] As KDM5A-ZMYND8-NuRD sit at the intersection of epigenetics and DNA repair, these pathways may shed light on the interplay between the DDR and cancer epigenetics, which may inform the use and development of cancer therapies involving these pathways.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgements
We thank members of the Miller lab for useful discussions and comments on the review. The K.M.M. laboratory is supported by the NIH National Cancer Institute (R01CA198279 and RO1CA201268) and the American Cancer Society (RSG-16-042-01-DMC).
Additional information
Funding
References
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi:10.1038/nature08467. PMID:19847258
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi:10.1016/j.molcel.2010.09.019. PMID:20965415
- Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 2011;13:1161–9. doi:10.1038/ncb2344. PMID:21968989
- Miller KM, Jackson SP. Histone marks: repairing DNA breaks within the context of chromatin. Biochem Soc Trans. 2012;40:370–6. doi:10.1042/BST20110747. PMID:22435814
- Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell. 2013;49:795–807. doi:10.1016/j.molcel.2013.01.017. PMID:23416108
- Gong F, Miller KM. Mammalian DNA repair: HATs and HDACs make their mark through histone acetylation. Mutat Res. 2013;750:23–30. doi:10.1016/j.mrfmmm.2013.07.002. PMID:23927873
- Gong F, Chiu LY, Miller KM. Acetylation reader proteins: linking acetylation signaling to genome maintenance and cancer. PLoS Genet. 2016;12:e1006272. doi:10.1371/journal.pgen.1006272. PMID:27631103
- Agarwal P, Miller KM. The nucleosome: orchestrating DNA damage signaling and repair within chromatin. Biochem Cell Biol. 2016;94:381–95. doi:10.1139/bcb-2016-0017. PMID:27240007
- Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–33. doi:10.1101/gad.2021311. PMID:21363960
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–8. doi:10.1038/nrm2858. PMID:20177397
- Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annu Rev Biochem. 2011;80:473–99. doi:10.1146/annurev-biochem-061809-175347. PMID:21529160
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi:10.1016/j.cell.2007.02.005. PMID:17320507
- Weake VM, Workman JL. Histone ubiquitination: triggering gene activity. Mol Cell. 2008;29:653–63. doi:10.1016/j.molcel.2008.02.014. PMID:18374642
- Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–99. doi:10.1146/annurev.genet.032608.103928. PMID:19886812
- Polo SE, Almouzni G. DNA damage leaves its mark on chromatin. Cell Cycle. 2007;6:2355–9. doi:10.4161/cc.6.19.4756. PMID:17703111
- Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi:10.1146/annurev.biochem.76.052705.162114. PMID:17362198
- Ruthenburg AJ, Li H, Patel DJ, et al. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8:983–94. doi:10.1038/nrm2298. PMID:18037899
- Musselman CA, Lalonde ME, Cote J, et al. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19:1218–27. doi:10.1038/nsmb.2436. PMID:23211769
- Xu Y, Price BD. Chromatin dynamics and the repair of DNA double strand breaks. Cell Cycle. 2011;10:261–7. doi:10.4161/cc.10.2.14543. PMID:21212734
- Seeber A, Dion V, Gasser SM. Remodelers move chromatin in response to DNA damage. Cell Cycle. 2014;13:877–8. doi:10.4161/cc.28200. PMID:24552812
- Rogakou EP, Pilch DR, Orr AH, et al. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi:10.1074/jbc.273.10.5858. PMID:9488723
- Stucki M, Clapperton JA, Mohammad D, et al. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–26. doi:10.1016/j.cell.2005.09.038. PMID:16377563
- Botuyan MV, Lee J, Ward IM, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–73. doi:10.1016/j.cell.2006.10.043. PMID:17190600
- Fradet-Turcotte A, Canny MD, Escribano-Diaz C, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–4. doi:10.1038/nature12318. PMID:23760478
- Dawson MA, Kouzarides T, Huntly BJ. Targeting epigenetic readers in cancer. N Engl J Med. 2012;367:647–57. doi:10.1056/NEJMra1112635. PMID:22894577
- Fujisawa T, Filippakopoulos P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol. 2017;18:246–62. doi:10.1038/nrm.2016.143. PMID:28053347
- Filippakopoulos P, Picaud S, Mangos M, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–31. doi:10.1016/j.cell.2012.02.013. PMID:22464331
- Filippakopoulos P, Knapp S. The bromodomain interaction module. FEBS Lett. 2012;586:2692–704. doi:10.1016/j.febslet.2012.04.045. PMID:22710155
- Gong F, Chiu LY, Cox B, et al. Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 2015;29:197–211. doi:10.1101/gad.252189.114. PMID:25593309
- Muller S, Filippakopoulos P, Knapp S. Bromodomains as therapeutic targets. Expert Rev Mol Med. 2011;13:e29. doi:10.1017/S1462399411001992. PMID:21933453
- Barbieri I, Cannizzaro E, Dawson MA. Bromodomains as therapeutic targets in cancer. Brief Funct Genomics. 2013;12:219–30. doi:10.1093/bfgp/elt007. PMID:23543289
- Zhang G, Smith SG, Zhou MM. Discovery of Chemical Inhibitors of Human Bromodomains. Chem Rev. 2015;115:11625–68. doi:10.1021/acs.chemrev.5b00205. PMID:26492937
- Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi:10.1038/nature09504. PMID:20871596
- Dawson MA, Prinjha RK, Dittmann A, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–33. doi:10.1038/nature10509. PMID:21964340
- Andrieu G, Belkina AC, Denis GV. Clinical trials for BET inhibitors run ahead of the science. Drug Discov Today Technol. 2016;19:45–50. doi:10.1016/j.ddtec.2016.06.004. PMID:27769357
- Boi M, Gaudio E, Bonetti P, et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted Drugs. Clin Cancer Res. 2015;21:1628–38. doi:10.1158/1078-0432.CCR-14-1561. PMID:25623213
- Adhikary S, Sanyal S, Basu M, et al. Selective Recognition of H3.1K36 Dimethylation/H4K16 Acetylation Facilitates the Regulation of All-trans-retinoic Acid (ATRA)-responsive Genes by Putative Chromatin Reader ZMYND8. J Biol Chem. 2016;291:2664–81. doi:10.1074/jbc.M115.679985. PMID:26655721
- Shen H, Xu W, Guo R, et al. Suppression of Enhancer Overactivation by a RACK7-Histone Demethylase Complex. Cell. 2016;165:331–42. doi:10.1016/j.cell.2016.02.064. PMID:27058665
- Li N, Li Y, Lv J, et al. ZMYND8 Reads the Dual Histone Mark H3K4me1-H3K14ac to Antagonize the Expression of Metastasis-Linked Genes. Mol Cell. 2016;63:470–84. doi:10.1016/j.molcel.2016.06.035. PMID:27477906
- Savitsky P, Krojer T, Fujisawa T, et al. Multivalent Histone and DNA Engagement by a PHD/BRD/PWWP Triple Reader Cassette Recruits ZMYND8 to K14ac-Rich Chromatin. Cell Rep. 2016;17:2724–37. doi:10.1016/j.celrep.2016.11.014. PMID:27926874
- Spruijt CG, Luijsterburg MS, Menafra R, et al. ZMYND8 Co-localizes with NuRD on Target Genes and Regulates Poly(ADP-Ribose)-Dependent Recruitment of GATAD2A/NuRD to Sites of DNA Damage. Cell Rep. 2016;17:783–98. doi:10.1016/j.celrep.2016.09.037. PMID:27732854
- Gong F, Clouaire T, Aguirrebengoa M, et al. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J Cell Biol. 2017;216:1959–74. doi:10.1083/jcb.201611135. PMID:28572115
- Fossey SC, Kuroda S, Price JA, et al. Identification and characterization of PRKCBP1, a candidate RACK-like protein. Mamm Genome. 2000;11:919–25. doi:10.1007/s003350010174. PMID:11003709
- Malovannaya A, Lanz RB, Jung SY, et al. Analysis of the human endogenous coregulator complexome. Cell. 2011;145:787–99. doi:10.1016/j.cell.2011.05.006. PMID:21620140
- Eberl HC, Spruijt CG, Kelstrup CD, et al. A map of general and specialized chromatin readers in mouse tissues generated by label-free interaction proteomics. Mol Cell. 2013;49:368–78. doi:10.1016/j.molcel.2012.10.026. PMID:23201125
- Nishibuchi G, Shibata Y, Hayakawa T, et al. Physical and functional interactions between the histone H3K4 demethylase KDM5A and the nucleosome remodeling and deacetylase (NuRD) complex. J Biol Chem. 2014;289:28956–70. doi:10.1074/jbc.M114.573725. PMID:25190814
- Xia L, Huang W, Bellani M, et al. CHD4 Has Oncogenic Functions in Initiating and Maintaining Epigenetic Suppression of Multiple Tumor Suppressor Genes. Cancer Cell. 2017;31:653–68 e7. doi:10.1016/j.ccell.2017.04.005.
- Sun Y, Jiang X, Price BD. Tip60: connecting chromatin to DNA damage signaling. Cell Cycle. 2010;9:930–6. doi:10.4161/cc.9.5.10931. PMID:20160506
- Tang J, Cho NW, Cui G, et al. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol. 2013;20:317–25. doi:10.1038/nsmb.2499. PMID:23377543
- Larsen DH, Poinsignon C, Gudjonsson T, et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J Cell Biol. 2010;190:731–40. doi:10.1083/jcb.200912135. PMID:20805324
- Smeenk G, Wiegant WW, Vrolijk H, et al. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J Cell Biol. 2010;190:741–9. doi:10.1083/jcb.201001048. PMID:20805320
- Chou DM, Adamson B, Dephoure NE, et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A. 2010;107:18475–80. doi:10.1073/pnas.1012946107. PMID:20937877
- Polo SE, Kaidi A, Baskcomb L, et al. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010;29:3130–9. doi:10.1038/emboj.2010.188. PMID:20693977
- Pan MR, Hsieh HJ, Dai H, et al. Chromodomain helicase DNA-binding protein 4 (CHD4) regulates homologous recombination DNA repair, and its deficiency sensitizes cells to poly(ADP-ribose) polymerase (PARP) inhibitor treatment. J Biol Chem. 2012;287:6764–72. doi:10.1074/jbc.M111.287037. PMID:22219182
- Li DQ, Kumar R. Mi-2/NuRD complex making inroads into DNA-damage response pathway. Cell Cycle. 2010;9:2071–9. doi:10.4161/cc.9.11.11735. PMID:20505336
- Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, et al. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2010;141:970–81. doi:10.1016/j.cell.2010.04.038. PMID:20550933
- Ohle C, Tesorero R, Schermann G, et al. Transient RNA-DNA Hybrids Are Required for Efficient Double-Strand Break Repair. Cell. 2016;167:1001–13 e7. doi:10.1016/j.cell.2016.10.001.
- Gaillard H, Aguilera A. Transcription as a Threat to Genome Integrity. Annu Rev Biochem. 2016;85:291–317. doi:10.1146/annurev-biochem-060815-014908. PMID:27023844
- Aronica L, Kasparek T, Ruchman D, et al. The spliceosome-associated protein Nrl1 suppresses homologous recombination-dependent R-loop formation in fission yeast. Nucleic Acids Res. 2016;44:1703–17. doi:10.1093/nar/gkv1473. PMID:26682798
- Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi:10.1016/j.cell.2007.05.009. PMID:17512414
- Nishioka K, Chuikov S, Sarma K, et al. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002;16:479–89. doi:10.1101/gad.967202. PMID:11850410
- Zegerman P, Canas B, Pappin D, et al. Histone H3 lysine 4 methylation disrupts binding of nucleosome remodeling and deacetylase (NuRD) repressor complex. J Biol Chem. 2002;277:11621–4. doi:10.1074/jbc.C200045200. PMID:11850414
- Li X, Liu L, Yang S, et al. Histone demethylase KDM5B is a key regulator of genome stability. Proc Natl Acad Sci U S A. 2014;111:7096–101. doi:10.1073/pnas.1324036111. PMID:24778210
- Conomos D, Reddel RR, Pickett HA. NuRD-ZNF827 recruitment to telomeres creates a molecular scaffold for homologous recombination. Nat Struct Mol Biol. 2014; doi:10.1038/nsmb.2877. PMID: 25150861
- Panagopoulos I, Micci F, Thorsen J, et al. Fusion of ZMYND8 and RELA genes in acute erythroid leukemia. PLoS One. 2013;8:e63663. doi:10.1371/journal.pone.0063663. PMID: 23667654
- Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi:10.1038/nature10166. PMID: 21720365
- Basu M, Khan MW, Chakrabarti P, et al. Chromatin reader ZMYND8 is a key target of all trans retinoic acid-mediated inhibition of cancer cell proliferation. Biochim Biophys Acta. 2017;1860:450–9. doi:10.1016/j.bbagrm.2017.02.004. PMID: 28232094
- Basu M, Sengupta I, Khan MW, et al. Dual histone reader ZMYND8 inhibits cancer cell invasion by positively regulating epithelial genes. Biochem J. 2017;474:1919–34. doi:10.1042/BCJ20170223. PMID: 28432260
- Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer. 2011;11:588–96. doi:10.1038/nrc3091. PMID: 21734722
- Le Gallo M, O'Hara AJ, Rudd ML, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012;44:1310–5. doi:10.1038/ng.2455. PMID: 23104009
- Teng YC, Lee CF, Li YS, et al. Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer Res. 2013;73:4711–21. doi:10.1158/0008-5472.CAN-12-3165. PMID: 23722541
- Hou J, Wu J, Dombkowski A, et al. Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer. Am J Transl Res. 2012;4:247–56. PMID: 22937203
- Sharma SV, Lee DY, Li B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi:10.1016/j.cell.2010.02.027. PMID: 20371346
- Rodriguez R, Miller KM. Unravelling the genomic targets of small molecules using high-throughput sequencing. Nat Rev Genet. 2014;15:783–96. doi:10.1038/nrg3796. PMID: 25311424
- Dawson MA. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science. 2017;355:1147–52. doi:10.1126/science.aam7304. PMID: 28302822
- Brown JS, O'Carrigan B, Jackson SP, et al. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017;7:20–37. doi:10.1158/2159-8290.CD-16-0860. PMID: 28003236
- Jackson SP, Helleday T. DNA REPAIR. Drugging DNA repair Science. 2016;352:1178–9.
- Zacharioudakis E, Agarwal P, Bartoli A, et al. Chromatin Regulates Genome Targeting with Cisplatin. Angew Chem Int Ed Engl. 2017;56:6483–7. doi:10.1002/anie.201701144. PMID: 28474855