
ABSTRACT
‘BRCAness’ is a term used to describe cancer cells that behave similarly to tumors with BRCA1 or BRCA2 mutations. The BRCAness phenotype is associated with hypersensitivity to chemotherapy agents including PARP inhibitors, which are a promising class of recently-licensed anti-cancer treatments. This hypersensitivity arises because of a deficiency in the homologous recombination (HR) pathway for DNA double-strand break repair. To gain further insight into how genetic modifiers of HR contribute to the BRCAness phenotype, we created a new mouse model of BRCAness by generating mice that are deficient in BLM helicase and the Exo1 exonuclease, which are involved in the early stages of HR. We find that cells lacking BLM and Exo1 exhibit a BRCAness phenotype, with diminished HR, and hypersensitivity to PARP inhibitors. We further tested how 53BP1, an important regulator of HR, affects repair efficiency in our BRCAness model. We find that deletion of 53BP1 can relieve several of the repair deficiencies observed in cells lacking BLM and Exo1, just as it does in cells lacking BRCA1. These results substantiate the importance of BRCAness as a concept for classification of cancer cases, and further clarify the role of 53BP1 in regulation of DNA repair pathway choice in mammalian cells.
Introduction
In recent years, a number of compounds forming a new generation of anti-cancer chemotherapy agents, the PARP inhibitors, have been licensed for clinical use [Citation1]. These molecules inhibit the normal activity of poly(ADP-ribose) polymerase (PARP), an enzyme that modifies proteins in the vicinity of DNA damage sites by addition of chains of ADP-ribose monomers. PARP inhibitors produce a significant increase in the rate of DNA double-strand break (DSB) formation. These DSBs can normally be repaired by cellular DNA repair activities, but cells which are deficient in DSB repair are hypersensitive to PARP inhibition, leading to cell death in the presence of PARP inhibitors. Cells from tumors carrying BRCA1 or BRCA2 mutations are especially sensitive to PARP inhibitors, because the homologous recombination (HR) pathway for DSB repair is deficient in these cells [Citation2,Citation3]. PARP inhibitors were therefore originally licensed for treatment of patients with BRCA1/2-associated ovarian cancer.
Other mutations affecting components of DNA repair pathways also confer PARP inhibitor hypersensitivity. The term ‘BRCAness’ has been used to describe a deficiency in HR and PARP inhibitor hypersensitivity, as is observed with BRCA1/2-deficiency [Citation4,Citation5]. By identifying additional genetic signatures associated with ‘BRCAness’ in cancer cells, it may be possible to expand the range of cases that can be treated using PARP inhibitor therapy. Estimates for the proportion of tumors that may exhibit ‘BRCAness’ reach as high as ∼50% for incidences of ovarian carcinoma [Citation6]. One challenge to expanding the range of clinical cases that can be treated with PARP inhibitors is in predicting which tumors are likely to exhibit ‘BRCAness’. BRCAness is generally defined as a deficiency in HR, but the exact contribution of different genetic modifiers of HR in mammalian cells is not fully understood. BRCA1 has also been reported to have roles in other cellular processes, such as regulation of gene expression, which may contribute to the particular repair deficiencies seen in BRCA1-deficient cells [Citation7–9].
One important regulator of HR is p53-Binding Protein 1 (53BP1), which was originally identified as an interacting partner of p53, but which has subsequently been revealed to have a role in repair of DSBs [Citation10]. 53BP1 is recruited to chromatin in the vicinity of DSBs, based on the interaction of its Tudor domain and Ubiquitin Dependent Region with modified histones [Citation11,Citation12]. We previously observed that deletion of the 53bp1 gene in Brca1-deficient mice largely rescues the pathological effects of Brca1 loss-of-function [Citation13–15]. In particular, embryonic lethality associated with homozygous loss of Brca1, or tumor susceptibility arising in cells with conditional knockout of Brca1, are both rescued by co-deletion of 53bp1. Based on measurements of altered HR efficiency in Brca1/53bp1 double-knockout cells, we showed that 53BP1 affects survival of Brca1-deficient cells by limiting the efficiency of HR. Other genetic modifiers of HR efficiency in Brca1-deficient cells, including REV7 and Polθ, have subsequently been identified [Citation16,Citation17].
According to current models, 53BP1 regulates the ability of cells to perform HR at least in part by modulating the extent of resection at DSBs [Citation10,Citation18]. Resection involves processing of a blunt DSB by nuclease activity on the 5’ ends of the DNA fragments, producing 3’ single-stranded DNA overhangs. The single-stranded DNA regions formed by DSB resection are bound by replication protein A (RPA) and subsequently by RAD51 and its paralogs, forming a recombinogenic nucleoprotein filament [Citation7]. The nucleoprotein filament can pair with a homologous DNA sequence, such as homologous sequence on a sister chromatid, leading to formation of a Holliday Junction and template-based repair of the DSB by HR. We proposed that BRCA1 is necessary to overcome a blocking effect of 53BP1 on DSB resection, and BRCA1-deficient cells have a defect in HR because they cannot overcome this inhibitory effect [Citation14,Citation19]. In the absence of 53BP1, Brca1/53bp1 double-knockout cells grow relatively normally, because the requirement for BRCA1 is relieved. This model is supported by several lines of evidence indicating that DSB resection is increased in the absence of 53BP1 [Citation13,Citation14,Citation20,Citation21]. On the other hand, this model also predicts that DSB resection should be deficient in Brca1-deficient cells when 53BP1 is present, but the evidence for a block in DSB resection associated with loss of BRCA1 is inconclusive [Citation8,Citation22]. Using a modified DNA combing assay, Cruz-Garcia et al demonstrated that the normal pace of DSB resection requires the interaction of BRCA1 and CtIP, although the BRCA1-CtIP interaction is not absolutely essential for resection to proceed [Citation23–25]. A cellular system to measure resection at AsiSI endonuclease cut sites also showed no evidence of a defect in resection after BRCA1 knockdown [Citation26]. In our own studies, we have observed no obvious defect in RPA loading at DSB sites induced by ionizing radiation (IR) in Brca1-deficient cells [Citation27].
To better understand the effect of 53BP1 in controlling DSB repair in cells with deficient HR, we have generated a mouse model of ‘BRCAness’, in which DSB resection is predicted to be deficient. The exonuclease, EXO1, and the RECQ helicase, BLM, have been reported to function in parallel pathways for ‘long-range’ resection of DSBs [Citation28–30]. Using Exo1−/− mice in which Blm is conditionally-inactivated, we have generated cells that lack long-range resection activity, and exhibit ‘BRCAness’, as demonstrated by PARP inhibitor hypersensitivity and reduced markers of HR. Co-deletion of the 53bp1 gene is sufficient to substantially rescue HR and partially relieves the cell growth defect of cells lacking both BLM and EXO1. 53bp1 deletion does not cause a significant increase in DSB resection in our ‘BRCAness’ cell model, however, supporting the idea that the effect of 53BP1 in regulating HR depends on other factors in addition to regulation of resection.
Results
Generation of a ‘BRCAness’ mouse model by deletion of Blm and Exo1
Repair of DSBs by HR requires DSB resection, which proceeds by an initial Mre11/CtIP-dependent step followed by ‘long-range’ resection of the DSB by the exonuclease, EXO1, and a complex of BLM/Dna2[Citation7,Citation28–30]. We reasoned that combined targeting of Exo1 and Blm would cause a defect in DNA double-strand break repair, and PARP inhibitor hypersensitivity, by limiting DSB resection. We therefore crossed Exo1+/− mice [Citation31] to Blmfl/fl mice [Citation32], which carry a conditional allele of Blm. Deletion of the Blmfl/fl alleles to generate BlmΔ/Δ cells was achieved by breeding to CD19-Cre knockin mice, which express Cre recombinase in the B lymphocyte lineage only [Citation33]. Primary BlmΔ/Δ Exo1−/− B cells were isolated from the spleens of appropriate mice, and placed in culture with LPS to induce cell proliferation. After two days of cell growth, we treated the cells either with olaparib, a PARP inhibitor, or mitomycin C (MMC), which produces interstrand DNA crosslinks. Following drug treatment, genomic instability was measured in the different genotypes using telomere PNA-FISH to reveal chromosomal abnormalities in metaphase spreads (A) [Citation34]. Although single-knockout BlmΔ/Δ or Exo1−/− metaphase spreads did not show substantial olaparib or MMC sensitivity, BlmΔ/Δ Exo1−/− double-knockout B cells showed a significantly higher rate of chromosome aberrations after drug exposure (B–D). BlmΔ/Δ Exo1−/− cells therefore appear to exhibit features of ‘BRCAness’.
Figure 1. Deletion of 53BP1 restores genomic integrity and cell proliferation in BlmΔ/ΔExo1−/− B cells.
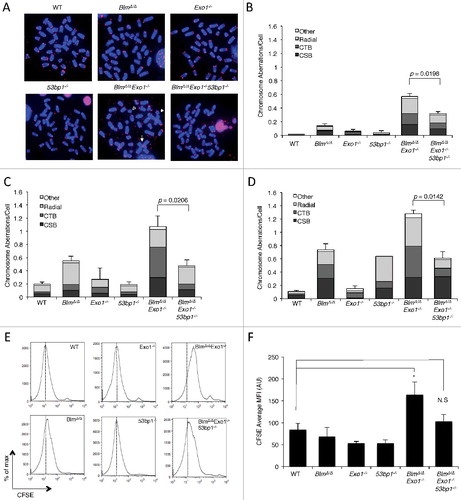
Deletion of 53bp1 rescues genomic instability in BlmΔ/Δ Exo1−/− Model of ‘BRCAness’
As 53bp1 deletion rescues many phenotypes of Brca1-deficient cells [Citation14,Citation35], we tested the contribution of 53BP1 to DNA repair efficiency in cells exhibiting ‘BRCAness’ by crossing BlmΔ/Δ Exo1+/− to 53bp1−/− mice to generate BlmΔ/Δ Exo1−/− 53bp1−/− ‘triple-knockout’ B cells. Despite deletion of these three repair factors, we found that the triple-knockout cells did not show a significant growth defect relative to WT. Interestingly, co-deletion of 53bp1 was sufficient to achieve a significant reduction in the frequency of chromosome aberrations in BlmΔ/Δ Exo1−/− cells treated with either olaparib or MMC (A–C). This result indicates that 53bp1 deletion can result in an improvement in genomic integrity in certain genetic backgrounds associated with ‘BRCAness’, as has previously been observed in Brca1-deficient cells.
To test the impact of combined deletion of Blm and Exo1 on cell growth in the presence of PARP inhibitors, we performed a CFSE dilution assay, which measures the ability of B cells to undergo cell division in vitro. Consistent with the chromosome instability results, olaparib treatment caused a significant reduction in the ability of BlmΔ/Δ Exo1−/− B cells to proliferate in vitro (E and F). BlmΔ/Δ Exo1−/− 53bp1−/− cells proliferated slightly less than WT or single-knockout cells, but this difference did not rise to the level of statistical significance. Together, these results support the idea that genomic instability arising with deletion of long-range resection activities leads to reduced cell growth. The presence of 53BP1 contributes significantly to the repair deficiencies of the BRCAness cells.
We measured levels of phosphorylated Kap1 in cells lacking BLM, EXO1 or 53BP1 after ionizing radiation, but did not find any substantial differences between the various genotypes (Figure S1A). This result suggests that phenotypes of BLM/EXO1-deficient cells are not dependent on reduced ATM signaling. We also measured class switch recombination of B lymphocytes lacking BLM, EXO1 or 53BP1 (Fig S1B-C). As expected, 53BP1−/− cells showed a substantial defect in class switching to IgG1, and this rate was further lowered by combined deletion of BLM and EXO1. Targeting resection activities therefore does not rescue the class switch recombination phenotype of 53BP1−/− cells.
53BP1 blocks homologous recombination in cells lacking long-range resection activities
Next, we tested whether the genomic instability phenotype of BlmΔ/Δ Exo1−/− cells was associated with a defect in homologous recombination. First, we quantified the appearance of nuclear foci of RAD51, a key component of the HR pathway, at IR-induced DNA damage sites (A and B). RAD51 foci are dependent on DSB resection, and deletion of resection factors, such as CtIP, reduces RAD51 foci formation [Citation24]. BlmΔ/Δ Exo1−/− cells showed a significant defect in accumulation of RAD51 foci, which was more pronounced than the defect evident in cells that were deficient in either Blm or Exo1 individually. The genomic instability observed in the double-mutant cells therefore correlates with a defect in HR, consistent with the known role of BLM and EXO1 in long-range resection of DSBs during HR-mediated repair. When 53bp1 was also deleted, to make BlmΔ/Δ Exo1−/− 53bp1−/− B cells, we found that RAD51 foci were substantially restored relative to BlmΔ/Δ Exo1−/− cells. This result suggests that the improved genomic integrity and cell growth after olaparib treatment observed with co-deletion of 53bp1 in BlmΔ/Δ Exo1−/− cells is related to improved HR. This result is similar to that seen with Brca1Δ11/Δ11 cells, which show genomic instability and reduced cell growth after PARP inhibitor treatment that can be reversed by co-deletion of 53bp1 [Citation14,Citation15,Citation35]. Using knockdown of BLM, EXO1 and 53BP1 in U2OS osteosarcoma cells, we were furthermore able to show that of deletion of these factors gave an equivalent effect on RAD51 foci formation in human cells (Fig S2A-C).
Figure 2. Deletion of 53BP1 restores HR in BLM and EXO1 double-deficient cells.
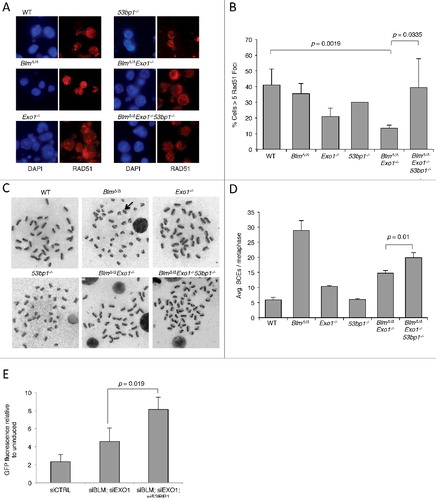
As a second measure of HR, we assayed the frequency of sister chromatid exchanges in B cells after overnight treatment with the PARP inhibitor, olaparib. Sister chromatid exchanges form as a result of crossover HR involving homologous sequences on sister chromatids in late S/G2 phase. As such, the rate of sister chromatid exchanges can be used as a measure of one product of HR [Citation36]. BlmΔ/Δ cells are known to have a high rate of sister chromatid exchanges, because BLM mediates an alternative pathway leading to non-crossover resolution of HR intermediates (C and D) [Citation7,Citation37]. We nonetheless found that BlmΔ/Δ Exo1−/− cells show significantly lower rates of sister chromatid exchanges relative to BlmΔ/Δ cells, suggesting that in the absence of long-range DSB resection, the high rates of crossover HR normally seen in BlmΔ/Δ cells cannot be sustained. This result points to a reduced rate of HR in BlmΔ/Δ Exo1−/− cells. The rate of SCEs is increased in BlmΔ/Δ Exo1−/− 53bp1−/− B cells (D), again demonstrating that 53BP1 exerts a block on HR in the BlmΔ/Δ Exo1−/− model of ‘BRCAness’. These results are reminiscent of observations made upon co-deletion of 53bp1 in Brca1Δ11/Δ11 cells. We also found that deletion of 53bp1 increases the rate of SCEs in BlmΔ/Δ B cells, as has previously been found using knockdown approaches in BLM-deficient cells [Citation38]. As a third measure of HR, we used EJ-DR reporter cells, which express GFP following HR-mediated repair of induced I-SceI-mediated DSBs (E). Knockdown of 53BP1 correlated with a higher frequency of HR in cells treated with siBLM and siEXO1 oligos, indicating that 53BP1 restricts the use of HR in cells lacking key DSB resection factors.
Reduced DSB resection in mouse ‘BRCAness’ model
To test whether the reduced genomic integrity and HR efficiency of our BlmΔ/Δ Exo1−/− cells was related to reduced DSB resection in these cells, we used a flow cytometry approach to detect RPA bound at single-stranded DNA regions (A) [Citation39]. RPA loading in this assay is linked to single-stranded DNA regions that arise during replication in S phase, and also single-stranded DNA that is formed at break sites through resection in the late S and G2 phases of the cell cycle. By quantifying RPA staining in late S and G2 phase cells, we were able to discern an increase in RPA intensity following DNA damage in WT cells, consistent with an increase in DSB resection (B and C). RPA intensity following DNA damage was not substantially reduced relative to the WT in BlmΔ/Δ, Exo1−/− or 53bp1−/− single-knockout cells. RPA intensity was significantly reduced, however, in BlmΔ/Δ Exo1−/− double-knockout cells. This result demonstrates that in primary B cells, normal DSB resection is dependent on redundant contributions from both BLM and EXO1, as has been seen previously using knockdowns in cell line models [Citation28]. The defect in DSB resection in double-knockout cells correlates with and likely accounts for the defective HR and repair phenotypes observed in these cells. We hypothesized that co-deletion of 53BP1 would rescue the resection defect evident in BlmΔ/Δ Exo1−/− cells. Surprisingly, in BlmΔ/Δ Exo1−/− 53bp1−/− cells, the rate of resection was not significantly different from BlmΔ/Δ Exo1−/− cells. This result suggests that rescue of genomic integrity by deletion of 53BP1 from BlmΔ/Δ Exo1−/− cells may not require a large increase in DSB resection.
Figure 3. Measurement of DSB resection by BLM and EXO1 in the absence of 53BP1.
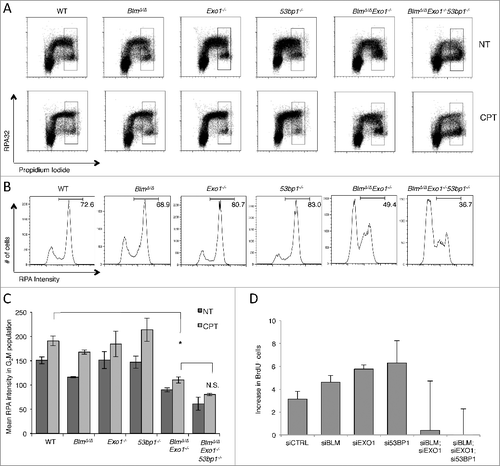
As a second measure of DSB resection in cells lacking BLM, EXO1 and 53BP1, we applied native BrdU immunofluorescence to measure exposed single-stranded DNA regions after ionizing radiation-induced DNA damage [Citation13]. After knockdown of BLM, EXO1 and/or 53BP1, cells were grown for 48 hrs in BrdU medium. Following DNA damage, BrdU exposed at resected DNA break sites was detected using an anti-BrdU antibody and flow cytometry (Figure S3). Using this approach, we were able to detect an increase in exposed single-stranded DNA, consistent with DSB resection, in control cells and cells with single knockdown of BLM, EXO1 or 53BP1, following ionizing radiation. Cells with combined knockdown of BLM and EXO1, or ‘triple-knockdown cells’ lacking BLM, EXO1 and 53BP1, did not show an equivalent increase in exposed BrdU signal, indicating that resection is defective in these cells (D). This result is consistent with our findings using RPA FACS, which showed that deletion of 53BP1 does not cause an increase in resection sufficient to be detected by these methods.
Lack of long-range DSB resection activities leads to defective G2M checkpoint
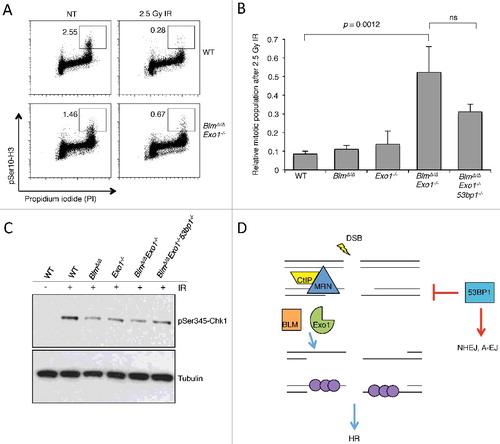
In addition to a defect in HR, Brca1-deficient cells show defective cell cycle checkpoints [Citation40]. The G2M checkpoint normally prevents entry of cells carrying chromosome breaks into mitosis, and is induced by DNA damage signaling involving ATM-Chk2 or ATR-Chk1 [Citation41]. The activation of the ATR-Chk1 signaling axis requires association of the ATR kinase with its partners, TOPBP1 or ETAA1, at RPA-bound single-stranded DNA regions [Citation42]. RPA loading requires resection, and defects in DSB resection, such as that caused by deletion of CtIP, reduce ATR activation and lead to a failure of the G2M checkpoint [Citation43,Citation44]. To test if the reduced DSB resection that we observed in BlmΔ/Δ Exo1−/− B cells is associated with a defect in signaling pathways leading to induction of the G2M checkpoint, we measured the proportions of mitotic cells after DNA damage induced by ionizing radiation. We found that deletion of either Blm or Exo1 singly does not cause a significant G2M checkpoint defect, but combined deletion of Blm and Exo1 caused a failure in the G2M checkpoint (A and B). To test if 53bp1 deletion modified the G2M checkpoint defect of BlmΔ/Δ Exo1−/− cells, we also tested BlmΔ/Δ Exo1−/− 53bp1−/− cells after IR treatment. 53bp1 deletion did not completely reverse the G2M checkpoint defect of BlmΔ/Δ Exo1−/− cells. BlmΔ/Δ Exo1−/− 53bp1−/− cells also showed a mitotic index after IR that was elevated relative to WT controls. Levels of Chk1 phosphorylation, which are indicative of ATR activation, were reduced in BLM/EXO1-deficient cells, and only partially rescued by 53BP1 deletion (C). Taken together, this evidence suggests that 53BP1 deletion is not sufficient to completely rescue ATR activation or restore a normal G2M checkpoint defect in BlmΔ/Δ Exo1−/− cells. Notably, 53bp1 deletion also does not rescue the G2M checkpoint defect of Brca1-deficient cells [Citation45].
Figure 4. Analysis of cell cycle progression after ionizing radiation in the absence of BLM, EXO1 and 53BP1.
Discussion
The concept of ‘BRCAness’ has been used to describe tumors that behave like BRCA1- or BRCA2-associated cancer cases by having a defect in HR and hypersensitivity to chemotherapy agents such as olaparib and cisplatin. To attempt to better test our understanding of ‘BRCAness’, we set out to create a mouse model that we hypothesized would show cellular phenotypes similar to those seen in cancer cells harboring BRCA1 or BRCA2 mutations. BLM and EXO1 have been shown to have parallel roles in ‘long-range’ DSB resection using biochemical approaches and in cell lines, but the importance of ‘long-range’ resection for HR in primary mammalian cells has not previously been tested [Citation28–30]. We find that combined deletion of these factors results in a ‘BRCAness’ phenotype. First, BlmΔ/Δ Exo1−/− cells are hypersensitive to PARP inhibition, showing elevated genomic instability and reduced cell growth after olaparib treatment. Second, they lack hallmarks of HR, such as DNA damage-induced RAD51 foci. Third, DSB resection is reduced in BlmΔ/Δ Exo1−/− cells, a phenotype that may also be associated with loss of BRCA1. Finally, BlmΔ/Δ Exo1−/− cells show a defect in the G2M checkpoint, as is seen in BRCA1- and BRCA2-deficient cells. These observations support the idea that BlmΔ/Δ Exo1−/− cells are a valid model for ‘BRCAness’.
HR initiates by an initial resection step involving CtIP and Mre11 [Citation7]. Our data is congruent with previous studies which show that, in the presence of 53BP1, this initial resection step is not sufficient for efficient HR [Citation28–30]. Additional resection mediated by BLM and Exo1 appears to be essential to support normal HR-mediated repair. Several other factors have been proposed to act in long-range resection in mammalian cells, including the RECQ family helicases, WRN and RECQL4 [Citation46–48]. Our results do not rule out the idea that these factors contribute to long-range resection, but they suggest that these additional ‘long-range resection’ factors cannot substitute fully for BLM and EXO1. Some level of HR is nonetheless possible in the absence of BLM and EXO1, as can be seen by the sustained rate of crossover HR events observed using the sister chromatid exchange assay. In contrast, knockdown of CtIP leads to a lower rate of SCE formation than in control cells [Citation49]. The initial stage of resection mediated by CtIP is therefore of more fundamental importance to HR than subsequent long-range resection by BLM and Exo1.
Our ‘BRCAness’ model involving combined deletion of Blm and Exo1 allows us to further test the importance of 53BP1 as a factor regulating DSB repair efficiency in cells with HR defects. 53BP1 expression correlates with survival in BRCA1-dependent breast cancer [Citation35,Citation50], and 53BP1 may also be an important modulator of tumor cell survival or chemoresistance in the context of other genetic mutations. In previous work, we found that deletion of 53BP1 largely rescues the repair deficiency of Brca1-deficient cells, but has little or no effect in cells with deficiencies in the HR genes Brca2, Palb2, and Xrcc2, or the Fanconi Anemia gene, Fancd2 [Citation13,Citation14,Citation35,Citation51]. We proposed that 53BP1 inhibits HR by reducing DSB resection. According to this model, 53bp1 deletion rescues the loss of HR in mutants in which there is a resection defect, but does not rescue the loss of HR in mutants that affect later stages of the recombination process (such as BRCA2 or XRCC2). The results of the present study are partially consistent with this model. BlmΔ/Δ Exo1−/− cells showed genomic instability linked to an HR defect, which correlated with reduced DSB resection. The HR defect and genomic instability phenotype showed a significant rescue when 53BP1 was additionally deleted. Interestingly, however, we did not observe a measurable increase in DSB resection in BlmΔ/Δ Exo1−/− cells when 53bp1 was deleted (B). This result suggests one of two possibilities (D). First, the effect of 53BP1 on resection dynamics may be most relevant at early timepoints after DSB induction. Deletion of 53bp1 may allow the initial CtIP/Mre11-mediated stage of resection to proceed more quickly, increasing the recombinogenic potential of the DSB, even though we could not detect a difference using our RPA loading assay. Alternatively, our results may indicate that 53BP1 has an additional important role in mediating choice of DSB repair pathways besides control of DSB resection.
In recent years, several functions of 53BP1 have been identified, which may contribute to its role in regulation of repair of DSBs. 53BP1 recruits its downstream interacting partners, RIF1 and PTIP, which regulate resection and DSB joining [Citation52–57]. PTIP associates with the endonuclease, Artemis, to cleave single-stranded DNA regions, potentially reducing the apparent rate of resection, but creating mutations by loss of DNA sequence surrounding the break site [Citation58]. At heterochromatic regions, 53BP1 recruitment is reported to enable heterochromatin relaxation, to facilitate DNA repair [Citation59,Citation60]. 53BP1 antagonizes mutagenic single-strand annealing, that may occur as a consequence of extensive double-strand break resection [Citation61]. Finally, 53BP1 also appears to contribute to DSB repair by nonhomologous end-joining (NHEJ) by mediating the joining of distant DNA ends, and by increasing the mobility of DSBs within the nucleus [Citation62–65]. These pro-NHEJ effects of 53BP1 are most likely to contribute to mutation in cells exhibiting ‘BRCAness’, because NHEJ has a propensity to introduce mutations at the repair junction, and to produce translocations [Citation19].
Ultimately, understanding the exact requirements for resection leading to productive HR will require a more holistic analysis of the different factors that contribute to this process. Notably, the role of BRCA1 in mediating resection or displacing 53BP1 remains unclear, although recent results indicate that the E3 ubiquitin ligase activity of BRCA1 may mediate 53BP1 repositioning [Citation66]. Nonetheless, our results further demonstrate the usefulness of ‘BRCAness’ as a concept for characterization of cancer cases. Mutation or loss of expression of resection factors such as BLM and Exo1 can clearly lead to mutagenic outcomes at the cellular level, which could contribute to malignancy. Identifying these changes would allow greater use of the PARP inhibitor drug family for anti-cancer therapy.
Methods
Animal husbandry
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Rutgers University, protocol 12–024.
Metaphase spreads
B cells were arrested in metaphase using 100 ng/ml of colcemid for 1 hour and fixed in 3:1 methanol:acetic acid fixative. PNA-FISH was performed as previously described [Citation34].
G2M cell cycle checkpoint
B cells were grown for 48 hours, treated with 2.5 Gy of ionizing radiation from a 237Cs gamma source, and collected 1 hour after irradiation. B cells were fixed in 70% ethanol and permeabilized in 0.25% Triton X-100 in PBS. Cells were stained with 0.75 μg of anti-phosphoSer10- histone H3 antibody (Millipore 06–570) and a 1:200 dilution of goat anti-rabbit Alexa-488 antibody. Cells were suspended in 1 mg/ml propidium iodide and 0.1 mg of RNase A in PBS and incubated at 37°C in the dark for 30 minutes. Mitotic cells were measured using a Becton Dickinson FACSCalibur and data were analyzed by FlowJo software.
Western blot and immunofluorescence
For western blotting, primary antibodies were used at the following dilutions: anti-tubulin (1:50,000, Sigma Aldrich) and rabbit anti-pChk1 Ser317 (1:500, Cell Signaling 2344S). For immunofluorescence, B lymphocyte cells were activated and treated with 10 Gy IR and 4 hour recovery prior to being dropped onto slides coated with Cell-Tak (BD). Cells were pre-extracted in 0.5% TX-100 in PBS, fixed in 2% paraformaldehyde in PBS, and permeabilized in 0.5% TX-100 in PBS and incubated in antibody. Primary antibody anti-Rad51 was used at a 1:100 dilution (Santa Cruz H-92) and detected with anti-rabbit Alexa-546 antibody at a 1:200 dilution (Thermo Fisher). Cells were counterstained with DAPI and mounted in Mowiol solution.
RPA FACS
B cells were treated, fixed, and stained as previously described [Citation39]. Primary antibody anti-rat RPA-32 (Cell Signaling 2208S) was used at a 1:500 dilution. B cells were suspended in 1 mg/ml propidium idodide and 0.1 mg of RNase A and incubated at 37°c for 30 minutes in the dark. RPA+ cells were measured using a Becton Dickinson FACSCalibur and data were analyzed by FlowJo software.
Sister chromatid exchange assay
B cells were grown in 10 μM BrdU for 48 hours prior to fixation. Slides were stained with 10 μg/ml Hoechst 33258 in PBS. Stained slides were rinsed in McIlvaine's solution (164 mM Na2HPO4, 16 mM citric acid, pH 7.0) and then UV irradiated using a transilluminator for 45 minutes on a low setting. Slides were incubated with pre-warmed 1x SSC at 55°c followed by ddH20. Slides were stained in a 1:12 Giemsa stain containing 3% methanol for 30 minutes. Slides were rinsed with ddH20 and then dehydrated by placing in a Coplin jar containing xylenes for 15 seconds, air dried, and mounted with Permount.
U20S EJ-DR assay
DNA repair assay was performed as previously described using U2OS EJ-DR cells [Citation67]. EJDR reporter cells were plated at 200,000 cells per wells in DMEM with 10% charcoal-stripped FBS (100-119; Gemini), antibiotic/antimycotic and transfected the next day with siRNA using Lipofectamine RNAiMAX (13778-150: invitrogen). 2 days after siRNA transfection, we replaced the media with DMEM containing 10% Tetracycline-free FBS (100-800; Gemini) and antibiotic/antimycotic. Incorporated I-Sce1 was induced with Shield1 (632189; Clontech) and triamcinolone (T6510; Sigma Aldrich) for 24 hrs. Relative NHEJ and HR activity were assessed 48 hrs after induction by quantification of the percentages of DsRed- and GFP-positive cells on BD FACS Calibur system and analyzed on FlowJo (Tree Star).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KCCY_A_1456295.pdf
Download PDF (1.3 MB)Acknowledgements
This work was funded by NIH R01CA190858 (to SFB). SMM, DSP and JH are supported by the New Jersey Commission on Cancer Research Pre-Doctoral and Post-Doctoral Fellowships. SMM and DSP also received support from the NIH Ruth L. Kirschstein National Research Service Award T32 GM8339.
Additional information
Funding
Related Research Data
References
- Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science. 2017;355:1152–1158. doi:10.1126/science.aam7344. PMID:28302823
- Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi:10.1038/nature03443. PMID:15829966
- Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi:10.1038/nature03445. PMID:15829967
- Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16:110–120. doi:10.1038/nrc.2015.21. PMID:26775620
- Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–819. doi:10.1038/nrc1457. PMID:15510162
- Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi:10.1038/nature10166. PMID:21720365
- Kowalczykowski SC. An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb Perspect Biol. 2015; 7:a016410.
- Prakash R, Zhang Y, Feng W, et al. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7:a016600. doi:10.1101/cshperspect.a016600. PMID:25833843
- Starita LM, Parvin JD. The multiple nuclear functions of BRCA1: transcription, ubiquitination and DNA repair. Curr Opin Cell Biol. 2003;15:345–350. doi:10.1016/S0955-0674(03)00042-5. PMID:12787778
- Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15:7–18. doi:10.1038/nrm3719. PMID:24326623
- Botuyan MV, Lee J, Ward IM, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi:10.1016/j.cell.2006.10.043. PMID:17190600
- Fradet-Turcotte A, Canny MD, Escribano-Diaz C, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–54. doi:10.1038/nature12318. PMID:23760478
- Bunting SF, Callen E, Kozak ML, et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell. 2012;46:125–135. doi:10.1016/j.molcel.2012.02.015. PMID:22445484
- Bunting SF, Callen E, Wong N, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi:10.1016/j.cell.2010.03.012. PMID:20362325
- Cao L, Xu X, Bunting SF, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell. 2009;35:534–541. doi:10.1016/j.molcel.2009.06.037. PMID:19716796
- Mateos-Gomez PA, Gong F, Nair N, et al. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature. 2015;518:254–257. doi:10.1038/nature14157. PMID:25642960
- Xu G, Chapman JR, Brandsma I, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521:541–544. doi:10.1038/nature14328. PMID:25799992
- Daley JM, Sung P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol. 2014;34:1380–1388. doi:10.1128/MCB.01639-13. PMID:24469398
- Bunting SF, Nussenzweig A. End-joining, translocations and cancer. Nat Rev Cancer. 2013;13:443–454. doi:10.1038/nrc3537. PMID:23760025
- Dorsett Y, Zhou Y, Tubbs AT, et al. HCoDES reveals chromosomal DNA end structures with single-nucleotide resolution. Mol Cell. 2014;56:808–818. doi:10.1016/j.molcel.2014.10.024. PMID:25435138
- Hakim O, Resch W, Yamane A, et al. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature. 2012;484:69–74. PMID:22314321
- Aparicio T, Baer R, Gautier J. DNA double-strand break repair pathway choice and cancer. DNA Repair. 2014;19:169–175. doi:10.1016/j.dnarep.2014.03.014. PMID:24746645
- Cruz-Garcia A, Lopez-Saavedra A, Huertas P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 2014;9:451–459. doi:10.1016/j.celrep.2014.08.076. PMID:25310973
- Polato F, Callen E, Wong N, et al. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J Exp Med. 2014;211:1027–1036. doi:10.1084/jem.20131939. PMID:24842372
- Reczek CR, Szabolcs M, Stark JM, et al. The interaction between CtIP and BRCA1 is not essential for resection-mediated DNA repair or tumor suppression. J Cell Biol. 2013;201:693–707. doi:10.1083/jcb.201302145. PMID:23712259
- Zhou Y, Caron P, Legube G, et al. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res. 2014;42:e19. doi:10.1093/nar/gkt1309. PMID:24362840
- Patel DS, Misenko SM, Her J, et al. BLM helicase regulates DNA repair by counteracting RAD51 loading at DNA double-strand break sites. J Cell Biol. 2017;216:3521–3534. doi:10.1083/jcb.201703144. PMID:28912125
- Gravel S, Chapman JR, Magill C, et al. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi:10.1101/gad.503108. PMID:18923075
- Nimonkar AV, Genschel J, Kinoshita E, et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–362. doi:10.1101/gad.2003811. PMID:21325134
- Nimonkar AV, Ozsoy AZ, Genschel J, et al. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci U S A. 2008;105:16906–16911. doi:10.1073/pnas.0809380105. PMID:18971343
- Wei K, Clark AB, Wong E, et al. Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 2003;17:603–614. doi:10.1101/gad.1060603. PMID:12629043
- Chester N, Babbe H, Pinkas J, et al. Mutation of the murine Bloom's syndrome gene produces global genome destabilization. Mol Cell Biol. 2006;26:6713–6726. doi:10.1128/MCB.00296-06. PMID:16914751
- Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25:1317–1318. doi:10.1093/nar/25.6.1317. PMID:9092650
- Misenko SM, Bunting SF. Rapid analysis of chromosome aberrations in mouse B lymphocytes by PNA-FISH. J Vis Exp: JoVE. 2014;90:e51806. doi:10.3791/51806. PMID:25177909
- Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–695. doi:10.1038/nsmb.1831.
- Simpson LJ, Sale JE. Sister chromatid exchange assay. Subcell Biochem. 2006;40:399–403. PMID: 17623929
- Chaganti RS, Schonberg S, German J. A manyfold increase in sister chromatid exchanges in Bloom's syndrome lymphocytes. Proc Natl Acad Sci U S A. 1974;71:4508–4512. doi:10.1073/pnas.71.11.4508. PMID: 4140506
- Tripathi V, Nagarjuna T, Sengupta S. BLM helicase-dependent and -independent roles of 53BP1 during replication stress-mediated homologous recombination. J Cell Biol. 2007;178:9–14. doi:10.1083/jcb.200610051. PMID: 17591918
- Forment JV, Walker RV, Jackson SP. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2012;81:922–928. doi:10.1002/cyto.a.22155. PMID: 22893507
- Xu B, Kim S, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol. 2001;21:3445–3450. doi:10.1128/MCB.21.10.3445-3450.2001. PMID: 11313470
- Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 2013;5:a012716.
- Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. 2017.
- Kousholt AN, Fugger K, Hoffmann S, et al. CtIP-dependent DNA resection is required for DNA damage checkpoint maintenance but not initiation. J Cell Biol. 2012;197:869–876. doi:10.1083/jcb.201111065. PMID: 22733999
- Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol. 2004;24:9478–9486. doi:10.1128/MCB.24.21.9478-9486.2004. PMID: 15485915
- Li M, Cole F, Patel DS, et al. 53BP1 ablation rescues genomic instability in mice expressing ‘RING-less’ BRCA1. EMBO reports. 2016;17:1532–1541. doi:10.15252/embr.201642497. PMID: 27670884
- Lu H, Shamanna RA, Keijzers G, et al. RECQL4 Promotes DNA end resection in repair of DNA double-strand breaks. Cell Rep. 2016;16:161–173. doi:10.1016/j.celrep.2016.05.079. PMID: 27320928
- Sturzenegger A, Burdova K, Kanagaraj R, et al. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J Biol Chem. 2014;289:27314–27326. doi:10.1074/jbc.M114.578823. PMID: 25122754
- Tomimatsu N, Mukherjee B, Deland K, et al. Exo1 plays a major role in DNA end resection in humans and influences double-strand break repair and damage signaling decisions. DNA repair. 2012;11:441–448. doi:10.1016/j.dnarep.2012.01.006. PMID: 22326273
- Feng Z, Zhang J. A dual role of BRCA1 in two distinct homologous recombination mediated repair in response to replication arrest. Nucleic Acids Res. 2012;40:726–738. doi:10.1093/nar/gkr748. PMID: 21954437
- Bi J, Huang A, Liu T, et al. Expression of DNA damage checkpoint 53BP1 is correlated with prognosis, cell proliferation and apoptosis in colorectal cancer. Int J Clin Exp Pathol. 2015;8:6070–6082. PMID: 26261485
- Bowman-Colin C, Xia B, Bunting S, et al. Palb2 synergizes with Trp53 to suppress mammary tumor formation in a model of inherited breast cancer. Proc Natl Acad Sci U S A. 2013;110:8632–8637. doi:10.1073/pnas.1305362110. PMID: 23657012
- Callen E, Di Virgilio M, Kruhlak MJ, et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell. 2013;153:1266–1280. doi:10.1016/j.cell.2013.05.023. PMID: 23727112
- Chapman JR, Barral P, Vannier JB, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–871. doi:10.1016/j.molcel.2013.01.002. PMID: 23333305
- Di Virgilio M, Callen E, Yamane A, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711–715. doi:10.1126/science.1230624. PMID: 23306439
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872–883. doi:10.1016/j.molcel.2013.01.001. PMID: 23333306
- Feng L, Fong KW, Wang J, et al. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem. 2013;288:11135–11143. doi:10.1074/jbc.M113.457440. PMID: 23486525
- Zimmermann M, Lottersberger F, Buonomo SB, et al. 53BP1 regulates DSB repair using Rif1 to control 5' end resection. Science. 2013;339:700–704. doi:10.1126/science.1231573. PMID: 23306437
- Wang J, Aroumougame A, Lobrich M, et al. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 2014;28:2693–2698. doi:10.1101/gad.252478.114. PMID: 25512557
- Hansen RK, Mund A, Poulsen SL, et al. SCAI promotes DNA double-strand break repair in distinct chromosomal contexts. Nat Cell Biol. 2016;18:1357–1366. doi:10.1038/ncb3436. PMID: 27820601
- Noon AT, Shibata A, Rief N, et al. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat Cell Biol. 2010;12:177–184. doi:10.1038/ncb2017. PMID: 20081839
- Ochs F, Somyajit K, Altmeyer M, et al. 53BP1 fosters fidelity of homology-directed DNA repair. Nat Struct Mol Biol. 2016;23:714–721. doi:10.1038/nsmb.3251.
- Bothmer A, Robbiani DF, Feldhahn N, et al. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med. 2010;207:855–865. doi:10.1084/jem.20100244. PMID: 20368578
- Difilippantonio S, Gapud E, Wong N, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533. doi:10.1038/nature07476. PMID: 18931658
- Dimitrova N, Chen YC, Spector DL, et al. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi:10.1038/nature07433. PMID: 18931659
- Lottersberger F, Karssemeijer RA, Dimitrova N, et al. 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell. 2015;163:880–893. doi:10.1016/j.cell.2015.09.057. PMID: 26544937
- Densham RM, Garvin AJ, Stone HR, et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol. 2016;23:647–655. doi:10.1038/nsmb.3236.
- Bindra RS, Goglia AG, Jasin M, et al. Development of an assay to measure mutagenic non-homologous end-joining repair activity in mammalian cells. Nucleic Acids Res. 2013;41:e115. doi:10.1093/nar/gkt255. PMID: 23585275