
Over the course of tumor progression, tumor cells in primary lesions spawn their progenies to initiate metastatic dissemination and colonization, resulting in successful tumor metastasis. More than 90% of cancer patients succumb to death due to tumor metastasis. Thus, the establishment of molecular mechanism-based therapies for tumor metastasis is a prime objective [Citation1]. Stress-responsive signaling cascades function as a rheostat for appropriate cellular responses; thus, their dysfunction culminates in tumor initiation and progression. The most important stress-responsive signaling cascades are mitogen-activated protein kinase (MAPK) pathways, composed of extracellular signal-regulated kinase (ERK), p38 MAPK, and c-Jun N-terminal kinase (JNK). Apoptosis signal-regulating kinase 1 (ASK1) is a Ser/Thr kinase that is activated in response to various stressors, such as cytokines and oxidative stress and is responsible for activation of p38 and JNK [Citation2]. Although ASK1 reportedly exerts both tumor-promoting and tumor-suppressive effects on tumor progression [Citation3], it remains unclear whether ASK1 modulates tumor metastasis and/or serves as an upstream regulator of p38 and JNK. In a recent study [Citation4], we reported drastic attenuation of lung tumor metastasis in ASK1 knockout mice. Intriguingly, tumor metastasis was also attenuated in wild-type mice that received bone marrow from ASK1 knockout mice but not myeloblast-specific (i.e., granulocytes, monocytes, and macrophages) ASK1 knockout mice. We, therefore, focused on platelets among the non-myeloblastic cell types.
Platelets are shed from megakaryocytes and positively regulate bloodborne tumor metastasis, as well as hemostasis and thrombosis, through aggregation and adhesion. ASK1 knockout mice had a normal platelet count; however, they exhibited unstable hemostasis and impaired thrombosis. Importantly, platelet-specific ASK1 knockout mice exhibited attenuated tumor metastasis, as well as unstable hemostasis and impaired thrombosis, indicating the involvement of platelet-intrinsic ASK1. Additional analysis of in vitro platelet aggregation suggested that ASK1 regulates adenosine diphosphate (ADP) signaling in platelets. ADP signaling is mediated by two G protein-coupled receptors (GPCRs), P2Y1 and P2Y12, in platelets [Citation5]. We newly identified the phosphorylation site (Thr345 in mice) of P2Y12 as a target of ASK1-JNK/p38 axis. This site is located at the C-terminal putative postsynaptic density 95/discs large/zonula occludens-1 (PDZ)-binding motif and is required for sustained Akt activation. Hence, the results in this study [Citation4] offer insight into the positive regulation of ADP signaling through P2Y12 phosphorylation and ASK1-mediated MAPK signaling in platelets. Another recent study from Naik's group [Citation6] revealed that ASK1-p38 axis contributes to the secretion of granules in platelets. Thus, there may be some crosstalk between ADP signaling and granule secretion under the control of ASK1-(JNK/) p38 axis. In addition, ASK1 may have other effectors in addition to JNK and p38 to regulate hemostasis, thrombosis, and bloodborne tumor metastasis (, upper right).
Figure 1. Schematic models of (upper right) platelet-intrinsic ASK1 regulation of hemostasis, thrombosis, and platelet-dependent tumor metastasis, (lower right) putative functions of P2Y12 phosphorylation, and (left) potential roles of ASK1 in X-ray-resistant cell types and immune cells. AC, adenylate cyclase; GRK, G protein-coupled receptor kinase; NK, natural killer; RE, recycling endosome.
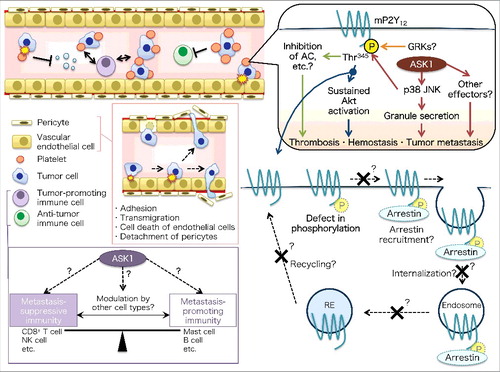
Further study will be needed to investigate the precise molecular mechanism by which the newly identified P2Y12 phosphorylation site affects P2Y12 function, as well as other downstream pathways, apart from Akt (e.g., inhibition of adenylate cyclase). In general, GPCR phosphorylation by GPCR kinases (GRKs) leads to desensitization, arrestin recruitment, and internalization followed by either receptor recycling or degradation [Citation7]. P2Y12 was shown to be phosphorylated by GRK2 and GRK6, resulting in desensitization; however, the phosphorylation sites targeted by these kinases have not been identified [Citation5]. Thus, GRKs may also phosphorylate Thr345 (in mice) or other sites of P2Y12. It would be interesting to examine the relationship between ASK1-JNK/p38 axis and GRKs in the regulation of P2Y12 phosphorylation and function. Moreover, it remains unknown whether P2Y12 Thr345-phosphorylation is relevant to arrestin recruitment, receptor internalization, and/or resensitization, similar to the bleeding-patient mutation (P341A) at the corresponding PDZ motif of human P2Y12 (, lower right) [Citation5].
Of note, ASK1 knockout mice that received bone marrow from wild-type mice also showed attenuated tumor metastasis, suggesting that ASK1 may regulate tumor metastasis in X-ray-resistant cells, such as pericytes and/or endothelial cells (ECs). ASK1 may mediate pericyte detachment from ECs, EC death induced by direct interaction with tumor cells, and adhesion followed by tumor cell transmigration. Furthermore, we have promising results that suggest metastasis-suppressive immunity is relatively enhanced in ASK1 knockout mice compared with wild-type mice (unpublished observation). The responsible cell types and the precise regulatory mechanism of tumor metastasis therein are now under investigation (, left).
In sum, our findings reveal a previously unidentified function of MAPK pathway in GPCR fine-tuning in platelets [Citation4]. This knowledge will greatly contribute to the development of novel molecular mechanism-based strategies against tumor metastasis and thrombosis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi:10.1126/science.1203543. PMID:21436443
- Ichijo H, Nishida E, Irie K, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi:10.1126/science.275.5296.90. PMID:8974401
- Ryuno H, Naguro I, Kamiyama M. ASK family and cancer. Adv Biol Regul. 2017;S2212–4926:30114–30118. doi:10.1016/j.jbior.2017.05.003. PMID:28552579
- Kamiyama M, Shirai T, Tamura S, et al. ASK1 facilitates tumor metastasis through phosphorylation of an ADP receptor P2Y12 in platelets. Cell Death Differ. 2017;24:2066–2076. doi:10.1038/cdd.2017.114. PMID:28753204
- Cunningham MR, Aungraheeta R, Mundell SJ. Pathophysiological consequences of receptor mistraffic: tales from the platelet P2Y12 receptor. Mol Cell Endocrinol. 2017;5:74–81. doi:10.1016/j.mce.2017.02.016. PMID:28212842
- Naik MU, Patel P, Derstine R, et al. Ask1 regulates murine platelet granule secretion, thromboxane A2 generation, and thrombus formation. Blood. 2016;129:1197–1209. doi:10.1182/blood-2016-07-729780. PMID:28028021
- Ritter SL, Hall RA. Fine-tuning of GPCR activity by receptor-interacting proteins. Nat Rev Mol Cell Biol. 2009;10:819–830. doi:10.1038/nrm2803. PMID:19935667