
ABSTRACT
The transcriptional repressor Capicua (CIC) has emerged as an important rheostat of cell growth regulated by RAS/MAPK signaling. Cic was originally discovered in Drosophila, where it was shown to be inactivated by MAPK signaling downstream of the RTKs Torso and EGFR, which results in signal-dependent responses that are required for normal cell fate specification, proliferation and survival of developing and adult tissues. CIC is highly conserved in mammals, where it is also negatively regulated by MAPK signaling. Here, we review the roles of CIC during mammalian development, tissue homeostasis, tumor formation and therapy resistance. Available data indicate that CIC is involved in multiple biological processes, including lung development, liver homeostasis, autoimmunity and neurobehavioral processes. Moreover, CIC has been shown to be involved in tumor development as a tumor suppressor, both in human as well as in mouse models. Finally, several lines of evidence implicate CIC as a determinant of sensitivity to EGFR and MAPK pathway inhibitors, suggesting that CIC may play a broader role in human cancer than originally anticipated.
KEYWORDS:
Introduction
Capicua (CIC) is a transcriptional repressor of the HMG-box family which binds specific DNA sites in target genes. Cic was first identified in Drosophila, where it was shown to control embryonic pattern formation downstream of the receptor tyrosine kinase (RTK)/RAS/ERK signaling pathway [Citation1]. Since then, Cic has been extensively studied in other Drosophila processes regulated by RTK signaling such as cell fate specification, proliferation and tissue homeostasis () [Citation2]. In addition, a recent study has identified an RTK-independent activity of Cic downstream of Toll/Interleukin-1 signaling [Citation3].
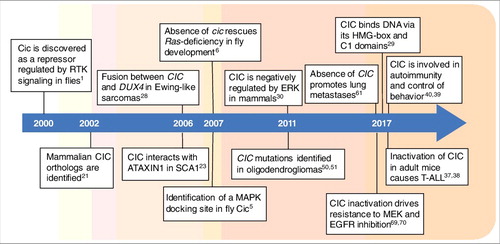
Figure 1. Timeline of key discoveries pertaining to CIC research. Cic was discovered in 2000 in Drosophila, and two years later it was identified in mammals. Its implication in cancer was originally reported in 2006 as a component of oncogenic fusions, but inactivating point mutations were not found until 2011. The last year has been particularly prolific in new findings about CIC function in mammalian development and cancer, including its roles in metastasis formation and therapy resistance. References are indicated in each box.
CIC proteins are highly conserved in mammals and, in recent years, this repressor has also emerged as a tumor suppressor whose function is directly controlled by RTK/RAS/ERK signaling, one of the most important pathways associated with cellular growth and cancer () [Citation2]. Mutations that promote excess RAS signaling are associated with a wide range of human tumors, but how these signals drive cellular transformation and tumorigenesis remains unclear after decades of study. To a large extent, this can be attributed to the fact that RAS/ERK signaling leads to phosphorylation of over 100 substrates, which can in turn interact with other signaling and regulatory inputs. Therefore, defining these targets and their activities is an important step towards understanding the biology and pathobiology of RAS signaling.
Cic restricts cellular growth in Drosophila
Cic was discovered almost two decades ago as a regulator of embryonic patterning in Drosophila [Citation1]. Fly embryos devoid of maternally contributed Cic activity lack most of their trunk and abdominal regions while maintaining the presumptive head and telson; hence the name capicua, a term derived from the words “head” (cap) and “tail” (cua) in Catalan. Drosophila Cic acts as a default repressor of genes regulated by RTK/Ras signaling. In the absence of signaling, Cic binds to and represses those genes, whereas activation of the pathway leads to phosphorylation and inactivation of Cic via degradation or relocalization from the nucleus to the cytoplasm (A) [Citation1–8]. For example, Cic is fully degraded in response to RTK activation at the anterior and posterior poles of the embryo, creating local gradients of Cic nuclear concentration that are complementary to the input gradients of ERK activity [Citation1,Citation5,Citation9]. In contrast, RTK activation in ovarian follicle cells promotes nuclear export of Cic and its partial redistribution to the cytoplasm [Citation5]. As a result of these inhibitory effects, Cic-mediated repression is prevented, allowing activation of its target genes by tissue-specific or ubiquitous transcription factors. This transcriptional switch operates downstream of at least two different RTKs, Torso and EGFR, resulting in signal-dependent responses that are required for normal cell fate specification, proliferation and survival of developing and adult tissues. In particular, EGFR-dependent signaling is essential for growth of larval tissues that will form adult structures such as the wings and eyes. Similarly, EGFR signaling promotes the proliferation of intestinal stem cells that is needed for regeneration of the adult midgut epithelium. In both cases, EGFR signaling acts, at least in part, by downregulating Cic [Citation6,Citation8,Citation10]. Indeed, loss of Cic activity via mutation enables cell proliferation in both contexts even in the absence of a functional EGFR signal, whereas overexpression of wild-type or phosphorylation-insensitive forms of Cic blocks EGFR/Ras-induced proliferation [Citation6,Citation8]. Cic appears to exert these effects by directly repressing a battery of target genes encoding cell cycle regulators and factors involved in DNA replication such as String/Cdc25 and Cyclin E [Citation8,Citation10,Citation11].
Figure 2. Role of Cic in Ras-MAPK signaling and growth control. (A) Regulation of Cic repressor activity via MAPK signaling in Drosophila. In the absence of RTK/RAS/MAPK signaling, Cic acts as a default repressor by binding to specific Cic-binding sites (CBS) in its target genes (left). Upon RTK activation, RAS proteins become GTP-bound and initiate a phosphorylation cascade via the kinases Draf (RAF ortholog), Dsor (MEK ortholog) and the MAPK Rolled (ERK ortholog) causing phosphorylation of Cic, which in turn results in its degradation and/or nuclear exclusion (right). As a consequence, Cic target genes are transcriptionally induced (derepressed) at specific times and places during development. This induction depends on transcriptional activators (A) which remain only partially characterized. Two confirmed activators of Cic targets are Dorsal and Zelda, which activate the intermediate neuroblasts defective gene in the early embryo [Citation7,Citation12,Citation13]. Zelda also appears to activate tailless, [Citation14] another embryonic Cic target [Citation1,Citation12,Citation15]. See also panel B and [Citation2] and [Citation11]. (B) Summary of Cic regulatory interactions in Drosophila growth control. In addition to its role downstream of Ras signaling, Cic mediates cross-interactions with the Hippo (Hpo) pathway and other regulatory inputs. For example, both Cic and the Sd:Yki co-activator complex regulate a common set of target genes, which become induced upon simultaneous reduction of Hpo signaling (leading to Sd:Yki upregulation) and Cic repressor activity. Some of these targets, including the Ets transcription factor Pnt [Citation8,Citation11] and the bantam microRNA, [Citation10,Citation16,Citation71,Citation72] are directly controlled by both Cic and Sd/Yki, whereas the input of Sd:Yki on other targets appears to be indirect, possibly via JAK/STAT signaling [Citation11]. This latter set of targets includes negative feedback regulators of Ras signaling such as Argos and Sprouty, whose activity is represented by a dashed loop. bantam has also been proposed to function in a negative feedback loop to downregulate Cic expression levels. Finally, recent evidence linking Mnb kinase activity to both Cic [Citation18] and Hpo signaling [Citation19] (not included in the model) implies the existence of additional layers of crosstalk. Cic and Sd are DNA binding proteins and are represented by ovals. The correspondence between Drosophila proteins illustrated in the diagram and their mammalian orthologs is indicated on the right. See main text for further details.
![Figure 2. Role of Cic in Ras-MAPK signaling and growth control. (A) Regulation of Cic repressor activity via MAPK signaling in Drosophila. In the absence of RTK/RAS/MAPK signaling, Cic acts as a default repressor by binding to specific Cic-binding sites (CBS) in its target genes (left). Upon RTK activation, RAS proteins become GTP-bound and initiate a phosphorylation cascade via the kinases Draf (RAF ortholog), Dsor (MEK ortholog) and the MAPK Rolled (ERK ortholog) causing phosphorylation of Cic, which in turn results in its degradation and/or nuclear exclusion (right). As a consequence, Cic target genes are transcriptionally induced (derepressed) at specific times and places during development. This induction depends on transcriptional activators (A) which remain only partially characterized. Two confirmed activators of Cic targets are Dorsal and Zelda, which activate the intermediate neuroblasts defective gene in the early embryo [Citation7,Citation12,Citation13]. Zelda also appears to activate tailless, [Citation14] another embryonic Cic target [Citation1,Citation12,Citation15]. See also panel B and [Citation2] and [Citation11]. (B) Summary of Cic regulatory interactions in Drosophila growth control. In addition to its role downstream of Ras signaling, Cic mediates cross-interactions with the Hippo (Hpo) pathway and other regulatory inputs. For example, both Cic and the Sd:Yki co-activator complex regulate a common set of target genes, which become induced upon simultaneous reduction of Hpo signaling (leading to Sd:Yki upregulation) and Cic repressor activity. Some of these targets, including the Ets transcription factor Pnt [Citation8,Citation11] and the bantam microRNA, [Citation10,Citation16,Citation71,Citation72] are directly controlled by both Cic and Sd/Yki, whereas the input of Sd:Yki on other targets appears to be indirect, possibly via JAK/STAT signaling [Citation11]. This latter set of targets includes negative feedback regulators of Ras signaling such as Argos and Sprouty, whose activity is represented by a dashed loop. bantam has also been proposed to function in a negative feedback loop to downregulate Cic expression levels. Finally, recent evidence linking Mnb kinase activity to both Cic [Citation18] and Hpo signaling [Citation19] (not included in the model) implies the existence of additional layers of crosstalk. Cic and Sd are DNA binding proteins and are represented by ovals. The correspondence between Drosophila proteins illustrated in the diagram and their mammalian orthologs is indicated on the right. See main text for further details.](/cms/asset/65807064-2cd8-4a90-a0f5-1049f058a678/kccy_a_1450029_f0002_oc.jpg)
Additional studies in Drosophila also suggest a more complex role of Cic at the intersection between Ras signaling and other growth control pathways. For instance, two targets regulated by Cic, Cyclin E and the microRNA gene bantam, are also regulated by the Retinoblastoma fly ortholog and by the Hippo pathway, respectively (B) [Citation8,Citation10,Citation20]. Indeed, Cic converges with Hippo signaling on a larger set of cell proliferation genes to downregulate their expression, allowing instead for the establishment of cellular differentiation programs [Citation11]. In turn, bantam appears to regulate Cic expression levels producing a negative feedback loop [Citation10]. These observations suggest the existence of elaborate control mechanisms in which Cic activity cooperates with other inputs to regulate cell cycle progression during fly development. In fact, Cic might itself integrate some of these signals directly, since recent data shows that Cic is phosphorylated and downregulated by Minibrain/DYRK1A, a kinase involved in growth control that would affect Cic in parallel with ERK-mediated inhibition [Citation18].
Conserved and unique features of CIC in mammals
CIC proteins are highly conserved across mammals (). Human and murine orthologs were identified in 2002 as novel Sox-related genes expressed during neural development [Citation21]. However, CIC expression in mammals is not restricted to the brain and is found in a variety of organs including thymus and lung [Citation22]. At the molecular level, mammalian and fly CIC proteins show the highest similarity in their HMG-box and C-terminal domains (). In addition, similar to Drosophila, both humans and mice express at least two isoforms, CIC-L (long) and CIC-S (short), with different N-terminal regions [Citation23]. Interestingly, the exons encoding the N-terminal regions of the Drosophila and mammalian CIC-S isoforms appear to have originated independently during evolution, suggesting that they may exert at least some distinct molecular functions [Citation24]. For instance, Drosophila Cic-S harbors a unique N-terminal motif, only present in dipteran insects, that allows its association with the Groucho (Gro) corepressor, an activity therefore unlikely to be present in its mammalian counterpart () [Citation24].
Figure 3. Structure and conservation of CIC orthologs from Drosophila and humans. Both species express CIC-L and CIC-S isoforms with alternative N-terminal regions. These isoforms display overlapping distributions in multiple tissues (particularly in mammals [Citation22,Citation25]), although very little is known about the mechanisms controlling these patterns of expression both in Drosophila and in mammals. Studies in Drosophila have revealed a key difference between Cic-S and Cic-L: Cic-S contains a specific motif called N2 which is critical for Gro-mediated repression in the embryo [Citation24]. Conversely, the Cic-L isoforms share a domain of unknown function (N1) in their N-terminal regions [Citation23,Citation24]. Other than that, the functional differences between these isoforms remain poorly understood. All short and long isoforms share the HMG-box and C1 domains involved in DNA binding. Although the ATXN1 binding domain (BD) characterized in mammalian CIC proteins [Citation23,Citation26] is only moderately conserved in Drosophila Cic [Citation23] (not shown), Ataxin-1 has been identified in a screen for Cic interactors in Drosophila embryos [Citation18]. The C2 MAPK docking site of Drosophila Cic and a distinct ERK BD of human CIC are also indicated [Citation5,Citation27].
![Figure 3. Structure and conservation of CIC orthologs from Drosophila and humans. Both species express CIC-L and CIC-S isoforms with alternative N-terminal regions. These isoforms display overlapping distributions in multiple tissues (particularly in mammals [Citation22,Citation25]), although very little is known about the mechanisms controlling these patterns of expression both in Drosophila and in mammals. Studies in Drosophila have revealed a key difference between Cic-S and Cic-L: Cic-S contains a specific motif called N2 which is critical for Gro-mediated repression in the embryo [Citation24]. Conversely, the Cic-L isoforms share a domain of unknown function (N1) in their N-terminal regions [Citation23,Citation24]. Other than that, the functional differences between these isoforms remain poorly understood. All short and long isoforms share the HMG-box and C1 domains involved in DNA binding. Although the ATXN1 binding domain (BD) characterized in mammalian CIC proteins [Citation23,Citation26] is only moderately conserved in Drosophila Cic [Citation23] (not shown), Ataxin-1 has been identified in a screen for Cic interactors in Drosophila embryos [Citation18]. The C2 MAPK docking site of Drosophila Cic and a distinct ERK BD of human CIC are also indicated [Citation5,Citation27].](/cms/asset/24398db6-bc4b-4e7f-9b4a-b15bee1b17ca/kccy_a_1450029_f0003_oc.jpg)
As in Drosophila, mammalian CIC proteins bind to highly conserved octameric sites in target genes via their HMG-box and C1 domains [Citation12,Citation15,Citation28,Citation29]. In all cases analyzed so far, such binding leads to repression of CIC targets, which include members of the PEA3 family (see below) [Citation22,Citation28,Citation30]. Similarly, CIC is also negatively regulated by ERK-mediated phosphorylation in mammalian cells, which prevents binding of the importin KPNA3 to CIC and ultimately leads to derepression of CIC targets [Citation30]. Recently, photocrosslinking studies have identified an ERK docking site in human CIC that is different from the site characterized in the Drosophila protein [Citation5,Citation27]. In addition, the direct ERK substrate p90RSK can phosphorylate CIC on residues adjacent to the HMG box, thereby creating docking sites for 14-3-3 proteins, which, in turn, appear to decrease the interaction of CIC with DNA [Citation30]. Yet, the regulation of CIC function remains poorly defined in mammals (see below), and it is not clear whether there are additional ERK-dependent or -independent mechanisms controlling CIC stability, localization or DNA binding in mammalian cells.
Specific roles of CIC in mammalian development and homeostasis
Mammalian CIC proteins form nuclear protein complexes with ATAXIN-1 (ATXN1) and its related factor ATAXIN-1-LIKE (ATXN1L) [Citation23]. ATXN1 is best known for its role in Spinocerebellar Ataxia Type 1 (SCA1), which is caused by the expansion of a polyglutamine tract in this protein [Citation31]. While the functional significance of CIC-ATXN1/ATXN1L complexes is not fully understood, several lines of evidence suggest that ATXN1 and ATXN1L help stabilize the CIC protein and also serve as CIC corepressors [Citation22,Citation23,Citation32,Citation33,Citation71]. For instance, reducing Atxn1/Atxn1L gene dosage in mice caused a decline in CIC protein levels and derepression of CIC target genes, [Citation22] although the mechanism by which ATXN1/ATXN1L proteins control CIC stability is currently unknown. Also, the interaction of ATXN1 with CIC is required for disease manifestation, since ATXN1S776A, a mutant that cannot bind CIC, is not pathogenic [Citation23]. Moreover, disruption of the ATXN1-CIC complex has been shown to have a therapeutic effect in SCA1 [Citation34]. This was demonstrated by breeding the SCA1 gain-of-function mouse model Atxn1154Q with a Cic-L–/– strain (). This strain carries a genetrap cassette introduced downstream of exon 1A, thereby selectively eliminating the CIC-L isoform, while at the same time reducing the expression of CIC-S [Citation22]. The majority of Cic-L–/– mice died before weaning, but reduced CIC activity in Cic-L+/– mice bred with Atxn1154Q mutants was sufficient to ameliorate the SCA1 phenotypes [Citation34]. This improvement was also observed by subjecting the mice to an exercise routine, which led to a reduction of CIC levels through activation of EGFR signaling in the brainstem [Citation34]. Remarkably, glutamine-expanded ATXN1 tends to form more organized and less toxic fibrillar aggregates in the context of reduced CIC expression levels [Citation35]. Moreover, a screen for ATXN1 regulators that could provide potential therapeutic options against SCA1 yielded several components of the MAPK pathway [Citation36]. ATXN1 protein stability was shown to be directly controlled by the MAPK pathway via MSK1-mediated phosphorylation, and downregulation of RAS-ERK-MSK1 activity decreased the levels and toxicity of glutamine-expanded ATXN1 [Citation36]. Thus, although further research is needed to fully elucidate the complex interplay between RAS-ERK signaling, ATXN1 and CIC, these and other data discussed below offer promising prospects for developing treatments against SCA1 and at least some cancers.
Table 1. Phenotypes of Cic-deficient mouse models.
Furthermore, while the interaction of CIC with gain-of-function ATXN1 contributes to SCA1, wildtype CIC-ATXN1/ATAXIN1L complexes are required to prevent neurobehavioral defects [Citation39]. Zoghbi and co-workers assessed the effects of deleting Cic in different regions of the brain using a Cic conditional allele carrying loxP sites flanking exons 9–11, whose deletion causes truncated CIC proteins due to incorporation of a premature stop codon (). Cic ablation driven by the Emx1-Cre strain, which expresses Cre activity in the forebrain, resulted in hyperactivity as well as learning and memory defects. In contrast, when conditional Cic ablation was driven by the Opt-Cre allele, which is active in the hypothalamus and medial amygdala, mice developed defects in social interaction related to autism spectrum disorders [Citation39]. Consistent with these phenotypes, CIC mutations have been associated with mental and developmental retardation as well as intellectual disability in humans [Citation39,Citation43,Citation44].
As mentioned above, CIC expression is not restricted to the brain and it exerts additional developmental and physiological roles in other tissues. Ablation of full-length Cic isoforms using the same conditional strain in cells of hematopoietic lineage (by crossing with Vav1-Cre) or in T lymphocytes (by crossing with Cd4-Cre) increases the population of follicular helper T cells (TFH) via derepression of Etv5 along with the subsequent induction of its target gene Maf [Citation40]. The increase in TFH cells also caused an expansion of germinal center B-cells and revealed autoimmunity phenotypes such as enlarged secondary lymphoid organs and infiltration of immune cells into tissues [Citation40].
CIC has also been implicated in liver homeostasis, as surviving 18-day-old Cic-L–/– mice show increased levels of bile acid in the liver and enhanced inflammatory responses owing to increased hepatic interleukin-6 and TNFα levels [Citation25]. Absence of CIC-L did not translate in hepatic damage by itself but cooperated with a 1% cholic acid diet to produce hepatic injury [Citation25]. Whether this phenotype is a consequence of reduced total CIC protein levels or a property selectively attributed to the CIC-L isoform remains to be determined. In addition, Cic-L–/– mice displayed lung alveolarization defects accompanied by MMP9 overexpression at P20 [Citation22]. A similar phenotype was found in Atxn1L null mice and it has been proposed that loss of either ATXN1L or CIC causes derepression of ETV4, a PEA3 family activator of matrix metalloprotease genes such as Mmp9 that would affect the alveolarization process.
Finally, germline expression of CIC isoforms lacking the HMG-box in mice (CicΔ2-6/Δ2-6 strain) leads to perinatal lethality and abnormal terminal differentiation of the respiratory epithelium during late embryonic development () [Citation37]. The observed defects in these mice correlate with a dramatic increase in proliferating cells and persistent TTF-1 expression. Furthermore, these mice display a reduction in the numbers of type II alveolar cells, suggesting that CIC loss-of-function embryos are incapable of producing enough surfactant for postnatal survival. These observations and those made in Cic-L–/– mice suggest that CIC activity is particularly relevant for late stage lung development, with phenotypes becoming apparent at the relatively advanced saccular and alveolar stages in CicΔ2-6/Δ2-6 and Cic-L–/– embryos, respectively [Citation22,Citation37]. Additionally, these CicΔ2-6/Δ2-6 embryos displayed omphalocele with high frequency, a defect also shared by Atxn1/Atxn1L double KO embryos, suggesting that the ATXN1/ATXN1L-CIC complexes may be needed for retraction of the gut from the umbilical cord [Citation37]. Collectively, these evidences indicate that ATXN1 and ATXN1L are key factors modulating the levels and activity of CIC in multiple settings, which emphasizes the need for further mechanistic studies dissecting the roles of CIC-ATXN1/ATXN1L complexes in normal and pathological conditions.
Role of CIC in RAS-driven proliferation in mammalian cells
Cic has been shown to play a key role in Ras-driven proliferation in flies. Ablation of the single Ras locus in Drosophila results in small, poorly growing cell clones in their imaginal discs. Concomitant inactivation of the cic gene restored this defect leading to the generation of normal clones indistinguishable from those expressing wild-type Ras [Citation6]. Likewise, depletion of Ras prevents mitotic divisions in intestinal stem cells and simultaneous inhibition of Cic expression rescues the proliferation defects [Citation8]. These observations suggest that, at least in these fly tissues, Cic is the key mediator of Ras-driven cell proliferation. In contrast, inactivation of mammalian CIC in mouse embryonic fibroblasts lacking the three RAS isoforms, Hras, Nras and Kras, [Citation45–47] failed to induce proliferation, indicating a more complex interpretation of RAS signaling in these mammalian cells [Citation37]. In this regard, it has been shown that ERK proteins phosphorylate and inactivate additional repressors such as ERF or ETV6 (also known as TEL) [Citation48,Citation49]. Therefore, it will be interesting to determine whether the combined inactivation of CIC, ERF and ETV6 may be sufficient for RAS-independent cell proliferation, or whether additional factors are also involved.
CIC is a tumor suppressor
More recently, CIC has been found to be involved in cancer development. Oligodendrogliomas, a type of low-grade brain tumor, had long been recognized to harbor loss of heterozygosity (LOH) of chromosome arms 1p and 19q. In a large-scale sequencing effort to identify potential tumor suppressor genes in these chromosomal deletions, Bettegowda and colleagues identified recurrent mutations in CIC (70% of cases) in the remaining allele on chromosome 19q, suggesting that CIC acts as a tumor suppressor gene [Citation50]. In most cases, these mutations co-occur with mutations in IDH1 and/or, less frequently, in FUBP1 or the TERT promoter [Citation50–52]. IDH1 mutations are known to change the catalytic properties of the mitochondrial enzyme isocitrate dehydrogenase (IDH). Whereas the wild-type enzyme is implicated in metabolic processes by converting isocitrate into α-ketoglutarate, the mutated version acquires a new catalytic activity and further converts α-ketoglutarate into 2-hydroxyglutarate (2HG) [Citation53]. More recently, 2HG has been recognized as an “oncometabolite”, thus contributing to oncogenic transformation by inhibiting 2-oxoglutarate-dependent dioxygenases [Citation54]. Ectopic expression of wild-type or mutant CIC-S proteins has been shown to cooperate with mutant IDH1 to further increase the production of 2HG [Citation55]. Surprisingly, CIC-S proteins have also been detected in the cytoplasm associated with mitochondria, suggesting that this localization could somehow modulate 2HG production by mutant IDH1 [Citation55]. However, further studies are required to clarify this intriguing possibility.
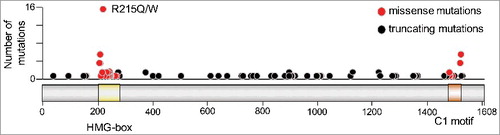
Interestingly, the majority of CIC missense mutations in oligodendroglioma cluster in two well-defined domains, the HMG-box and a C-terminal motif known as C1, both of which are implicated in DNA binding and, hence, repression of CIC targets (). In addition, a variety of other mutations in these tumors causes premature stop codons, altered splice sites, and frameshift insertions or deletions that are likely to disable CIC's repressor activity (). More recent studies have identified distinct CIC missense mutations within a single tumor, suggesting that selective pressure to inactivate CIC function causes several subclones to acquire distinct mutations independently, thereby contributing to intratumoral heterogeneity [Citation56]. These CIC mutations are not always maintained in recurrent oligodendrogliomas, adding further support to the concept that some of these mutations may be subclonal secondary events [Citation57]. Finally, the presence of mutated CIC alleles correlates with a more aggressive phenotype when compared to tumors that only harbor the 1p/19q co-deletion, indicating that complete inactivation of CIC function actively contributes to tumor progression [Citation58].
Figure 4. CIC mutations in CNS/brain tumors. Number of mutations are plotted along the length of the CIC-S protein (depicted with the HMG-box and C1 domains highlighted in yellow and brown, respectively). Missense mutations are indicated by red circles and truncating mutations by black circles. Mutation data were obtained from cBioPortal Version v1.8.3, selecting only CNS/brain datasets.
In mice, elimination of CIC activity in the entire brain by crossing a Ciclox/lox strain carrying loxP sites flanking exons 2–6 with mice expressing a Cre recombinase under the control of the GFAP promoter did not result in any significant alterations at the histopathological level for up to one year of age () [Citation37]. Likewise, eliminating CIC activity by targeting exons 9–11 in Ciclox/lox mice with the Emx1- (forebrain) or the Opt-Cre (hypothalamus and medial amygdala) strains did not result in brain tumor formation either [Citation39]. These results suggest that inactivation of CIC is not an initiating event in glioma development. However, Yang and colleagues have recently identified an aberrantly proliferating neural population in a germline Cic-deficient mouse model in which some mice survived until P4 () [Citation42]. Similarly, the authors also tested whether absence of CIC modulates glioma formation in mice. To this end, they used a well-characterized model of oligodendroglioma that is driven by overexpression of PDGFB. Neurospheres from Cic+/+ or Ciclox/lox mice were transduced with retroviruses expressing PDGFB and Cre-expressing adenoviruses to remove the Cic conditional alleles and create Cic–/– neurospheres. These Cic–/– neurospheres, when implanted into the brain of immunocompromised mice, gave rise to PDGFB-driven gliomas with significantly lower latency than Cic+/+ neurospheres, indicating that absence of CIC potentiates PDGFB-driven glioma formation [Citation42]. Taken together, these studies indicate that loss of CIC activity is likely not sufficient on its own to initiate brain tumorigenesis, but it may accelerate the growth of brain tumors driven by other cancer drivers. Whether the aberrantly proliferating neural cell population identified by Yang and colleagues in Cic-deficient mice can play a role in tumor initiation awaits further clarification.
CIC mutations have subsequently been identified in a variety of other cancers such as stomach adenocarcinomas (12.9%), endometrial carcinomas (6.9%), colorectal carcinomas (6.1%), or melanomas (5.2%) [Citation59,Citation60]. Yet, the contribution of CIC mutations to these cancers has not been thoroughly investigated. Moreover, inactivation of CIC has been implicated in metastasis formation. In particular, CIC mutations were associated with advanced stage lung adenocarcinomas and the inactivation of CIC promoted metastasis in an in vivo orthotopic model of lung cancer [Citation61,Citation62]. Interestingly, a variety of missense mutations identified in advanced stage lung adenocarcinomas affect codons that encode residues of yet uncharacterized protein regions. Likewise, loss of CIC has been implicated in metastatic progression of prostate cancer [Citation63]. More recently, absence of CIC was found to promote progression and metastasis of hepatocellular carcinomas induced by the chemical carcinogen diethylnitrosamine [Citation41]. Here, CIC proteins were deleted specifically from the liver by crossing Ciclox/lox mice with the Alb-Cre strain ().
From the molecular point of view, most of the available data suggest that CIC exerts its tumor suppressive functions by repressing its specific target genes. Mutations in exons encoding the HMG-box or the C1 motif clearly disrupt DNA binding of CIC [Citation29]. Since both types of mutations are frequently found in a variety of tumors (usually in combination with LOH of chromosome arm 19q), the loss of DNA binding (and presumably repressor activity) appears to be a key mechanism of CIC tumorigenesis. However, it remains unclear whether, and through which mechanisms, other missense mutations identified outside the HMG-box and C1 domains also contribute to tumor progression.
Additionally, CIC has been shown to be part of chromosomal translocations that result in oncogenic fusion proteins carrying domains of DUX4 or FOXO4 in Ewing-like sarcomas, or of NUTM1 in brain tumors [Citation28,Citation64]. These chimeric proteins usually retain most parts of CIC, including the C1 domain, attached to the various C-terminal regions of their fusion partners. The CIC-DUX4 fusion proteins, the best characterized so far, are believed to recognize CIC binding elements in target promoters and activate instead of repress gene expression via the DUX4 activation domain [Citation28,Citation29].
Role of CIC in T-ALL
Systemic inactivation of CIC proteins in adult mice using the conditional strain carrying loxP sites flanking exons 2–6 led to the development of acute T-cell lymphoblastic lymphoma (T-ALL) before one year of age [Citation37]. No other tissue in these mice displayed detectable alterations. Further characterization of the tumors revealed a high degree of malignancy, such as dissemination to other organs, transplantability of the disease, or clonal expansions of T cell populations. Transcriptomal analysis of these tumors by RNA sequencing revealed a variety of highly derepressed CIC targets including the transcription factors ETV4 and, to a lesser extent, ETV5. Notably, inactivation of CIC in an Etv4-deficient background dramatically reduced the incidence of T-ALL, indicating that ETV4 is a key effector of T-ALL development. Subsequent studies in which ETV4 expression was downregulated in human T-ALL cell lines also revealed a dependence of this tumor type on ETV4 expression [Citation37]. Induction of T-ALL by expression of an H-RASG12V oncoprotein from the mouse Kras locus revealed highly related transcriptional profiles with those induced by inactivation of CIC proteins [Citation37,Citation65] Similar transcriptional profiles were also observed in human T-ALLs carrying mutations that predict activation of the RAS/MAPK pathway [Citation37]. These results suggest that CIC is a key effector of RAS/MAPK driven T-ALL.
A more recent study utilizing the Ciclox/lox strain with loxP sites flanking exons 9–11 obtained similar results [Citation38]. When the authors systemically eliminated CIC from adult mice, but also specifically in hematopoietic progenitors by crossing with the Tek-Cre strain, mice developed fully penetrant T-ALL (). These important observations indicate that CIC's tumor suppressor activity is inherent to the hematopoietic lineage, since a potential implication of non-hematopoietic tissues cannot be ruled out upon systemic CIC elimination. In this regard, the same study also observed T-ALL formation using the Vav1-Cre line (also active in hematopoietic cells), although much more delayed and with incomplete penetrance. This slower, milder effect may explain why a previous study did not observe lymphoma development using the same Cre recombinase line [Citation40].
All these data taken together strongly support the notion that CIC's transcriptional repressor activity is crucial to suppress tumorigenesis. However, in contrast to Drosophila, inactivation of CIC in mammals does not seem to induce cell proliferation directly. This raises the question of how CIC inactivation contributes to cancer progression. In the majority of the cases described so far, derepression of the PEA3 family of transcription factors was key to tumor development. Thus, it is conceivable that CIC mutations impact on other cancer traits that are distinct from mere cell proliferation. Furthermore, at least two lines of evidence support the idea that CIC inactivation particularly affects the late stages of cancer progression. First, CIC mutations themselves appear to be a late event in tumor formation, suggesting that they do not play a major role in cancer initiation [Citation61]. Second, PEA3 transcription factors are well-known to control expression of matrix metalloproteases which in turn have been implicated in cancer cell invasion [Citation22,Citation61]. However, it should not be ruled out that other, yet unidentified mechanisms also contribute to tumor growth. Interestingly, CIC mutations can also occur in tumors carrying mutations in the RAS pathway, [Citation59,Citation60] suggesting that RAS inputs on other factors can enhance the CIC inactivation phenotype, at least in certain tumors.
CIC and therapy resistance
Despite the generally low frequency of RAS mutations in T-ALL, it has been suggested that up to 50% of these tumors display aberrant RAS signaling [Citation66]. Moreover, RAS/MAPK activating mutations are much more prevalent in relapsed cases, suggesting that targeting RAS/MAPK signaling could be a therapeutic option in a significant percentage of T-ALL patients [Citation67]. Indeed, experiments using mouse models suggest that T-ALLs driven by Ras oncogenes are susceptible to MEK inhibition [Citation68]. However, T-ALL cells from tumors obtained upon CIC inactivation do not exhibit increased MAPK activity and are completely resistant to the MEK kinase inhibitor trametinib [Citation37]. Moreover, inactivation of CIC from trametinib-sensitive human T-ALL cell lines using CRISPR/Cas9 also makes these cells insensitive to MEK inhibition. These observations suggest that inactivation of CIC may render MAPK inhibition inefficient in human cancer. In agreement with these observations, a genetic screen for genes whose absence causes resistance to MEK inhibition in lung and gastric cancer cell lines resulted in the identification of CIC [Citation69]. Similarly, CIC was also identified as a determinant of sensitivity to blocking EGFR signaling in neural stem cells or NSCLC cell lines [Citation42,Citation70]. Taken together, these studies suggest that absence of CIC derepresses a significant fraction of the gene expression program induced by activation of the EGFR/RAS/MAPK pathway. Therefore, the levels of CIC activity should be a key biomarker to predict the sensitivity of RAS/MAPK-driven tumors to MEK or ERK inhibitors. In sum, the relevance of CIC for cancer progression and therapy resistance is only just emerging and we believe that a better understanding of CIC functions could indeed make a significant impact on cancer therapies in the future.
Open questions
The involvement of CIC in several human diseases has stimulated substantial interest in understanding its multiple biological functions. Yet, despite the rapid progress made in recent years, the mechanisms of CIC activity and regulation are far from being fully understood. Thus, apart from the points discussed earlier, many open questions remain that need to be addressed in the future. For example, the functional differences between both CIC isoforms, CIC-L and CIC-S, remain largely uncharacterized. Related to this question, it will also be important to determine the precise molecular mechanisms of CIC repression and the role of ATXN proteins in this context. Most CIC functions appear to rely on this repressor activity, but does CIC possess activator or even transcription-independent functions? In this regard, further studies are also needed to clarify the potential role of CIC-S in mitochondria. Furthermore, from a biological perspective, it is evident that CIC has evolved independent functions in flies and mammals, which often depend on distinct sets of target genes, but is there a common, ancestral function shared across species? Questions also remain concerning the roles of CIC in cancer. It is striking that CIC mutations cluster into the HMG-box and the C1 motif only in oligodendroglioma. What is then the significance of the more even distribution of CIC mutations throughout the entire coding region in other cancers? Are all of these mutations merely passenger events or could they reflect tissue-specific differences in protein activity or sensitivity to mutagenesis? One initial study already suggested that all tested CIC mutations found in lung cancer, independently of where they occur, produce loss-of-function proteins [Citation61,Citation62]. Moreover, it has been suggested that CIC haploinsufficiency might cause predisposition to cancer and could represent in itself a RASopathy syndrome, [Citation38] an idea that warrants further investigation. Furthermore, it is unclear how loss of CIC function contributes to resistance to MAPK pathway inhibition, beyond causing derepression of the PEA3 family of transcription factors. Finally, it is expected that answers to these questions will provide a first step towards the bigger challenge of translating all that knowledge into novel therapies for CIC-related diseases.
Disclosure of potential conflicts of interest
The authors report no conflict of interest.
Additional information
Funding
References
- Jiménez G, Guichet A, Ephrussi A, et al. Relief of gene repression by Torso RTK signaling: role of capicua in Drosophila terminal and dorsoventral patterning. Genes Dev. 2000;14:224–231. PMID: 10652276.
- Jiménez G, Shvartsman SY, Paroush Z. The Capicua repressor – a general sensor of RTK signaling in development and disease. J Cell Sci. 2012;125:1383–1389. PMID: 22526417. doi:10.1242/jcs.092965.
- Papagianni A, Forés M, Shao W, et al. Capicua controls Toll/IL-1 signaling targets independently of RTK regulation. Proc Natl Acad Sci USA. 2018;115:1807–1812. PMID: 29432195. doi:10.1073/pnas.1713930115.
- Roch F, Jiménez G, Casanova J. EGFR signaling inhibits Capicua-dependent repression during specification of Drosophila wing veins. Development. 2002;129:993–1002. PMID: 11861482.
- Astigarraga S, Grossman R, Díaz-Delfín J, et al. A MAPK docking site is critical for downregulation of Capicua by Torso and EGFR RTK signaling. EMBO J. 2007;26:668–677. PMID: 17255944. doi:10.1038/sj.emboj.7601532.
- Tseng AS, Tapon N, Kanda H, et al. Capicua regulates cell proliferation downstream of the receptor tyrosine kinase/ras signaling pathway. Curr Biol. 2007;17:728–733. PMID: 17398096. doi:10.1016/j.cub.2007.03.023.
- Lim B, Samper N, Lu H, et al. Kinetics of gene derepression by ERK signaling. Proc Natl Acad Sci USA. 2013;110:10330–10335. PMID: 23733957. doi:10.1073/pnas.1303635110.
- Jin Y, Ha N, Forés M, et al. EGFR/Ras signaling controls Drosophila intestinal stem cell proliferation via Capicua-regulated genes. PLoS Genet. 2015;11:e1005634. PMID: 26683696. doi:10.1371/journal.pgen.1005634.
- Grimm O, Sanchez Zini V, Kim Y, et al. Torso RTK controls Capicua degradation by changing its subcellular localization. Development. 2012;139:3962–3968. PMID: 23048183. doi:10.1242/dev.084327.
- Herranz H, Hong X, Cohen SM. Mutual repression by bantam miRNA and Capicua links the EGFR/MAPK and Hippo pathways in growth control. Curr Biol. 2012;22:651–657. PMID: 22445297. doi:10.1016/j.cub.2012.02.050.
- Pascual J, Jacobs J, Sansores-Garcia L, et al. Hippo reprograms the transcriptional response to Ras signaling. Dev Cell. 2017;42:667–680. PMID: 28950103. doi:10.1016/j.devcel.2017.08.013.
- Ajuria L, Nieva C, Winkler C, et al. Capicua DNA-binding sites are general response elements for RTK signaling in Drosophila. Development. 2011;138:915–924. PMID: 21270056. doi:10.1242/dev.057729.
- Samee MA, Lim B, Samper N, et al. A systematic ensemble approach to thermodynamic modeling of gene expression from sequence data. Cell Syst. 2015;1:396–407. PMID: 27136354. doi:10.1016/j.cels.2015.12.002.
- Liang HL, Nien CY, Liu HY, et al. The zinc-finger protein Zelda is a key activator of the early zygotic genome in Drosophila. Nature. 2008;456:400–3. PMID: 18931655. doi:10.1038/nature07388.
- Löhr U, Chung HR, Beller M, et al. Antagonistic action of Bicoid and the repressor Capicua determines the spatial limits of Drosophila head gene expression domains. Proc Natl Acad Sci USA. 2009;106:21695–21700. PMID: 19959668. doi:10.1073/pnas.0910225106.
- Nolo R, Morrison CM, Tao C, et al. The bantam microRNA is a target of the hippo tumor-suppressor pathway. Curr Biol. 2006;16:1895–904. PMID: 16949821. doi:10.1016/j.cub.2006.08.057.
- Oh H, Irvine KD. Cooperative regulation of growth by Yorkie and Mad through bantam. Dev Cell. 2011;20:109–22. PMID: 21238929. doi:10.1016/j.devcel.2010.12.002.
- Yang L, Paul S, Trieu KG, et al. Minibrain and Wings apart control organ growth and tissue patterning through down-regulation of Capicua. Proc Natl Acad Sci USA. 2016;113:10583–10588. PMID: 27601662. doi:10.1073/pnas.1609417113.
- Degoutin JL, Milton CC, Yu E, et al. Riquiqui and minibrain are regulators of the hippo pathway downstream of Dachsous. Nat Cell Biol. 2013;15:1176–85. PMID: 23955303. doi:10.1038/ncb2829.
- Krivy K, Bradley-Gill MR, Moon NS. Capicua regulates proliferation and survival of RB-deficient cells in Drosophila. Biol Open. 2013;2:183–190. PMID: 23429853. doi:10.1242/bio.20123277.
- Lee CJ, Chan WI, Cheung M, et al. CIC, a member of a novel subfamily of the HMG-box superfamily, is transiently expressed in developing granule neurons. Brain Res Mol Brain Res. 2002;106:151–156. PMID: 12393275. doi:10.1016/S0169-328X(02)00439-4.
- Lee Y, Fryer JD, Kang H, et al. ATXN1 protein family and CIC regulate extracellular matrix remodeling and lung alveolarization. Dev Cell. 2011;21:746–757. PMID: 22014525. doi:10.1016/j.devcel.2011.08.017.
- Lam YC, Bowman AB, Jafar-Nejad P, et al. ATXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–1347. PMID: 17190598. doi:10.1016/j.cell.2006.11.038.
- Forés M, Ajuria L, Samper N, et al. Origins of context-dependent gene repression by capicua. PLoS Genet. 2015;11:e1004902. PMID: 25569482. doi:10.1371/journal.pgen.1004902.
- Kim E, Park S, Choi N, et al. Deficiency of Capicua disrupts bile acid homeostasis. Sci Rep. 2015;5:8272. PMID: 25653040. doi:10.1038/srep08272.
- Kim E, Lu HC, Zoghbi HY, et al. Structural basis of protein complex formation and reconfiguration by polyglutamine disease protein Ataxin-1 and Capicua. Genes Dev. 2013;27:590–5. PMID: 23512657. doi:10.1101/gad.212068.112.
- Futran AS, Kyin S, Shvartsman SY, et al. Mapping the binding interface of ERK and transcriptional repressor Capicua using photocrosslinking. Proc Natl Acad Sci USA. 2015;112:8590–8595. PMID: 26124095. doi:10.1073/pnas.1501373112.
- Kawamura-Saito M, Yamazaki Y, Kaneko K, et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet. 2006;15:2125–2137. PMID: 16717057. doi:10.1093/hmg/ddl136.
- Forés M, Simón-Carrasco L, Ajuria L, et al. A new mode of DNA binding distinguishes Capicua from other HMG-box factors and explains its mutation patterns in cancer. PLoS Genet. 2017;13:e1006622. PMID: 28278156. doi:10.1371/journal.pgen.1006622.
- Dissanayake K, Toth R, Blakey J, et al. ERK/p90(RSK)/14-3-3 signalling has an impact on expression of PEA3 Ets transcription factors via the transcriptional repressor capicua. Biochem J. 2011;433:515–525. PMID: 21087211. doi:10.1042/BJ20101562.
- Banfi S, Servadio A, Chung MY, et al. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat Genet. 1994;7:513–520. PMID: 7951322. doi:10.1038/ng0894-513.
- Crespo-Barreto J, Fryer JD, Shaw CA, et al. Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet. 2010;6:e1001021. PMID: 20628574. doi:10.1371/journal.pgen.1001021.
- Tsai CC, Kao HY, Mitzutani A, et al. Ataxin 1, a SCA1 neurodegereative disorder protein, is functionally linked to the silencing mediator of retinoid and thyroid hormone receptors. Proc Natl Acad Sci USA. 2004;101:4047–4052. PMID: 15016912. doi:10.1073/pnas.0400615101.
- Fryer JD, Yu P, Mandel-Brehm C, et al. Exercise and genetic rescue of SCA1 via the transcriptional repressor Capicua. Science. 2011;334:690–693. PMID: 22053053. doi:10.1126/science.1212673.
- Lasagna-Reeves CA, Rousseaux MW, Guerrero-Muñoz MJ, et al. A native interactor scaffolds and stabilizes ATAXIN-1 oligomers in SCA1. Elife. 2015;4:e07558. doi:10.7554/eLife.07558. PMID: 25988806.
- Park J, Al-Ramahi I, Tan Q, et al. RAS-MAPK-MSK1 pathway modulates ataxin 1 protein levels and toxicity and SCA1. Nature. 2013;498:325–331. PMID: 23719381. doi:10.1038/nature12204.
- Simón-Carrasco L, Graña O, Salmón M, et al. Inactivation of Capicua in adult mice causes T-cell lymphoblastic lymphoma. Genes Dev. 2017;31:1456–1468. PMID: 28827401. doi:10.1101/gad.300244.117.
- Tan Q, Brunetti L, Rousseaux MWC, et al. Loss of Capicua alters early T cell development and predisposes mice to T cell lymphoblastic leukemia/lymphoma. Proc Natl Acad Sci USA. 2018;115:E1511–E1519. PMID: 29382756. doi:10.1073/pnas.1716452115.
- Lu HC, Tan Q, Rousseaux MW, et al. Disruption of the ATXN1-CIC complex causes a spectrum of neurobehavioral phenotypes in mice and humans. Nat Genet. 2017;49:527–536. PMID: 28288114. doi:10.1038/ng.3808.
- Park S, Lee S, Lee CG, et al. Capicua deficiency induces autoimmunity and promotes follicular helper T cell differentiation via derepression of ETV5. Nat Commun. 2017;8:16037. PMID: 28855737. doi:10.1038/ncomms16037.
- Kim E, Kim D, Lee JS, et al. Capicua suppresses hepatocellular carcinoma progression by controlling ETV4-MMP1 axis. Hepatology. 2017;Dec 18;doi:10.1002/hep.29738. PMID: 29251790. doi:10.1002/hep.29738.
- Yang R, Chen LH, Hansen LJ, et al. Cic loss promotes gliomagenesis via aberrant neural stem cell proliferation and differentiation. Cancer Res. 2017;doi:10.1158/0008-5472.CAN-17-1018. PMID: 28939681.
- Vissers LE, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–1112. PMID: 21076407. doi:10.1038/ng.712.
- Athanasakis E, Licastro D, Faletra F, et al. Next generation sequencing in nonsyndromic intellectual disability: from a negative molecular karyotype to a possible causative mutation detection. Am J Med Genet A. 2014;164A:170–176. PMID: 24307393. doi:10.1002/ajmg.a.36274.
- Drosten M, Dhawahir A, Sum EY, et al. Genetic analysis of Ras signaling pathways in cell proliferation, migration and survival. EMBO J. 2010;29:1091–1104. PMID: 20150892. doi:10.1038/emboj.2010.7.
- Drosten M, Sum EY, Lechuga CG, et al. Loss of p53 induces cell proliferation via Ras-independent activation of the Raf/Mek/Erk signaling pathway. Proc Natl Acad Sci USA. 2014;111:15155–15160. PMID: 25288756. doi:10.1073/pnas.1417549111.
- Lechuga CG, Simón-Carrasco L, Jacob HK, et al. Genetic validation of cell proliferation via Ras-independent activation of the Raf/Mek/Erk pathway. Methods Mol Biol. 2017;1487:269–276. PMID: 27924574. doi:10.1007/978-1-4939-6424-6_20.
- Le Gallic L, Sgouras D, Beal G Jr, et al. Transcriptional repressor ERF is a Ras/mitogen-activated protein kinase target that regulates cellular proliferation. Mol Cell Biol. 1999;19:4121–4133. PMID: 10330152. doi:10.1128/MCB.19.6.4121.
- Maki K, Arai H, Waga K, et al. Leukemia-related transcription factor TEL is negatively regulated through extracellular signal-regulated kinase-induced phosphorylation. Mol Cell Biol. 2004;24:3227–3237. PMID: 15060146. doi:10.1128/MCB.24.8.3227-3237.2004.
- Bettegowda C, Agrawal N, Jiao Y, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science. 2011;333:1453–1455. PMID: 21817013. doi:10.1126/science.1210557.
- Yip S, Butterfield YS, Morozova O, et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol. 2012;226:7–16. PMID: 22072542. doi:10.1002/path.2995.
- Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013;110:6021–6026. PMID: 23530248. doi:10.1073/pnas.1303607110.
- Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. PMID: 19935646. doi:10.1038/nature08617.
- Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. PMID: 21251613. doi:10.1016/j.ccr.2010.12.014.
- Chittaranjan S, Chan S, Yang C, et al. Mutations in CIC and IDH1 cooperatively regulate 2-hydroxyglutarate levels and cell clonogenicity. Oncotarget. 2014;5:7960–7979. PMID: 25277207. doi:10.18632/oncotarget.2401.
- Suzuki H, Aoki K, Chiba K, et al. Mutational landscape and clonal architecture in grade II and grade III gliomas. Nat Genet. 2015;47:458–468. PMID: 25848751. doi:10.1038/ng.3273.
- Aihara K, Mukasa A, Nagae G, et al. Genetic and epigenetic stability of oligodendrogliomas at recurrence. Acta Neuropathol Commun. 2017;5:18. PMID: 28270234. doi:10.1186/s40478-017-0422-z.
- Gleize V, Alentorn A, Connen de Kérillis L, et al. CIC inactivating mutations identify aggressive subset of 1p19q codeleted gliomas. Ann Neurol. 2015;78:355–374. PMID: 26017892. doi:10.1002/ana.24443.
- Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. PMID: 22588877. doi:10.1158/2159-8290.CD-12-0095.
- Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical prolfiles using cBioPortal. Sci Signal. 2013;6:l1. PMID: 23550210. doi:10.1126/scisignal.2004088.
- Okimoto RA, Breitenbuecher F, Olivas VR, et al. Inactivation of Capicua drives cancer metastasis. Nat Genet. 2017;49:87–96. PMID: 27869830. doi:10.1038/ng.3728.
- Okimoto RA, Bivona TG. Metastasis: From head to tail. Cell Cycle. 2017;16:487–488. PMID: 28055306. doi:10.1080/15384101.2016.1271636.
- Seim I, Jeffery PL, Thomas PB, et al. Whole-genome sequence of the metastatic PC3 and LNCaP human prostate cancer cell lines. G3 (Bethesda). 2017;7:1731–1741. PMID: 28413162.
- Sturm D, Orr BA, Toprak UH, et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell. 2016;164:1060–1072. PMID: 26919435. doi:10.1016/j.cell.2016.01.015.
- Drosten M, Simón-Carrasco L, Hernández-Porras I, et al. H-Ras and K-Ras oncoproteins induce different tumor spectra when driven by the same regulatory sequences. Cancer Res. 2017;77:707–718. PMID: 27872088. doi:10.1158/0008-5472.CAN-16-2925.
- Von Lintig FC, Huvar I, Law P, Diccianni MB, et al. Ras activation in normal white blood cells and childhood acute lymphoblastic leukemia. Clin Cancer Res. 2000;6:1804–1810. PMID: 10815901.
- Oshima K, Khiabanian H, da Silva-Almeida AC, et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2016;113:11306–11311. PMID: 27655895. doi:10.1073/pnas.1608420113.
- Dail M, Li Q, McDaniel A, et al. Mutant Ikzf1, KrasG12D, and Notch1 cooperate in T lineage leukemogenesis and modulate responses to targeted agents. Proc Natl Acad Sci USA. 2010;107:5106–5111. PMID: 20194733. doi:10.1073/pnas.1001064107.
- Wang B, Krall EB, Aguirre AJ, et al. ATXN1L, CIC, and ETS transcription factors modulate sensitivity to MAPK pathway inhibition. Cell Rep. 2017;18:1543–1557. PMID: 28178529. doi:10.1016/j.celrep.2017.01.031.
- Liao S, Davoli T, Leng Y, et al. A genetic interaction analysis identifies cancer drivers that modify EGFR dependency. Genes Dev. 2017;31:184–196. PMID: 28167502. doi:10.1101/gad.291948.116.
- Mizutani A, Wang L, Rajan H, et al. Boat, an AXH domain protein, suppresses the cytotoxicity of mutant ataxin-1. EMBO J. 2005;24:3339–3351. PMID: 16121196. doi:10.1038/sj.emboj.7600785.
- Thompson BJ, Cohen SM. The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell. 2006;126:767–74. doi:10.1016/j.cell.2006.07.013.