
ABSTRACT
Mutagenesis is a hallmark and enabling characteristic of cancer cells. The E3 ubiquitin ligase RAD18 and its downstream effectors, the ‘Y-family’ Trans-Lesion Synthesis (TLS) DNA polymerases, confer DNA damage tolerance at the expense of DNA replication fidelity. Thus, RAD18 and TLS polymerases are attractive candidate mediators of mutagenesis and carcinogenesis. The skin cancer-propensity disorder xeroderma pigmentosum-variant (XPV) is caused by defects in the Y-family DNA polymerase Pol eta (Polη). However it is unknown whether TLS dysfunction contributes more generally to other human cancers. Recent analyses of cancer genomes suggest that TLS polymerases generate many of the mutational signatures present in diverse cancers. Moreover biochemical studies suggest that the TLS pathway is often reprogrammed in cancer cells and that TLS facilitates tolerance of oncogene-induced DNA damage. Here we review recent evidence supporting widespread participation of RAD18 and the Y-family DNA polymerases in the different phases of multi-step carcinogenesis.
Introduction
The discovery of error-prone ‘Y-family’ Trans-Lesion Synthesis (TLS) DNA polymerases was a major advance that provided a molecular mechanism for mutagenesis and carcinogenesis. DNA Polymerase eta (Polη), the prototypical member of the Y-family DNA Polymerase family in humans, was first identified as the mutated XPV gene product of skin cancer-prone xeroderma pigmentosum-variant patients [Citation1]. Defining the molecular etiology of XPV syndrome demonstrated that normal expression and activity of TLS pathway components is necessary for tumor-suppression and that TLS pathway imbalance (for example due to Polη-deficiency) can lead to mutations and skin cancer.
However, the extent to which the Y-family polymerases are altered in neoplastic cells and whether TLS dysfunction impacts tumorigenesis more broadly is not known. Recent studies show that many cancers have hallmarks of Polη-mediated mutagenesis [Citation2], that the TLS pathway is pathologically activated in many cancer cells [Citation3,Citation4] and that TLS can sustain DNA replication and viability of cells experiencing oncogene-induced DNA damage [Citation5]. Moreover, it is well established that TLS confers chemoresistance to a variety of therapeutic agents [Citation6,Citation7]. Taken together, these findings indicate that TLS may have diverse roles in cancer etiology and therapy. Here we consider the potential impact of the TLS pathway on cancer with a particular emphasis on newer roles of Y-family polymerases and their proximal activator RAD18 in cancer cells.
Background
Imbalanced TLS pathway function causes skin carcinogenesis
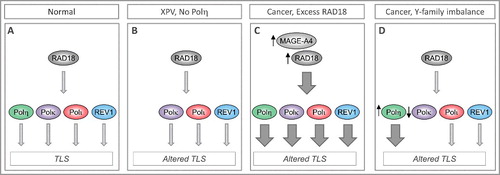
Seminal work by Masutani and colleagues showed that the sunlight-sensitivity and skin cancer-propensity syndrome xeroderma pigmentosum-Variant (XPV) is caused by inactivating mutations in Polη [Citation1]. Polη is a specialized DNA polymerase that is error-prone when replicating undamaged templates but can perform efficient and error-free replicative bypass of templates harboring UV-induced cyclobutane pyrimidine dimers or CPD [Citation8]. Thus Polη sustains replication of UV-damaged genomes and confers cell viability (termed ‘DNA damage tolerance’). In the absence of Polη, compensatory and error-prone TLS of CPD by alternative Y-family DNA Polymerases, Pol kappa (Polκ) and Pol iota (Polι) leads to hypermutability [Citation9,Citation10] (A, B), thereby explaining the cancer predisposition phenotype of individuals with XPV. Similar to Polη, the other TLS polymerases are also error-prone on undamaged templates yet are relatively error-free when copying DNA templates harboring damage from their cognate lesions. For example Polκ is unable to bypass cis-syn T-T dimers, yet replicates templates containing benzo[a]pyrene-adducted guanines by inserting the correct C opposite the bulky lesion [Citation11].
Figure 1. Imbalanced expression/ activity of RAD18 and Y-family DNA polymerases dictates choice of TLS polymerases and can influence replication fidelity and genome stability. (A) In normal cells TLS polymerases are activated sparingly and used selectively to minimize error-prone DNA synthesis and ensure genome stability. (B) In XPV cells lacking functional Polh, compensatory bypass of Polh-cognate lesions by inappropriate DNA polymerases leads to mutagenesis. (C) In many cancer cells RAD18 is expressed at high levels (sometimes owing to stabilization by its binding partner MAGE-A4), leading to increased TLS pathway activation. It is not known whether all Y-family DNA polymerases are equally dependent on RAD18 for activation. Differential activation of Y-family TLS polymerases by over-active RAD18 would constitute a mechanism of imbalance and altered TLS. (D) Over-expression or reduced expression of individual TLS polymerases has been reported in many cancers and represents another mechanism of TLS pathway imbalance and mutagenesis.
RAD18, ubiquitin signaling and the TLS polymerase switch
Human XPV syndrome illustrates the detrimental impact of imbalanced TLS polymerase activity on genome stability. Clearly, to maintain balance and prevent mutagenesis, TLS polymerases must be used sparingly only when needed to replicate damaged templates. Consequently, there has been immense interest in elucidating the mechanisms that regulate TLS polymerase activities and determine whether DNA replication is mutagenic. Ubiquitin signaling is a major mechanism of TLS polymerase recruitment to replisomes. In response to DNA damage, the E3 ubiquitin ligase RAD18 mono-ubiquitinates the DNA polymerase processivity factor PCNA at a conserved lysine residue (K164) [Citation12,Citation13].
DNA damage-induced RAD18 activation and PCNA mono-ubiquitination are dependent on RPA-coated ssDNA [Citation14], a species that can arise due to helicase/polymerase uncoupling at stalled DNA replication forks or as an intermediate during DNA repair [Citation15]. Thus PCNA mono-ubiquitination occurs in response to DNA replication stalling in S-phase, and during the course of BER, MMR, and NER in non-replicating cells [Citation16–18]. Y-family DNA polymerases have Ubiquitin-binding Zinc Finger (UBZ) and Ubiquitin-Binding Motif (UBM) domains [Citation19]. The UBZ and UBM domains, together with PCNA-interacting Peptide (PIP) motifs common to many DNA polymerases, mediate the association of Y-family members with the mono-ubiquitinated form of PCNA (PCNA-Ub) [Citation19].
Association of TLS pols with PCNA-Ub engages TLS pols at the replisomes and facilitates replicative bypass of DNA lesions. There is also some evidence that TLS polymerases themselves participate in PCNA mono-ubiquitination. For example, Polη exists in a complex with RAD18 [Citation20], facilitates the association of RAD18 with PCNA, and promotes PCNA mono-ubiquitination [Citation21]. In ectopic expression experiments even modest (∼2-fold) increases in levels of RAD18, Polη, or other Y-family DNA polymerases can stimulate PCNA mono-ubiquitination and lead to inappropriate and error-prone DNA replication by Y-family polymerases [Citation21]. Therefore, imbalanced expression or activity of TLS pathway components has the potential to cause mutagenic outcomes.
Is TLS altered in cancer cells?
Owing to their error-propensity, TLS polymerases are very attractive candidate mediators of mutagenesis in cancer. the question then arises – is there any evidence that TLS proteins are inappropriately utilized in neopastic cells, that altered TLS shapes the genomic landscape of cancer cells, or that TLS contributes to tumorigenic phenotypes? These issues will be considered in turn below.
(i) TLS-mediated mutational signatures and mutable motifs in cancer genomes. The biochemical properties of the Y-family polymerases and their replication fidelities on damaged and undamaged templates have been studied for many years. From in vitro and in vivo studies there is now a wealth of information on the mutagenic consequences and DNA context specificities (mutable motifs and mutational signatures) of error-prone DNA replication by the TLS polymerases [Citation22]. Cancer genome sequencing efforts are now revealing numerous mutations, some of which probably result from TLS polymerase activities.
Cancer genome studies of somatic mutations necessitate working with large amounts of data. The inherent challenges of analyzing such large data sets were largely resolved by using the ‘mutational signature’ technique [Citation23–26]. Because it is usually not possible to define the DNA strand on which a mutation occurred (for example, distinguishing C>A mutations from G>T mutations on the opposite strand), there are essentially only six types of substitutions to be analyzed [Citation26]. Similarly, there are 96 context-dependent mutation frequencies that consider two nucleotides in the flanking 5’ and 3’ positions of the mutated nucleotide [Citation26]. Mutational signature is an important concept for describing individual mutagenic factors and for quantifying their contribution to mutational spectra in cancer samples. Several computational methods have been proposed for solving this decomposition problem [Citation22–26]. Cancer genome sequencing efforts have delineated at least 30 mutational patterns that are commonly found in human cancers (http://cancer.sanger.ac.uk/cosmic/signatures) [Citation26]. Two of the 30 delineated mutational signatures, namely Signatures 13 and 9 () are attributed to TLS DNA polymerases.
Figure 2. The potential DNA polymerase η mutational signature (Signature 9, http://cancer.sanger.ac.uk/cosmic/signatures). The mutation context (+1 and −1 positions) are shown below frequency bars. This signature (together with more than 20 other distinct mutational signatures) was extracted using a classification analysis of 4,938,362 mutations from 7,042 cancers [Citation26].
![Figure 2. The potential DNA polymerase η mutational signature (Signature 9, http://cancer.sanger.ac.uk/cosmic/signatures). The mutation context (+1 and −1 positions) are shown below frequency bars. This signature (together with more than 20 other distinct mutational signatures) was extracted using a classification analysis of 4,938,362 mutations from 7,042 cancers [Citation26].](/cms/asset/e11d10db-5f8a-43f4-bf8e-2ee9c7e8ee35/kccy_a_1456296_f0002_oc.jpg)
Mutational signature 13 is thought to arise due to the concerted actions of APOBEC (apolipoprotein B mRNA editing enzyme, catalytic) and the Y-family TLS polymerase REV1. This annotation was based on previous analyses of APOBEC3A/B that employed known mutable motifs of APOBEC3A/B (TCW/WGA) [Citation27,Citation28]. The APOBECs deaminate Cytosine to Uracil. During APOBEC-mediated mutagenesis, the Uracils formed by cytosine deaminiation are excised to generate AP sites. Trans-lesion synthesis of the non-coding AP sites can lead to C>T mutations or C>G mutations. The C>G mutations are dependent on the catalytic activity of REV1. APOBEC-mediated mutations arise in clusters within unusually long and persistent stretches of ssDNA [Citation27,Citation28]. It is interesting to note that ssDNA is the common species generated by many forms of DNA damage and replication stress that activates RAD18 and TLS DNA polymerases [Citation14,Citation29]. Therefore ssDNA-accumulation may provide a biochemical basis for coordinate activities of APOBECs and the TLS machinery during mutagenesis. PCNA mono-ubiquitination was attenuated in cells engineered to express a catalytically-inactive mutant APOBEC3A (when compared with cells expressing WT APOBEC3A), potentially consistent with some form of crosstalk between APOBEC and TLS activities at ssDNA [Citation30].
Although decomposition into signatures is useful for interpreting the mutagenic processes, there are certain limitations of this approach [Citation22]. One limitation is the heuristic nature of associations between mutational signatures and molecular mechanisms – which often reflect expert opinions of researchers providing mutational signature annotations. For example, the Polη signature in COSMIC (; mutational Signature 9, http://cancer.sanger.ac.uk/cosmic/signatures) has been found in chronic lymphocytic leukemia and malignant B-cell lymphoma genomes. Interestingly, signature 9 has a higher frequency of T:A>G:C transversions compared with T:A>C:G transitions. Such a Polη-mediated mutational pattern has not been observed in vitro or in vivo (a clear excess of T:A>C:G transitions was found in all experiments), although context properties do resemble the Polη mutable motif WA/TW (W = A/T, the mutation hotspot position is underlined) [Citation22,Citation31]. In addition, the putative Polη mutational signature 9 was not found in follicular lymphoma although this cancer is associated with the activity of AID (an enzyme which cooperates with Polη to induce hypermutations during immunoglobulin class switch recombination) [Citation32]. These observations suggest that the signature 9 is a mixture of descriptions of Polη mutations and some yet unknown process with context properties resembling the Polη mutable motif. It is also possible that Signature 9 represents only a subset of the mutational patterns induced by Polη. Differences between mutable motifs and mutational signatures are reviewed elsewhere [Citation22].
A recent study suggested that Polη may cause somatic mutations in lymphoid cells [Citation33]; most of the characteristic clustered mutations were found in promoters, as with AID-initiated somatic hypermutation. In solid tumors, however, somatic mutations are likely to be associated with the other factors, including exogenous exposures, UV radiation or alcohol consumption [Citation33]. This study is based on mutational signature 9 () and suggested that Polη targets the H3K36me3 chromatin of active genes in a mismatch repair (MMR)-dependent manner. These regions normally have a low mutation rate because error-free MMR also targets H3K36me3 chromatin [Citation34]. Carcinogens and error-prone repair therefore redistribute mutations to the more important regions of the genome, contributing a substantial mutation load in many tumors, including driver mutations [Citation33]. These results somewhat contradict the recent study that found fewer somatic mutations in exons than expected from their sequence content [Citation35]. It was suggested that this trend is likely to be caused by higher conventional mismatch-repair activity in exonic than in intronic regions [Citation35].
Rogozin et al. have studied the possible involvement of Polη in the generation of somatic mutations in skin cancer, other cancers and in normal cells [Citation2]. Those workers found a highly significant correlation between Polη mutable motifs and somatic mutations in skin cancer cells. However, this correlation was not observed in normal skin samples. Traces of Polη-mediated mutagenesis were also found in various other cancers. A significant excess of somatic mutations in WA/TW motifs was present in 11 out of 14 solid tumors from various tissue types [Citation2]. Frequent tandem mutations are known to be an intrinsic property of Polη when copying undamaged DNA and they have the same context specificity as single mutations [Citation36]. Although tandem mutations occur much less frequently, there was nevertheless a significant excess of tandem mutations in the WA/TW context in 3 out of 8 cancer types [Citation2]. Taken together with the results of expression analysis, this study suggests the widespread participation of Polη in mutagenesis in cancer cells [Citation2]. Further studies of in vitro and in vivo mutation spectra are needed to identify mutational signatures and mutable motifs associated with TLS DNA polymerases.
(ii) Altered expression of function of TLS proteins in cancer cells. Changes in expression or activity of Y-family DNA polymerases could potentially create TLS pathway imbalance (C) causing replication infidelity and genetic changes (possibly including mutational signatures 13 and 9). Several studies have documented altered expression and function of TLS polymerases and of their proximal activator RAD18 in cancer. For example a steady state kinetic analyses of REV1 proteins harboring different polymorphisms (including variants associated with cancer) revealed moderate or slight effects on biochemical activities (such as kcat/Km for dCTP insertion, binding affinities to DNA substrates harboring various lesions) when compared with wild-type REV1 [Citation37]. Therefore, it is plausible that germline missense REV1 variations affect cancer susceptibility by modifying the fidelity and capacity for replication on undamaged and damaged DNA templates. Epidemiological data are fully consistent with the possibility that TLS polymerase polymorphisms affect tumorigenic outcomes. In a study that assessed associations of DNA repair genes with lung cancer risk, a REV1 variant showed associations with squamous cell carcinoma risk while a POLI polymorphism was associated with adenocarcinoma and squamous cell carcinoma risk [Citation38]. Another case-controlled study revealed association between genetic variants of POLK and REV1 and susceptibility and survival of lung cancer respectively [Citation39]. POLK genetic variants have also been associated with increased breast cancer risk [Citation40].
Altered expression of TLS polymerases may also affect mutagenesis and cancer susceptibility. Polι which has error rates as high as 10^0 on undamaged templates shows elevated expression in breast cancer cells [Citation41]. Moreover, this elevated Polι expression was associated with mutagenesis. This study strongly suggested that Polι contributes to generation of spontaneous and trans-lesion mutations. In a mouse intestinal tumorigenesis model, transgenic overexpression of Rev1 accelerated intestinal adenoma formation in response to N-methyl-N-nitrosourea (MNU) treatment [Citation42] (but did not affect spontaneous tumorigenesis). Therefore aberrant Rev1 levels may promote accumulation of mutations and accelerate tumorigenesis.
Albertella and colleagues also found that Polι was overexpressed in a range of tumor types [Citation43]. Polκ overexpression is often observed in lung cancer and in cell culture studies Polκ overexpression induces spontaneous mutagenesis, DNA breaks, genetic exchanges and aneuploidy [Citation44]. Polι overexpression is positively correlated with clinical tumor grade in bladder cancer [Citation45]. Both Polκ and Polι (but not Polη) are overexpressed in human gliomas and their overexpression is associated with shorter patient survival [Citation46]. Ziv and colleagues identified an interesting mechanism by which some Acute Myeloid Leukemia (AML) cells alter Polη expression. Those workers showed that NPM1 (Nucleophosmin, the product of a commonly mutated gene in AML) interacts with the catalytic core of Polη. Remarkably, the prevalent NPM1c+ mutation in AML patients leads to excessive Polη degradation and reduces error-free TLS of DNA templates containing CPD lesions [Citation47]. Thus, there is ample evidence that up- or down-regulated expression or activity of individual Y-family polymerases is common in tumors and represents a potential source of imbalanced TLS pathway activity and mutagenesis.
A recent study found that the proximal activator of all four Y-family TLS polymerases, the E3 ligase RAD18, is also aberrantly over expressed in many cancer cell lines [Citation3]. One mechanism of RAD18 overexpression in cancer cells appears to involve increased stability via an overexpressed binding partner, the Cancer/Testes Antigen (CTA), MAGE-A4. MAGE-A4 is absent from all normal somatic cells, thereby demonstrating a fundamental difference in the TLS pathway between untransformed cells and neoplastic cells. Even small fold-increases in RAD18 protein expression are sufficient to stimulate PCNA mono-ubiquitination and drive recruitment of Y-family TLS pols to replicating (undamaged) DNA [Citation21]. Therefore, elevated RAD18 could increase the activity of all TLS polymerases (C). Alternatively, if Y-family polymerases are differentially dependent on RAD18, then preferential activation of individual DNA polymerases by over-active RAD18 could be a source of TLS pathway imbalance that leads to error-prone DNA replication ( D).
Taken together, it is clear that expression of TLS polymerases and RAD18 is often deregulated in cancer cells and thus represents a potential source of genomic instability. It will be important to determine whether specific mutational signatures are associated with particular changes in relative activities of RAD18 and the Y-family DNA polymerases.
(iii) TLS facilitates tolerance of oncogenic stress. The elevated expression of TLS proteins in cancer, and the dependency of neoplastic cells on pathologically-activated RAD18 for DNA damage tolerance [Citation3] may suggest that TLS can confer a survival/selective advantage upon neoplastic cells. How then might TLS facilitate survival of neoplastic cells? Y-family DNA polymerases have long been implicated in bypass of chemically-induced DNA adducts. However, it is increasingly clear that these polymerases are recruited to replicating DNA in S-phase (even in absence of exogenously-induced damage) and play key roles in DNA synthesis at hard-to-replicate areas including common fragile sites [Citation48–51]. The question arises then – are there intrinsic DNA replication intermediates or species of endogenous DNA damage in cancer cells that depend on the TLS pathway for remediation?
In the last decade it has become apparent that neoplastic cells experience considerable DNA ‘replication stress’ and accumulate DNA damage (in the form of single- and double-stranded breaks) as a consequence of oncogene signaling. Seminal papers published in 2006 showed that ectopic overexpression of various oncogenes (including Cyclin E, CDC25A, KRAS, MOS, and MYC) in primary untransformed human cells induces replication-associated DNA damage, impaired replication fork progression and accumulation of aberrant DNA replication intermediates. Moreover, oncogene-induced DNA damage signaling via ATM/CHK2/p53 signaling leads to irreversible cell cycle arrest termed ‘Oncogene-Induced Senescence’ (OIS) [Citation52,Citation53]. The discovery that oncogenes induce DNA damage is highly significant because it provides a mechanism by which neoplastic cells destabilize their genomes. Moreover, OIS may be a mechanism for eliminating oncogene-expressing cells and thus serves as a barrier to tumorigenesis [Citation54–56]. However, discovery of the OIS pathway poses several critical questions: what is the nature of oncogene-induced stress/damage? How does oncogene signaling cause DNA damage? Do different oncogenes induce DNA damage via common mechanisms? How do neoplastic cells tolerate and adapt to oncogene-signaling? What is the extent to which (and putative mechanism whereby) oncogene-induced DNA damage helps shape the mutational landscape of cancer cells? Recent work suggests that the TLS pathway is activated in response to oncogene signaling and can facilitate tolerance of oncogene-induced DNA damage [Citation5]. Therefore, TLS may provide solutions to some of the key unanswered questions relating to OIS. Below we summarize proposed mechanisms by which oncogenes induce DNA replication defects and DNA damage. Then we consider the potential ways in which TLS proteins might allow cancer cells to tolerate those forms of oncogene-induced DNA replication stress.
Mechanisms of oncogene-induced DNA damage
(a) Re-replication. Some of the earliest papers describing OIS observed aberrant re-initiation of DNA synthesis multiple times each per cell cycle – a process usually termed ‘re-replication’ or ‘hyper-replication’ – in oncogene-expressing cells [Citation53]. It is unknown whether oncogene-induced re-replication is caused by inappropriate activation of DNA replication licensing factors, initiation factors, or deregulation of both licensing and initiation phases of DNA synthesis. Re-replication generates ‘onion skin’ DNA structures in which head-to-tail collisions between replication forks generate ssDNA gaps, replication fork reversal [Citation57] and eventually DSB formation and checkpoint activation [Citation58].
(b) Collisions between DNA replication machinery and transcription complexes. A significant amount of oncogene (Cyclin E)-induced replication slow-down and DNA damage is due to interference between replication and transcription [Citation59]. Indeed, overexpression of the general transcription factor TATA-box binding protein (TBP) even in the absence of an oncogene leads to increased transcription, DNA replication fork slowing and DNA damage [Citation60]. In a recent elegant study Cimprich and colleagues showed that the DNA damage response is critically determined by the relative orientation of colliding replication and transcription complexes. Thus in a defined episomal system, head-on (HO) collisions between replication forks and transcription complexes led to increased formation of R-loops (RNA-DNA hybrids formed when nascent transcripts reanneal to template DNA, displacing the non-template strand as ssDNA) and activated ATM/CHK2 signaling [Citation61]. Conversely, co-directional (CD) collisions led to decreased R-loop formation and did not trigger DNA damage signaling. Halazonetis and co-workers have demonstrated that the premature S-phase entry due to oncogenes (Cyclin E and MYC) induces inappropriate firing of DNA replication origins within transcribed genes, leading to replication/transcription conflicts and fork collapse. Remarkably, the ensuing DSB were also associated with chromosomal rearrangement breakpoints both in cultured cells and in a large cohort of human cancers. Therefore, there is now strong evidence that oncogene-induced replication/transcription conflicts are relevant to genomic instability in human cancer [Citation62].
(c) Reactive Oxygen Species (ROS). Oncogene expression often induces increased levels of ROS which act as mitogenic stimuli, yet also cause genotoxicity that may contribute to DNA damage signaling and senescence [Citation63–65]. For example, Vafa et al. found that c-MYC-induced DNA damage and p53 activation were attenuated by anti-oxidants [Citation66]. Cells expressing oncogenic RAS have increased mitochondrial mass and generated elevated levels of ROS [Citation67]. Moreover, oxidative DNA damage accompanies RAS-induced senescence and mitochondrial dysfunction alone induces senescence [Citation67]. However, C-MYC- and RAS expression cause DNA replication defects that are temporally separable from metabolic reprogramming and ROS generation. Therefore, individual oncogenes may cause DNA damage via ROS-mediated as well as ROS-independent mechanisms [Citation68].
(d) Nucleotide deficiency. Forced bypass of the RB-mediated G1/S restriction point by overexpressed Cyclin E or HPV E6/E7 oncogenes depleted cellular nucleotide levels and led to ‘replication stress’ [Citation69]. Importantly Cyclin E or HPV oncoprotein-induced replication stress and DNA damage signaling were rescued by exogenously added nucleotides. Interestingly, overexpression of c-MYC, an oncoprotein that induces transciption of nucleotide biosynthesis genes, restored the nucleotide pool and also prevented the DNA replication-induced DNA damage response.
The different proposed mechanisms of oncogene-induced DNA damage described above are not necessarily mutually exclusive. Moreover, different oncogenes may induce DNA damage via distinct mechanisms. For example, Petermann and colleagues showed that CDK inhibition did not relieve HRASV12-induced replication fork slowing [Citation60]. Those workers inferred that HRASV12 causes replication stress by a transcriptional mechanism that is different from CDK2-activating oncogenes such as Cyclin E and CDC25A. Conversely, overexpressed c-MYC results in elevated replication-fork stalling and collapse and independently of RNA transcription [Citation70].
How neoplastic cells tolerate oncogene-induced DNA damage
Understanding how different oncogenes induce DNA damage will be necessary if we are to determine how neoplastic cells tolerate oncogenic stress, breach the OIS barrier and progress to malignancy. It seems likely that multiple DNA damage tolerance/genome maintenance mechanisms could help sustain viability of oncogene-expressing cells and thereby facilitate tumorigenesis. The few studies to address mechanisms of oncogenic replication stress tolerance are consistent with roles for genome maintenance in supporting cell proliferation and tumorigenesis in neoplastic cells. For example, Halazonetis and colleagues performed siRNA-based screening to identify genes that facilitate tolerance of overexpressed Cyclin E [Citation71]. Those workers showed that the DNA polymerase delta subunits POLD3 and POLD4 facilitate S-phase progression in Cyclin E-overexpressing cells by supporting break induced DNA replication (BIR). Interestingly, POLD3/4 were dispensable for tolerance of hydroxyurea-induced DNA replication stress, suggesting that oncogene signaling induces unique DNA replication defects.
Cell culture experiments show that oncogenic stress activates the ATR/CHK1-mediated S-phase checkpoint pathway [Citation72]. Moreover, Atr can promote oncogene-induced carcinogenesis in vivo [Citation72]. Similar to ATR/CHK1 signaling, TLS is initiated by ssDNA [Citation14]. Moreover, TLS is pathologically activated in many cancer cells, leading Yang et al. to test a hypothetical role for the TLS pathway in tolerance of oncogene expression [Citation5]. Those workers demonstrated that acute expression of Cyclin E and oncogenic RAS (but not C-MYC) led to increased PCNA mono-ubiquitination and recruitment of TLS polymerases in untransformed cells. Pharmacological activation of CDK2 (using the WEE1 kinase inhibitor MK-1775) also triggered PCNA mono-ubiquitination. RAD18 and its downstream effector Polκ (but interestingly not Polη) were necessary to support DNA replication and prevent ssDNA accumulation in response to oncogenic stimuli [Citation5].
How then do oncogenic stimuli activate the TLS pathway and how does TLS confer tolerance of oncogene-induced DNA replication stress? All of the proposed mechanisms of oncogene-induced DNA damage described above have the potential to generate ssDNA species that activate TLS. However, re-replication intermediates alone may not explain TLS pathway activation by oncogenes since overexpression of the licensing factor CDT1 (which stimulates re-replication and DSB formation [Citation58]) does not induce PCNA mono-ubiquitination. TLS pathway activation in oncogene expressing cells is probably also not a consequence of limiting nucleotiode pools since exogenously-added nucleosides do not attenuate Cyclin E-induced PCNA mono-ubiquitination [Citation5].
Oxidative DNA damage activates RAD18 in replicating and non-replicating cells, and the TLS pathway clearly allows cells to tolerate ROS-induced genotoxicity [Citation16,Citation17,Citation73]. Therefore, replication and repair of oxidative DNA damage could explain how RAD18 allows cells to tolerate oncogene-expression. However, it is unknown whether all oncogenic stimuli induce ROS or whether tolerance of ROS-induced genotoxicity fully explains how the RAD18 pathway sustains DNA replication of oncogene-expressing cells. For example oncogenic RAS is known to elicit ROS formation, yet RAS-induced PCNA mono-ubiquitination is relatively modest when compared to the Cyclin E-induced PCNA monoubiquitination response [Citation5]. It will be necessary to test whether Cyclin E-CDK2 induces ROS, and whether ROS-scavengers ablate Cyclin E-induced TLS pathway activation before inferring a role for ROS in mediating TLS activation in response to additional oncogenic stimuli.
The role of transcriptional complexes in oncogene-induced TLS pathway activation has not been tested comprehensively. Polη may facilitate restart of forks that are stalled by co-directional collisions of replication and transcription complexes, possibly suggesting that TLS is activated when the DNA replication machinery and transcriptional complexes collide [Citation74]. However, available evidence suggests that transcriptional mechanisms do not fully account for oncogene-induced TLS pathway activation. Kotsantis and colleagues showed that CDK inhibition does not relieve HRASV12-induced replication fork slowing [Citation60]. Those workers suggest that HRASV12 causes replication stress by a transcriptional mechanism effects (presumably HO collisions of replication and transcriptional complexes) that is different from oncogenes such as Cyclin E and CDC25A [Citation60]. In a separate study RAS-induced PCNA mono-ubiquitination was less robust than the PCNA mono-ubiquitination response to overexpressed Cyclin E and was attenuated by CDK inhibition [Citation5]. Therefore RAS-induced DNA damage may occur primarily via collisions between replication and transcriptional complexes while Cyclin E-induced DNA replication stress might involve a distinct CDK2-induced DNA lesion that preferentially activates TLS. This conclusion should be viewed only as an untested hypothesis since it is inappropriate to over-interpret results obtained in different studies using different oncogenes and different experimental systems. Clearly there is a need to systematically compare the effects of different oncogenes on DNA replication dynamics, accumulation of different damaged DNA intermediates and DNA damage signaling in a common and well-defined experimental system. It is most likely that each oncogenic stimulus induces a spectrum of DNA lesions and replication defects and that there is partial overlap between the various types of DNA damage caused by different oncogenes.
Regardless of the primary DNA lesions or replication intermediates induced by different oncogenes, ssDNA-containing replication structures most probably mediate TLS pathway activation in response to diverse oncogenic stimuli. ssDNA is generated in response to aberrant CDK2 activity [Citation75] and probably other CDK2-activating oncogenes such as RAS [Citation76]. Suppressing ssDNA accumulation via post-replicative gap-filling is also the major role of TLS polymerases in the response to bulky DNA lesions. Post-replicative filling of ssDNA gaps (including ssDNA arising outside of S-phase) is a major role of the RAD18/TLS pathway [Citation16,Citation17,Citation77]. Genomes containing persistent ssDNA are vulnerable to nucleolytic attack and are likely to generate lethal DSB. Thus RAD18/TLS-mediated repair synthesis most prevents accumulation of oncogene/CDK2-induced ssDNA gaps and subsequent breaks in the genome.
It is interesting that Polκ but not Polη sustains DNA replication and cell survival in response to aberrant CDK2 activity [Citation5]. Polη is the most versatile Y-family DNA polymerases enzyme and is probably the default TLS polymerase recruited to most stalled DNA replication forks [Citation20]. Polη resides constitutively in replication factories during an unperturbed S-phase cells and facilitates DNA synthesis at fragile sites, telomeres, and elsewhere in the genome [Citation49,Citation51,Citation78]. Perhaps it is surprising that Polκ but not Polη is specifically required for tolerance of CDK-induced replication stress. However, Polκ and Polη preferentially perform replicative bypass of distinct cognate bulky DNA lesions. Therefore, it is likely that these Y-family DNA polymerases also have distinct roles in repair and tolerance of intrinsically-arising replication intermediates in oncogene-expressing cells. TLS polymerases are implicated in bypass of quadruplex DNA [Citation78] (a structure that is generated at persistent ssDNA [Citation79]). Polκ preferentially binds G4 when compared with non-G4 DNA, and displays enhanced activity when within 2–3 nucleotides of a G4 motif [Citation80]. Polκ can also freely exchange with Pol δ in vitro to facilitate replicative bypass at a [GT]10 microsatellite sequence more accurately than Polη [Citation81]. Further work is necessary to identify the putative oncogene-induced DNA lesions that require Polκ (but not Polη) for repair or replicative bypass.
Conclusions and perspectives
There is now considerable evidence that cancer genomes bear mutational signatures attributable to TLS. Altered expression and activity of Y-family DNA polymerases and RAD18 commonly occurs in cancer cells and is a potential cause of error-prone TLS. Moreover, TLS facilitates tolerance of oncogene-induced DNA replication stress. Taken together, these observations suggest multiple ways by which TLS impacts tumorigenesis: Error-prone replication of damaged DNA templates may contribute to the initiating mutations that incite malignancy (). Later in tumorigenesis, TLS may facilitate tolerance of oncogene-induced stress and bypass of the OIS barrier () in addition to promoting mutability. It is interesting to compare the potential roles of TLS and the ATR-CHK1 signaling pathway in different stages of multi-step tumorigenesis. Bartek and colleagues have suggested that early in tumorigenesis, ATR-CHK1 signaling contributes to OIS and serves a tumor-suppressive role [Citation82]. However, later during tumorigenesis, ATR-Chk1 signaling may support viability of cells that breach the OIS barrier in the malignant tumor. Thus, the TLS and ATR-CHK1 effector branches of the DDR, both of which are activated by a common proximal signal (RPA-ssDNA) may cooperate to support the later stages of malignant progression.
Figure 3. Proposed roles of RAD18 and TLS DNA polymerases in tumorigenesis. Mutagenic TLS of damaged DNA by Y-family DNA polymerases can activate oncogenes (e.g. KRASV12) that initiate carcinogenesis. Oncogene-expressing cells can be eliminated via a DNA damage-mediated senescence program (OIS) that serves as a barrier to tumorigenesis. RAD18/TLS and ATR-CHK1 help sustain damage-tolerant DNA synthesis and viability of cells that breach the OIS barrier. Error-prone TLS might also confer mutability that drives subsequent stages of multi-step tumorigenesis. The selective pressure for cancer cells to activate TLS also confers chemoresistance.
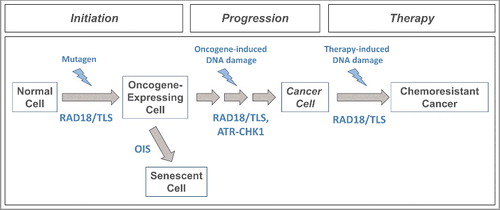
Clearly it is necessary to test the hypothetical effects of TLS on tumorigenesis (as proposed in and ). In particular it will be very important to determine the extent to which altered expression or activity of TLS genes (Rad18, Y-family DNA polymerases) in an in vivo setting influences mutational spectra and phenotypes of oncogene-induced tumors. Since Rad18 and Polκ support DNA replication in oncogene-expressing cells, Rad18−/− or Polk−/− mice may be refractory to oncogene-induced tumorigenesis (owing to reduced viability of pre-neoplastic cells).
Sequencing of oncogene-induced tumors from WT and Rad18 or Y-family DNA polymerase mutant mice could identify mutational signatures resulting from error-prone TLS. The final mutational ‘portrait’ of any tumor is a composite of multiple mutational signatures. Comparison of tumor portraits from WT and Rad18 or TLS polymerase-deficient mice might reveal subsets of signatures that are Rad18/TLS- dependent (i.e. absent from Rad18/TLS-deficient tumors). TLS-deficiency often leads to increased DSB formation [Citation83,Citation84]. Therefore, it is likely that Rad18/Y-family Polymerase-mediated mutations are replaced by alternative mutational signatures when TLS is compromised. For example, oncogene-induced tumors generated in Rad18/TLS-deficient mice might have increased evidence of DSB-induced genetic changes (such as indels arising via Polq-mediated TMEJ). It would also be interesting to model the consequences of overexpressed Rad18, its cancer-specific activator MAGE-A4, or individual TLS polymerases (commonly observed in tumors) on mutational spectra and tumorigenesis in vivo. Comparison of TLS-mediated mutational signatures obtained in mouse tumorigenesis studies with the 30 known mutational patterns in cancer genomes could help reveal the extent to which TLS shapes human cancer genomes.
Mechanistically, it is still unclear what species of oncogene-induced DNA damage or replication intermediate are tolerated or processed by RAD18/TLS. It is important to determine whether all or just a subset of oncogenes trigger forms of damage that are tolerated by RAD18/TLS. In a mouse model of Seckel Syndrome, reduced Atr levels attenuated the development of MYC-induced lymhomas and pancreatic tumors, but did not impact KRASG12V-driven pancreatic adenocarcinomas [Citation85]. It is likely that neoplastic cells harboring different oncogenic drivers will have differential dependencies on TLS-mediated DNA damage tolerance. Similar to studies that defined roles of Atr in tolerance of oncogenic stress It will be important to address mechanisms of TLS-mediated oncogenic stress tolerance using cell culture models and to define the effect TLS on tumorigenesis using mouse models.
The discussion here has considered the consequences of imbalanced expression/activity of RAD18 and TLS polymerases relative to each other (). However, the impact of TLS on genome stability and replication stress tolerance will doubtless also be dictated by the repertoire of other available genome maintenance mechanisms. For example, the processing of DNA breaks generated via TLS-deficiency will be dependent on the choice of error-prone and error-free DSB repair mechanisms (namely NHEJ, TMEJ and HR). The Polq and Brca1 gene products (which mediate TMEJ and HR) help sustain viability of cells harboring excessive CDK2 activity [Citation5]. Thus, TLS is a single component of a broader genome maintenance network that determines oncogenic stress tolerance. Similar to TLS proteins, core components of the TMEJ and HR pathways often have altered expression in cancer. For example, POLQ is upregulated in BRCA1 mutant (HR-compromised) breast and ovarian cancers [Citation86]. The extent to which neoplastic cells employ TLS, and the genome-destabilizing impact of TLS are likely determined by the integrity of other DNA repair pathways. It will eventually be important to systematically interrogate the contributions of TLS and other DNA repair pathways (including HR, TMEJ) in carcinogenesis in response to defined oncogenic drivers using mouse models.
An important byproduct of the selective pressures for mutagenesis and DNA damage tolerance during tumorigenesis might be the emergence of chemoresistant cancer cells. It is firmly established that Polη-mediated TLS allows replication of cisplatin-damaged DNA templates [Citation87–91] and the structural basis for Polη-mediated chemoresistance to cisplatin has been elucidated [Citation7,Citation92]. Numerous studies have demonstrated that TLS-deficient cells lacking Polη [Citation6,Citation93,Citation94] or RAD18 [Citation95,Citation96] fail to replicate cisplatin-damaged genomes and instead accumulate unfilled post-replicative gaps, collapsed replication forks and lethal DSB. High-level Polη expression is also correlated with poor survival of platinum-treated cancer patients [Citation97,Citation98].
If cancer cells commonly require ATR-CHK1, TLS, TMEJ or other genome maintenance pathways to remain viable, those mechanisms represent molecular vulnerabilities that could be exploited to sensitize cells to intrinsic or therapy-induced DNA damage. The rapid uncontrolled DNA synthesis and proliferation of tumor cells may make them particularly sensitive to additional therapy-induced DNA damage and DDR inhibition. The ATR/CHK1-mediated checkpoint pathway is particularly important for suppressing CDKs, coordinating cycle progression with DNA repair, and sustaining cell viability. Therefore, both ATR and CHK1 protein kinases are appealing therapeutic targets. Several small molecule inhibitors of ATR (including VX-970 and AZD6738) and of CHK1 (including MK8776) are being evaluated in clinical and pre-clinical trials [Citation99–101]. ATR inhibitors sensitize tumor cells to overexpressed Cyclin E [Citation102] and therapeutic genotoxins notably platinating agents [Citation103,Citation104]. CHK1 inhibitors are particularly effective in p53-deficient cells which lack the G1 checkpoint and have greater dependency on S-phase checkpoint mechanisms for DNA damage tolerance [Citation99].
It is interesting to consider the potential therapeutic impact of combining ATR/CHK1 inhibitors with TLS inhibitors. Historically, the UV-sensitivity phenotype of many XPV cell lines was revealed by caffeine treatment [Citation105]. It is now known that caffeine is an ATR inhibitor and that combined deficiencies of ATR and Polη lead to UV-hypersensitivity [Citation106]. It is possible therefore that combined inhibition of ATR/CHK1 and TLS pathways would similarly sensitize neoplastic cells to therapeutic genotoxins. Indeed, ablation of REV3 and REV7 (subunits of the TLS polymerase Polζ) sensitizes cancer cells to a combination of ATRi and cisplatin [Citation107].
Small molecule inhibition of WEE1 kinase (which de-represses CDK1/2) is being considered as a therapeutic strategy in cancer [Citation108]. Similar to ATR/CHK1 inhibitors, WEE1 inhibition causes cells to sustain CDK activities even in the face of DNA damage. TLS allows cancer cells to tolerate WEE1 inhibition [Citation5] and therefore the anti-cancer activity of WEE1 inhibitors might be improved with concurrent TLS inhibition.
Thus, defining roles of TLS and other genome maintenance pathways in tolerance of oncogene- and therapy-induced replication stress might translate into improved strategies for cancer therapy and precision medicine. Of potential concern, targeting pathways that can function both as tumor suppressors and tumor promoters might lead to adverse effects such as therapy-induced cytotoxicity to normal cells or genome instability and secondary malignancies. Therefore, there is a need for stratification of tumors (e.g. based on TLS/ATR CHK1 and other pathways) to identify subtypes of cancers that can be selectively killed (e.g. based on synthetic lethalities) using DDR or TLS inhibition.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgements
I.B.R. was supported by the Intramural Research Program of the National Library of Medicine at the National Institutes of Health and V.Y. was supported by Moravskoslezsky kraj research initiative grant 01211/2016/RRC. C.V. was supported by R01 CA215347 from the National Institutes of Health.
Additional information
Funding
References
- Masutani C, Kusumoto R, Yamada A, et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999;399:700–704. doi:10.1038/21447. PMID:10385124
- Rogozin IB, Goncearenco A, Lada AG, et al. DNA polymerase eta mutational signatures are found in a variety of different types of cancer. Cell Cycle. 2018;17(3):348–355. doi:10.1080/15384101.2017.1404208.
- Gao Y, Mutter-Rottmayer E, Greenwalt AM, et al. A neomorphic cancer cell-specific role of MAGE-A4 in trans-lesion synthesis. Nat Commun. 2016;7:12105. doi:10.1038/ncomms12105. PMID:27377895
- Gao Y, Tateishi S, Vaziri C. Pathological trans-lesion synthesis in cancer. Cell Cycle. 2016;15(22):3005–3006. doi:10.1080/15384101.2016.1214045.
- Yang Y, Gao Y, Mutter-Rottmayer L, et al. DNA repair factor RAD18 and DNA polymerase Polkappa confer tolerance of oncogenic DNA replication stress. J Cell Biol. 2017;216(10):3097. doi:10.1083/jcb.201702006.
- Albertella MR, Green CM, Lehmann AR, et al. A role for polymerase eta in the cellular tolerance to cisplatin-induced damage. Cancer Res. 2005;65:9799–9806. doi:10.1158/0008-5472.CAN-05-1095. PMID:16267001
- Zhao Y, Biertumpfel C, Gregory MT, et al. Structural basis of human DNA polymerase eta-mediated chemoresistance to cisplatin. Proc Natl Acad Sci USA. 2012;109:7269–7274. doi:10.1073/pnas.1202681109. PMID:22529383
- Masutani C, Araki M, Yamada A, et al. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. Embo J. 1999;18:3491–3501. doi:10.1093/emboj/18.12.3491. PMID:10369688
- Ziv O, Geacintov N, Nakajima S, et al. DNA polymerase zeta cooperates with polymerases kappa and iota in translesion DNA synthesis across pyrimidine photodimers in cells from XPV patients. Proc Natl Acad Sci USA. 2009;106:11552–11557. doi:10.1073/pnas.0812548106. PMID:19564618
- Shachar S, Ziv O, Avkin S, et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009;28:383–393. doi:10.1038/emboj.2008.281. PMID:19153606
- Ohmori H, Ohashi E, Ogi T. Mammalian Pol kappa: regulation of its expression and lesion substrates. Adv Protein Chem. 2004;69:265–278. doi:10.1016/S0065-3233(04)69009-7. PMID:15588846
- Ulrich HD, Jentsch S. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 2000;19:3388–3397. doi:10.1093/emboj/19.13.3388. PMID:10880451
- Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell. 2004;14:491–500. doi:10.1016/S1097-2765(04)00259-X. PMID:15149598
- Davies AA, Huttner D, Daigaku Y, et al. Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein a. Mol Cell. 2008;29:625–636. doi:10.1016/j.molcel.2007.12.016. PMID:18342608
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi:10.1126/science.1083430. PMID:12791985
- Yang Y, Durando M, Smith-Roe SL, et al. Cell cycle stage-specific roles of Rad18 in tolerance and repair of oxidative DNA damage. Nucleic Acids Res. 2013;41(4):2296–2312. doi:10.1093/nar/gks1325.
- Zlatanou A, Despras E, Braz-Petta T, et al. The hMsh2-hMsh6 complex acts in concert with monoubiquitinated PCNA and Pol eta in response to oxidative DNA damage in human cells. Mol Cell. 2011;43:649–662. doi:10.1016/j.molcel.2011.06.023. PMID:21855803
- Ogi T, Limsirichaikul S, Overmeer RM, et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell. 2010;37:714–727. doi:10.1016/j.molcel.2010.02.009. PMID:20227374
- Bienko M, Green CM, Crosetto N, et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–1824. doi:10.1126/science.1120615. PMID:16357261
- Watanabe K, Tateishi S, Kawasuji M, et al. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. Embo J. 2004;23:3886–3896. doi:10.1038/sj.emboj.7600383. PMID:15359278
- Durando M, Tateishi S, Vaziri C. A non-catalytic role of DNA polymerase eta in recruiting Rad18 and promoting PCNA monoubiquitination at stalled replication forks. Nucleic Acids Res. 2013;41(5):3079–3093. doi:10.1093/nar/gkt016.
- Rogozin IB, Pavlov YI, Goncearenco A, et al. Mutational signatures and mutable motifs in cancer genomes. Brief Bioinform. 2017;1–17. doi:10.1093/bib/bbx049. PMID:28498882
- Alexandrov LB, Stratton MR. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr Opin Gene Dev. 2014;24:52–60. doi:10.1016/j.gde.2013.11.014.
- Temiz NA, Donohue DE, Bacolla A, et al. The somatic autosomal mutation matrix in cancer genomes. Hum Genet. 2015;134:851–864. doi:10.1007/s00439-015-1566-1. PMID:26001532
- Goncearenco A, Rager SL, Li M, et al. Exploring background mutational processes to decipher cancer genetic heterogeneity. Nucleic Acids Res. 2017;45(W1):W514–W522. doi:10.1093/nar/gkx367. PMID:28472504
- Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi:10.1038/nature12477. PMID:23945592
- Roberts SA, Sterling J, Thompson C, et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell. 2012;46:424–435. doi:10.1016/j.molcel.2012.03.030. PMID:22607975
- Chan K, Gordenin DA. Clusters of multiple mutations: incidence and molecular mechanisms. Annu Rev Genet. 2015;49:243–67. doi:10.1146/annurev-genet-112414-054714. PMID:26631512
- Tsuji Y, Watanabe K, Araki K, et al. Recognition of forked and single-stranded DNA structures by human RAD18 complexed with RAD6B protein triggers its recruitment to stalled replication forks. Genes Cells. 2008;13:343–354. doi:10.1111/j.1365-2443.2008.01176.x. PMID:18363965
- Buisson R, Lawrence MS, Benes CH, et al. APOBEC3A and APOBEC3B Activities Render Cancer Cells Susceptible to ATR Inhibition. Cancer Res. 2017;77:4567–4578. doi:10.1158/0008-5472.CAN-16-3389. PMID:28698210
- Mayorov VI, Rogozin IB, Adkison LR, et al. DNA polymerase eta contributes to strand bias of mutations of A versus T in immunoglobulin genes. J Immunol. 2005;174:7781–7786. doi:10.4049/jimmunol.174.12.7781. PMID:15944281
- Rogozin IB, Lada AG, Goncearenco A, et al. Activation induced deaminase mutational signature overlaps with CpG methylation sites in follicular lymphoma and other cancers. Sci Rep. 2016;6:38133. doi:10.1038/srep38133. PMID:27924834
- Supek F, Lehner B. Clustered mutation signatures reveal that error-prone DNA repair targets mutations to active genes. Cell. 2017;170:534–547, e23. doi:10.1016/j.cell.2017.07.003. PMID:28753428
- Li F, Mao G, Tong D, et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. 2013;153:590–600. doi:10.1016/j.cell.2013.03.025. PMID:23622243
- Frigola J, Sabarinathan R, Mularoni L, et al. Reduced mutation rate in exons due to differential mismatch repair. Nat Genet. 2017;49:1684–1692. doi:10.1038/ng.3991. PMID:29106418
- Rogozin IB, Pavlov YI, Bebenek K, et al. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat Immunol. 2001;2:530–536. doi:10.1038/88732. PMID:11376340
- Yeom M, Kim IH, Kim JK, et al. Effects of twelve germline missense variations on DNA lesion and G-Quadruplex bypass activities of human DNA polymerase REV1. Chem Res Toxicol. 2016;29:367–379. doi:10.1021/acs.chemrestox.5b00513. PMID:26914252
- Sakiyama T, Kohno T, Mimaki S, et al. Association of amino acid substitution polymorphisms in DNA repair genes TP53, POLI, REV1 and LIG4 with lung cancer risk. Int J Cancer. 2005;114:730–737. doi:10.1002/ijc.20790. PMID:15609317
- Xu HL, Gao XR, Zhang W, et al. Effects of polymorphisms in translesion DNA synthesis genes on lung cancer risk and prognosis in Chinese men. Cancer Epidemiol. 2013;37:917–922. doi:10.1016/j.canep.2013.08.003. PMID:24012694
- Dai ZJ, Liu XH, Ma YF, et al. Association between single nucleotide polymorphisms in DNA polymerase kappa gene and breast cancer risk in Chinese han population: a STROBE-compliant observational study. Medicine. 2016;95:e2466. doi:10.1097/MD.0000000000002466. PMID:26765445
- Yang J, Chen Z, Liu Y, et al. Altered DNA polymerase iota expression in breast cancer cells leads to a reduction in DNA replication fidelity and a higher rate of mutagenesis. Cancer Res. 2004;64:5597–5607. doi:10.1158/0008-5472.CAN-04-0603. PMID:15313897
- Sasatani M, Xi Y, Kajimura J, et al. Overexpression of Rev1 promotes the development of carcinogen-induced intestinal adenomas via accumulation of point mutation and suppression of apoptosis proportionally to the Rev1 expression level. Carcinogenesis. 2017;38:570–578. doi:10.1093/carcin/bgw208. PMID:28498946
- Albertella MR, Lau A, O'Connor MJ. The overexpression of specialized DNA polymerases in cancer. DNA Repair (Amst). 2005;4:583–593. doi:10.1016/j.dnarep.2005.01.005. PMID:15811630
- Bavoux C, Leopoldino AM, Bergoglio V, et al. Up-regulation of the error-prone DNA polymerase {kappa} promotes pleiotropic genetic alterations and tumorigenesis. Cancer Res. 2005;65:325–330. PMID:15665310
- Yuan F, Xu Z, Yang M, et al. Overexpressed DNA polymerase iota regulated by JNK/c-Jun contributes to hypermutagenesis in bladder cancer. PloS One. 2013;8:e69317. doi:10.1371/journal.pone.0069317. PMID:23922701
- Wang H, Wu W, Wang HW, et al. Analysis of specialized DNA polymerases expression in human gliomas: association with prognostic significance. Neuro Oncol. 2010;12:679–686. doi:10.1093/neuonc/nop074. PMID:20164241
- Ziv O, Zeisel A, Mirlas-Neisberg N, et al. Identification of novel DNA-damage tolerance genes reveals regulation of translesion DNA synthesis by nucleophosmin. Nat Commun. 2014;5:5437. doi:10.1038/ncomms6437. PMID:25421715
- Despras E, Sittewelle M, Pouvelle C, et al. Rad18-dependent SUMOylation of human specialized DNA polymerase eta is required to prevent under-replicated DNA. Nat Commun. 2016;7:13326. doi:10.1038/ncomms13326. PMID:27811911
- Garcia-Exposito L, Bournique E, Bergoglio V, et al. Proteomic profiling reveals a specific role for translesion DNA polymerase eta in the alternative lengthening of telomeres. Cell Rep. 2016;17:1858–1871. doi:10.1016/j.celrep.2016.10.048. PMID:27829156
- Rey L, Sidorova JM, Puget N, et al. Human DNA polymerase eta is required for common fragile site stability during unperturbed DNA replication. Mol Cell Biol. 2009;29:3344–3354. doi:10.1128/MCB.00115-09. PMID:19380493
- Bergoglio V, Boyer AS, Walsh E, et al. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J Cell Biol. 2013;201:395–408. doi:10.1083/jcb.201207066. PMID:23609533
- Bartkova J, Rezaei N, Liontos M, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi:10.1038/nature05268. PMID:17136093
- Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi:10.1038/nature05327. PMID:17136094
- Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26:7773–7779. doi:10.1038/sj.onc.1210881. PMID:18066090
- Bartek J, Lukas J, Bartkova J. DNA damage response as an anti-cancer barrier: damage threshold and the concept of ‘conditional haploinsufficiency’. Cell Cycle. 2007;6:2344–2347. doi:10.4161/cc.6.19.4754. PMID:17700066
- Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi:10.1038/nature03482. PMID:15829956
- Neelsen KJ, Zanini IM, Mijic S, et al. Deregulated origin licensing leads to chromosomal breaks by rereplication of a gapped DNA template. Genes Dev. 2013;27:2537–2542. doi:10.1101/gad.226373.113. PMID:24298053
- Vaziri C, Saxena S, Jeon Y, et al. A p53-dependent checkpoint pathway prevents rereplication. Mol Cell. 2003;11:997–1008. doi:10.1016/S1097-2765(03)00099-6. PMID:12718885
- Jones RM, Mortusewicz O, Afzal I, et al. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene. 2013;32:3744–3753. doi:10.1038/onc.2012.387. PMID:22945645
- Kotsantis P, Silva LM, Irmscher S, et al. Increased global transcription activity as a mechanism of replication stress in cancer. Nat Commun. 2016;7:13087. doi:10.1038/ncomms13087. PMID:27725641
- Hamperl S, Bocek MJ, Saldivar JC, et al. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 2017;170:774–786, e19.
- Macheret M, Halazonetis TD. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature. 2018;555:112–116. doi:10.1038/nature25507. PMID:29466339
- Irani K, Xia Y, Zweier JL, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi:10.1126/science.275.5306.1649. PMID:9054359
- Lee AC, Fenster BE, Ito H, et al. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. 1999;274:7936–7940. doi:10.1074/jbc.274.12.7936. PMID:10075689
- Ogrunc M, Di Micco R, Liontos M, et al. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014;21:998–1012. doi:10.1038/cdd.2014.16. PMID:24583638
- Vafa O, Wade M, Kern S, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–1044. doi:10.1016/S1097-2765(02)00520-8. PMID:12049739
- Moiseeva O, Bourdeau V, Roux A, et al. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol. 2009;29:4495–507. doi:10.1128/MCB.01868-08. PMID:19528227
- Maya-Mendoza A, Ostrakova J, Kosar M, et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Molecular oncology. 2015;9:601–616. doi:10.1016/j.molonc.2014.11.001. PMID:25435281
- Bester AC, Roniger M, Oren YS, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi:10.1016/j.cell.2011.03.044. PMID:21529715
- Srinivasan SV, Dominguez-Sola D, Wang LC, et al. Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep. 2013;3:1629–1639. doi:10.1016/j.celrep.2013.04.002. PMID:23643534
- Costantino L, Sotiriou SK, Rantala JK, et al. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science. 2014;343:88–91. doi:10.1126/science.1243211. PMID:24310611
- Gilad O, Nabet BY, Ragland RL, et al. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res. 2010;70:9693–9702. doi:10.1158/0008-5472.CAN-10-2286. PMID:21098704
- Petta TB, Nakajima S, Zlatanou A, et al. Human DNA polymerase iota protects cells against oxidative stress. Embo J. 2008;27:2883–2895. doi:10.1038/emboj.2008.210. PMID:18923427
- Watanabe T, Marotta M, Suzuki R, et al. Impediment of replication forks by Long Non-coding RNA Provokes Chromosomal Rearrangements by Error-Prone Restart. Cell Rep. 2017;21:2223–2235. doi:10.1016/j.celrep.2017.10.103. PMID:29166612
- Neelsen KJ, Zanini IM, Herrador R, et al. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol. 2013;200:699–708. doi:10.1083/jcb.201212058. PMID:23479741
- Fikaris AJ, Lewis AE, Abulaiti A, et al. Ras triggers ataxia-telangiectasia-mutated and Rad-3-related activation and apoptosis through sustained mitogenic signaling. J Biol Chem. 2006;281:34759–34767. doi:10.1074/jbc.M606737200. PMID:16968694
- Daigaku Y, Davies AA, Ulrich HD. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 2010;465:951–955. doi:10.1038/nature09097. PMID:20453836
- Betous R, Rey L, Wang G, et al. Role of TLS DNA polymerases eta and kappa in processing naturally occurring structured DNA in human cells. Mol Carcinog. 2009;48:369–378. doi:10.1002/mc.20509. PMID:19117014
- Cea V, Cipolla L, Sabbioneda S. Replication of structured DNA and its implication in epigenetic stability. Front Genet. 2015;6:209. doi:10.3389/fgene.2015.00209. PMID:26136769
- Eddy S, Tillman M, Maddukuri L, et al. Human translesion polymerase kappa exhibits enhanced activity and reduced fidelity two nucleotides from G-quadruplex DNA. Biochemistry. 2016;55(37):5218–5229. doi:10.1021/acs.biochem.6b00374. PMID:27525498
- Hile SE, Wang X, Lee MY, et al. Beyond translesion synthesis: polymerase kappa fidelity as a potential determinant of microsatellite stability. Nucleic Acids Res. 2012;40:1636–1647. doi:10.1093/nar/gkr889. PMID:22021378
- Bartek J, Mistrik M, Bartkova J. Thresholds of replication stress signaling in cancer development and treatment. Nat Struct Mol Biol. 2012;19:5–7. doi:10.1038/nsmb.2220. PMID:22218289
- Bi X, Barkley LR, Slater DM, et al. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol Cell Biol. 2006;26:3527–3540. doi:10.1128/MCB.26.9.3527-3540.2006. PMID:16611994
- Bi X, Slater DM, Ohmori H, et al. DNA polymerase kappa is specifically required for recovery from the benzo[a]pyrene-dihydrodiol epoxide (BPDE)-induced S-phase checkpoint. J Biol Chem. 2005;280:22343–22355. doi:10.1074/jbc.M501562200. PMID:15817457
- Murga M, Campaner S, Lopez-Contreras AJ, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol. 2011;18:1331–1335. doi:10.1038/nsmb.2189. PMID:22120667
- Ceccaldi R, Liu JC, Amunugama R, et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature. 2015;518:258–262. doi:10.1038/nature14184. PMID:25642963
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. doi:10.1038/nrc2167. PMID:17625587
- Mamenta EL, Poma EE, Kaufmann WK, et al. Enhanced replicative bypass of platinum-DNA adducts in cisplatin-resistant human ovarian carcinoma cell lines. Cancer Res. 1994;54:3500–3505. PMID:8012973
- Kunz BA, Straffon AF, Vonarx EJ. DNA damage-induced mutation: tolerance via translesion synthesis. Mutat Res. 2000;451:169–185. doi:10.1016/S0027-5107(00)00048-8. PMID:10915871
- Lehmann AR. Replication of damaged DNA by translesion synthesis in human cells. FEBS Lett. 2005;579:873–876. doi:10.1016/j.febslet.2004.11.029. PMID:15680966
- Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–294. doi:10.1038/nature10760. PMID:22258607
- Ummat A, Rechkoblit O, Jain R, et al. Structural basis for cisplatin DNA damage tolerance by human polymerase eta during cancer chemotherapy. Nat Struct Mol Biol. 2012;19:628–632. doi:10.1038/nsmb.2295. PMID:22562137
- Alt A, Lammens K, Chiocchini C, et al. Bypass of DNA lesions generated during anticancer treatment with cisplatin by DNA polymerase eta. Science. 2007;318:967–970. doi:10.1126/science.1148242. PMID:17991862
- Chen YW, Cleaver JE, Hanaoka F, et al. A novel role of DNA polymerase eta in modulating cellular sensitivity to chemotherapeutic agents. Mol Cancer Res. 2006;4:257–265. doi:10.1158/1541-7786.MCR-05-0118. PMID:16603639
- Wagner JM, Karnitz LM. Cisplatin-induced DNA damage activates replication checkpoint signaling components that differentially affect tumor cell survival. Mol Pharmacol. 2009;76:208–214. doi:10.1124/mol.109.055178. PMID:19403702
- Yamashita YM, Okada T, Matsusaka T, et al. RAD18 and RAD54 cooperatively contribute to maintenance of genomic stability in vertebrate cells. Embo J. 2002;21:5558–5566. doi:10.1093/emboj/cdf534. PMID:12374756
- Ceppi P, Novello S, Cambieri A, et al. Polymerase eta mRNA expression predicts survival of non-small cell lung cancer patients treated with platinum-based chemotherapy. Clin Cancer Res. 2009;15:1039–1045. doi:10.1158/1078-0432.CCR-08-1227. PMID:19188177
- Teng KY, Qiu MZ, Li ZH, et al. DNA polymerase eta protein expression predicts treatment response and survival of metastatic gastric adenocarcinoma patients treated with oxaliplatin-based chemotherapy. J Transl Med. 2010;8:126. doi:10.1186/1479-5876-8-126. PMID:21110884
- Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med. 2011;17:88–96. doi:10.1016/j.molmed.2010.10.009. PMID:21087899
- Karnitz LM, Zou L. Molecular pathways: targeting ATR in cancer therapy. Clin Cancer Res. 2015;21:4780–4785. doi:10.1158/1078-0432.CCR-15-0479. PMID:26362996
- Brandsma I, Fleuren EDG, Williamson CT, et al. Directing the use of DDR kinase inhibitors in cancer treatment. Expert Opin Investig Drugs. 2017;26:1341–1355. doi:10.1080/13543784.2017.1389895. PMID:28984489
- Toledo LI, Murga M, Zur R, et al. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18:721–727. doi:10.1038/nsmb.2076. PMID:21552262
- Reaper PM, Griffiths MR, Long JM, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7:428–430. doi:10.1038/nchembio.573. PMID:21490603
- Huntoon CJ, Flatten KS, Wahner Hendrickson AE, et al. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013;73:3683–3691. doi:10.1158/0008-5472.CAN-13-0110. PMID:23548269
- Maher VM, Ouellette LM, Curren RD, et al. Caffeine enhancement of the cytotoxic and mutagenic effect of ultraviolet irradiation in a xeroderma pigmentosum variant strain of human cells. Biochem Biophys Res Commun. 1976;71:228–234. doi:10.1016/0006-291X(76)90272-2. PMID:962915
- Despras E, Daboussi F, Hyrien O, et al. ATR/Chk1 pathway is essential for resumption of DNA synthesis and cell survival in UV-irradiated XP variant cells. Hum Mol Genet. 2010;19:1690–1701. doi:10.1093/hmg/ddq046. PMID:20123862
- Mohni KN, Thompson PS, Luzwick JW, et al. A synthetic lethal screen identifies DNA repair pathways that sensitize cancer cells to combined ATR inhibition and cisplatin treatments. PloS One. 2015;10:e0125482. doi:10.1371/journal.pone.0125482. PMID:25965342
- Sakurikar N, Thompson R, Montano R, et al. A subset of cancer cell lines is acutely sensitive to the Chk1 inhibitor MK-8776 as monotherapy due to CDK2 activation in S phase. Oncotarget. 2016;7:1380–1394. doi:10.18632/oncotarget.6364. PMID:26595527