
ABSTRACT
DNA double-strand breaks (DSBs) generated by the SPO11 protein initiate meiotic recombination, an essential process for successful chromosome segregation during gametogenesis. The activity of SPO11 is controlled by multiple factors and regulatory mechanisms, such that the number of DSBs is limited and DSBs form at distinct positions in the genome and at the right time. Loss of this control can affect genome integrity or cause meiotic arrest by mechanisms that are not fully understood. Here we focus on the DSB-responsive kinase ATM and its functions in regulating meiotic DSB numbers and distribution. We review the recently discovered roles of ATM in this context, discuss their evolutionary conservation, and examine future research perspectives.
Introduction
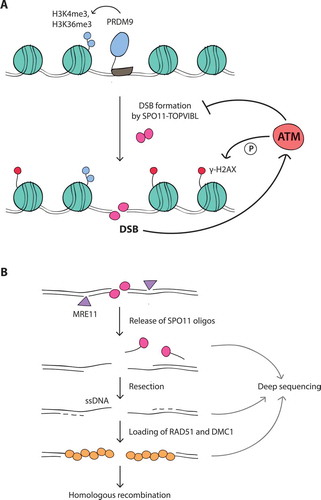
During meiosis, homologous chromosomes pair and undergo reciprocal exchange of DNA via homologous recombination, which is essential for their faithful segregation at the first meiotic division. In addition, meiotic recombination increases genetic diversity within species [Citation1–Citation3]. Meiotic recombination is initiated during early prophase I with the formation of programmed DNA double-strand breaks (DSBs) by the evolutionarily conserved SPO11 transesterase in complex with TOPVIBL () [Citation4,Citation5]. SPO11 generates DSBs at many genomic positions to promote homologous chromosome pairing and recombination [Citation6].
DSBs are dangerous, as failure to repair them can compromise genome integrity [Citation7–Citation9]. Therefore, meiotic cells control DSB numbers and mostly restrict DSB formation to a narrow window of time during prophase I [Citation10,Citation11]. Moreover, in most species, meiotic DSBs are not randomly distributed throughout the genome but are primarily (but not exclusively) localized to narrow genomic regions referred to as “hotspots” [Citation6,Citation12–Citation15]. DSB hotspots themselves are unevenly spread, so that different chromosomal regions manifest as DSB-“rich” and DSB-“poor” [Citation13,Citation16]. The shape of this “DSB landscape” is molded by a complex web of factors operating over different size scales (from single base pairs to whole chromosomes; expanded further in this review) [Citation16–Citation18]. In addition to its effects on genome diversity and evolution, dictating the position of recombination events likely promotes correct DSB repair [Citation16,Citation19]. Thus, understanding how meiotic DSBs are numerically and spatially patterned is an exciting and important area of current research.
In recent years, it has become evident that the DNA damage-responsive kinase ATM (Ataxia Telangiectasia Mutated) plays a pivotal role in regulating meiotic DSB formation in various species [Citation17,Citation20–Citation26]. In mouse, where the lack of ATM causes a particularly extreme excess of DSBs, this deregulated DSB formation appears to be a major cause of meiotic defects in ATM-deficient spermatocytes [Citation22,Citation27–Citation30]. It has been proposed that ATM, activated by DSBs, operates via a negative feedback loop to restrain SPO11 from further DSB formation [Citation10,Citation20,Citation22,Citation26,Citation31]. Because ATM is activated in the vicinity of the break, this model predicts that the additional DSBs formed in the absence of ATM will display an altered distribution [Citation22]. Indeed it was found that the budding yeast ATM ortholog, Tel1, inhibits formation of DSBs nearby one another on the same chromatid [Citation20] and also prevents cleavage of the same location on the sister chromatid and homolog chromatids [Citation25]. The Tel1-dependent process of distance-dependent DSB suppression, termed “DSB interference”, has the power to mold the DSB distribution [Citation12,Citation23]. Thus, ATM/Tel1-mediated negative feedback is thought to provide a conserved regulatory mechanism controlling the number and positions of DSBs.
Recent studies of genome-wide DSB distributions in mouse [Citation17] and yeast [Citation23] have revealed that the loss of ATM/Tel1 leaves distinct footprints on DSB distributions at different size scales. Here, we focus on research performed in mouse, which has uncovered the prominent role of ATM in shaping the genome-wide DSB distribution [Citation17]. We discuss this discovery in relation to parallel work in the budding yeast Saccharomyces cerevisiae demonstrating that Tel1 contributes to the global DSB distribution in that organism as well [Citation23]. We also highlight species-specific differences in how the DSB landscape responds to the loss of ATM/Tel1 at shorter and longer size scales. Together, these studies expand our view of the local and global roles of ATM/Tel1 in molding DSB distributions. We present an overview of advances made in understanding the spectrum of ATM/Tel1-mediated DSB control and we discuss potential underlying mechanisms.
ATM-dependent regulation of DSB numbers
The average number of meiotic DSBs per cell varies over a wide range in different species and is not correlatedwith genome size or chromosome number, suggesting the existence of species-specific (and thus genetically influenced) “set points”. For example, approximately ~ 200–250 DSBs are formed on the 20 mouse chromosome pairs and ~ 140–170 on the 16 budding yeast chromosome pairs, despite a ~ 225-fold difference in genome size between the two organisms [Citation6,Citation16,Citation32–Citation34]. On the other hand, meiotic cells of Arabidopsis and Tetrahymena, similarly experience ~ 200 DSBs but each has only 5 chromosome pairs [Citation35,Citation36]. However, DSB numbers also show substantial cell-to-cell variation even within a single individual, suggesting that the number of DSBs has a strong stochastic component [Citation6,Citation10,Citation32,Citation37,Citation38]. Nevertheless, meiotic cells generally form “just enough” DSBs to mediate homology search between chromosomes while seeming to avoid making “too many,” likely to minimize the risk of failure of DSB repair. This self-organization of meiotic DSB formation rests in large part on a network of negative feedback circuits that regulate SPO11 activity [Citation10]. ATM is key to this regulation.
ATM is a Ser/Thr kinase mutated in the cancer-prone disease ataxia telangiectasia [Citation39] and is primarily known for its function in the DNA damage response in somatic cells. ATM is activated by the MRE11 complex, which senses DSBs (for extensive review see ref [Citation40].). Once activated, ATM phosphorylates a large number of substrates (e.g., H2AX, CHK2) to regulate two interdependent processes: the cellular DNA damage response and DSB repair [Citation41–Citation44]. In contrast to mitotic cells, ATM is essential to mammalian meiotic cells, as ATM deficiency in humans and mice causes fully penetrant cell death during meiosis, resulting in infertility [Citation45–Citation47]. Although ATM may exert similar DNA damage signaling and repair functions in meiosis [Citation27,Citation30,Citation48,Citation49], it acquires an additional critical role (or co-opts an existing role) in controlling the number of SPO11-induced DSBs.
Testes from Atm–/– mice display > 10-fold higher levels of SPO11-oligonucleotide (oligo) complexes, a quantitative by-product of DSB formation (), than wild-type testes, and this increase is apparent as soon as DSBs first appear in Atm–/– juvenile testes [Citation22]. Furthermore, increasing Spo11 gene dosage has no effect in ATM-proficient mice, but increases SPO11-oligo complex levels in ATM-deficient mice [Citation22]. Conversely, while Spo11 heterozygosity in otherwise wild-type mice has little effect on DSB numbers (~ 20% reduction [Citation32,Citation50]), in an Atm–/– mutant background (i.e., in Spo11+/–Atm–/– mice) it reduces levels of SPO11-oligo complexes by half [Citation22]. These findings indicate that ATM keeps DSB numbers within a normal range even when Spo11 gene dosage is altered. The 2-fold reduction in DSB numbers in Spo11+/–Atm–/– mice has a dramatic effect on meiotic progression, such that spermatocytes complete recombination to progress to the end of prophase I [Citation22,Citation30,Citation51]. Thus, in wild-type meiotic cells, ATM is likely activated by DSBs to trigger a negative feedback loop that inhibits toxic numbers of DSBs (also see ref [Citation10].).
Figure 1. (A) Meiotic DSB formation. The SPO11-TOPVIBL complex induces DSBs at genomic locations enriched for H3K4me3 and H3K36me3, histone modifications deposited by PRDM9. DSBs activate the ATM kinase, which suppresses SPO11 from further DSB formation by an as yet unknown mechanism. In response to DSBs, ATM phosphorylates histone H2AX (γ-H2AX) in surrounding chromatin. (B) Meiotic DSB processing and detection. SPO11, covalently attached to DNA, is removed by endonucleolytic cleavage by the MRE11 complex. This reaction releases SPO11 bound to short oligonucleotides (SPO11-oligo complexes) and exposes short single-stranded DNA (ssDNA) tails at DSB sites. These tails are further extended by resection and become substrates for DMC1 and RAD51 strand exchange proteins, which mediate homologous recombination. SPO11 oligos, ssDNA, and DMC1/RAD51-coated ssDNA can be analyzed by deep sequencing to map genome-wide DSB positions (see main text for references)
This mechanism appears to be evolutionarily conserved. In Drosophila melanogaster female meiosis, temperature-sensitive mutants of tefu, the fly Atm homolog, show 1.5- to 3-fold elevated γ-H2AV (phosphorylated H2AV) signals in germaria at the restrictive temperature [Citation21]. H2AV is the equivalent of mammalian H2AX, and is phosphorylated by either ATM or the ATM-related kinase ATR (MEI-41 in flies) in response to meiotic DSBs [Citation21,Citation52]. Thus, this phenotype in flies is consistent with the role of ATM in the mouse germline.
In S. cerevisiae, tel1 mutant cells demonstrate a > 2-fold increase in Spo11-oligo complexes [Citation23], which is consistent with increases seen in recombination locally and genome-wide [Citation24,Citation25]. Three earlier studies using DNA electrophoresis-based assays in DSB repair-defective mutant backgrounds, i.e., rad50S and sae2, had also reported higher DSB frequency in tel1 mutants at several DSB hotspots and on at least one whole chromosome (not all assayed chromosomes showed this tendency) [Citation20,Citation25,Citation53]. However, two other studies using the electrophoresis-based assays reported reduced DSB frequency [Citation54,Citation55]. These conflicting results likely arise from the need to perform these assays in DSB-repair defective mutants, which may themselves have altered DSB formation [Citation10,Citation31,Citation34,Citation56]. In addition to their utility in otherwise wild-type cells, Spo11-oligo assays are likely more reliable because complexes display a long lifespan during prophase I and, as by-products of DSB formation, are relatively insensitive to DSB repair kinetics following their formation [Citation22,Citation56]. Thus, immunoprecipitation of Spo11-oligo complexes allows for a more robust assessment of Spo11 activity than detection of short-lived DSB intermediates in the chromosome. (The DSB lifespan issue, as well as the likely impact of Tel1 itself on that process, is discussed in more detail in ref [Citation23].).
Collectively, these data support the conclusion that ATM orthologs play a conserved role in regulating DSB numbers in mouse, flies, and budding yeast. Nonetheless, the consequence of ATM/Tel1 loss is different in the three species. Whereas ATM-deficient mouse spermatocytes arrest early in prophase I, tel1 budding yeast cells produce viable spores. Given the much smaller fold increase in DSB numbers in yeast than in mouse, this quantitative difference is probably sufficient to explain the more severe phenotype in mouse. The observation that lowering DSB numbers by reducing Spo11 gene dosage leads to partial rescue of meiotic progression in Atm-mutant spermatocytes also supports this interpretation.
However, in Drosophila females, tefu mutant eggs show hallmarks of DSB-repair defective female-sterile mutants, even though DSBs appear to increase similarly to yeast, and developing mutant embryos arrest. Of note, however, fly ATM is essential for normal cell proliferation as well [Citation21,Citation57], so perhaps yeast and flies are differentially responsive to additional DSBs in causing cell cycle arrest. For instance, Drosophila female meiosis might possess a lower intrinsic DSB tolerance threshold. Notably, in contrast to mouse and yeast, flies do not rely on DSB formation for homologous chromosome pairing [Citation58]. Consequently, the number of meiotic DSBs is low (~ 14 per oocyte), closer to the required number of crossovers [Citation59]. Alternatively, the fold change in DSB numbers in the absence of fly ATM might be underestimated because of the indirect quantification method used. For example, additional breaks formed nearby on the same molecule might be undetectable by measurements of γ-H2AV. Such multiple cuts might be as toxic (difficult to repair) as a large excess of well-dispersed DSBs. Whether Drosophila ATM regulates DSB distributions as in mouse and yeast remains an open question.
Regulation of DSB distribution by ATM
Control of the DSB distribution may be as important as regulation of DSB numbers in ensuring faithful genome transmission. Simultaneous DSBs at the same location on more than one chromatid could impede homologous recombination by damaging DNA templates needed for repair, while multiple DSBs nearby on the same chromatid carry the risk of deletion of intervening sequences. Further, in organisms like budding yeast and mouse that rely on DSB formation for chromosome pairing [Citation60], clustered DSB formation may interfere with the orderly and/or timely pairing of homologous chromosomes.
The final distribution of meiotic DSBs within a cell results from the interplay between factors acting at multiple levels and in multiple combinations [Citation6,Citation13,Citation14,Citation16–Citation18,Citation61–Citation63]. For example, the binding and/or activity of the DSB machinery are determined by local factors that predispose chromatin to DSB formation (e.g., histone modifications, nucleosome occupancy) in combination with factors that define higher-order chromosome structures (e.g., loop-axis organization, DSB-promoting chromosome structure proteins) [Citation4,Citation6]. Together, these properties of the DSB machinery and its chromosomal substrate have been described as “intrinsic” factors that pattern the DSB landscape. This is in contrast to another group of factors that are thought to act “extrinsically” by forming regulatory circuits to modulate DSB distribution and thus maintain DSB homeostasis [Citation4,Citation10,Citation12,Citation16]. The ATM/Tel1-dependent negative feedback loop is one such extrinsic DSB determinant [Citation12,Citation17,Citation23]. Below we discuss evidence from mouse and yeast work that ATM/Tel1 contributes to the spatial patterning of DSBs over different size scales.
Local DSB patterning (fine-scale distribution)
Despite the potential for SPO11 to act essentially anywhere in the genome, DSBs are preferentially formed at hotspots, discrete genomic positions of high DSB-forming potential. Nucleotide-resolution DSB maps obtained by deep sequencing of SPO11 oligos allows the most precise description of DSB hotspot structure [Citation16,Citation17,Citation23,Citation64], though, DSB hotspots have been mapped genome-wide by different approaches, including those that measure resected DNA () [Citation16,Citation34,Citation62,Citation65–Citation71].
In mouse (and humans), most hotspot locations are determined by PRDM9 (), a meiosis-specific histone methyltransferase with DNA binding specificity that marks nucleosomes flanking future DSB sites by tri-methylation of histone H3 on lysine 4 (H3K4me3) and on lysine 36 (H3K36me3) [Citation17,Citation62,Citation63,Citation66,Citation67,Citation69,Citation70,Citation72–Citation81]. Levels of PRDM9-dependent H3K4me3 and H3K36me3 are a moderate predictor of DSB hotspot activity, with variation of histone modification levels accounting for up to ~ 40% of variation in hotspot activity [Citation17,Citation62,Citation63,Citation79]. Interestingly, although H3K4me3 also marks active transcription promoters in mice [Citation82,Citation83], PRDM9-dependent H3K4me3 generally occurs outside of promoters, in both genic and intergenic regions, and SPO11 rarely cuts H3K4me3-enriched promoters [Citation67,Citation69]. But in the absence of PRDM9, e.g., in mice with targeted disruption of Prdm9 [Citation69] or in dogs, which naturally lack a functional PRDM9 [Citation84–Citation88], SPO11 preferentially targets H3K4me3-enriched regions that include promoters. This is likely true as well in finches, which like dogs, also lack a PRDM9 [Citation89].
Yeast hotspots are mostly in promoters [Citation16,Citation90], and hence are also enriched for H3K4me3 [Citation18,Citation91]. Moreover, a physical link between H3K4me3 and the DSB machinery has been established in budding yeast [Citation92,Citation93], and a potentially parallel mechanism may exist in mice [Citation94,Citation95]. However, H3K4me3 levels in wild-type yeast cells predict hotspot DSB frequencies poorly if at all [Citation18], and mutants lacking certain transcription factors experience changes in H3K4me3 levels at specific hotspots that are completely uncorrelated with changes in DSB frequency [Citation96].
Recent studies have explored the role of ATM/Tel1 in shaping the local distribution of DSBs in and near hotspots. Mouse DSB hotspots, as defined by clusters of SPO11-oligo sequence reads (), have a characteristic width. Most SPO11-oligo reads cluster in a prominent primary peak at the hotspot center that coincides with a nucleosome-depleted region where PRDM9 motifs are also usually found, but much weaker secondary peaks that coincide with the linkers between flanking nucleosomes can be clearly observed when SPO11-oligo maps are averaged [Citation17]. ATM deficiency affects this hotspot substructure, with an average SPO11-oligo profile that is detectably wider (143 bp in wild type versus 185 bp in Atm–/–) and has more prominent secondary peaks. At individual hotspots, this pattern can be seen as a wider distribution of SPO11-oligo signals around the hotspot center (). One explanation for this observation is that the activity of SPO11 becomes less biased toward hotspot centers. For example, ATM loss might affect chromosomal features (e.g., presence or positioning of methylated nucleosomes surrounding positions where DSBs form, see ) to cause less centered SPO11 activity. Alternatively, ATM might inhibit SPO11 from cutting the same chromatid more than once, as Tel1 does in budding yeast (see below). In this second scenario, multiple DSBs form on the same DNA molecule and spread outward from the normally centrally constrained positions. Both scenarios would imply that the ATM-mediated inhibition of DSB formation operates locally at small size scales encompassing a single hotspot.
Figure 2. (A) Local DSB patterning. (top) Examples of SPO11-DSB hotspots on chromosome 1 in Atm wild type (wt) and Atm null (data from ref [Citation17].). Note the emergence of a new hotspot in the absence of ATM. (bottom) Overlays of Atm wt and Atm null hotspot profiles show a wider distribution of SPO11 oligos around the center of hotspot B in Atm null. Methods: As previously described [Citation17], hotspots were defined as regions in smoothed SPO11-oligo maps with uniquely mapped reads exceeding 50 times the genome average. The hotspot center was defined as the coordinate of the maximum SPO11-oligo value within the hotspot. Here, SPO11-oligo maps were smoothed with a 101-bp Hann filter. RPM denotes reads per million.(B) Schematic of MRE11-independent formation of SPO11 oligos by introduction of nearby DSBs (see main text)
![Figure 2. (A) Local DSB patterning. (top) Examples of SPO11-DSB hotspots on chromosome 1 in Atm wild type (wt) and Atm null (data from ref [Citation17].). Note the emergence of a new hotspot in the absence of ATM. (bottom) Overlays of Atm wt and Atm null hotspot profiles show a wider distribution of SPO11 oligos around the center of hotspot B in Atm null. Methods: As previously described [Citation17], hotspots were defined as regions in smoothed SPO11-oligo maps with uniquely mapped reads exceeding 50 times the genome average. The hotspot center was defined as the coordinate of the maximum SPO11-oligo value within the hotspot. Here, SPO11-oligo maps were smoothed with a 101-bp Hann filter. RPM denotes reads per million.(B) Schematic of MRE11-independent formation of SPO11 oligos by introduction of nearby DSBs (see main text)](/cms/asset/65592ade-52ec-4fe5-8241-da9ffbdfb1f5/kccy_a_1464847_f0002_oc.jpg)
These two possibilities cannot be discriminated by population averaged SPO11-oligo analysis. However, the idea of multiple local cleavages might be partially supported by analysis of SPO11-oligo lengths. Normally, two prominent SPO11-oligo size classes are seen, ~ 15–27 and ~ 31–35 nt, but in the absence of ATM this bimodal length distribution is shifted toward longer oligos, leading to a substantial increase in the normally less abundant population of 40–70 nt oligos and an appearance of very long oligos (> 300 nt) [Citation17,Citation22]. These long SPO11 oligos could arise from multiple DSBs formed on the same chromatid, with the 3′ end of a SPO11 oligo created by another DSB nearby rather than MRE11 nicking (ref [Citation17,Citation23]. and M. Neale and F. Klein, personal communication) (). Thus, the observed alterations in mouse SPO11-oligo lengths and in the SPO11-hotspot width might both reflect multiple DSBs on the same DNA molecule.
Alternatively, but not mutually exclusively, long SPO11 oligos in the absence of ATM could arise from altered end processing (e.g., changes in the positions of endonucleolytic cleavage and/or decreased 3′→5′ exonucleolytic activity by MRE11). In yeast, the length distribution of Spo11 oligos in tel1 is altered in a fashion similar to those in Atm–/– spermatocytes [Citation23], and Tel1 is known to affect end processing. Tel1 phosphorylates Sae2 and Mre11 [Citation97,Citation98], which are both required for Spo11-oligo release [Citation99–Citation103], and is also important for full-length resection of DSB ends [Citation104,Citation105], However, yeast differs from mouse in that individual Spo11-oligo hotspots in tel1 yeast display a similar width to those in wild type.
In yeast, clear evidence has been provided for Tel1 suppression of multiple DSBs at closely-spaced hotspots on the same chromatid (cis interference) [Citation20]. In the presence of Tel1, the frequency of concurrent DSB formation at hotspots ~≤ 7.5 kb apart is similar to that expected by chance (no interference), whereas in the absence of Tel1, is higher than expected (negative interference). For example, the absence of Tel1 promotes the appearance of a ~ 2.4 kb DNA fragment consistent with Spo11 cutting at two DSB hotspots within the HIS4::LEU2 locus. The strength of this Tel1-mediated suppression of DSBs decays with distance but can be detected over lengths of 70–100 kb. These observations are consistent with the hypothesis that Tel1-mediated suppression primarily functions within a single chromatin loop (~ 10–20 kb in budding yeast) and to a lesser extent between adjacent loops (discussed in ref [Citation12].).
Although population averaged Spo11-oligo maps cannot predict DSBs in individual cells, maps in budding yeast provide support for the occurrence of cis interference of DSBs: for the two strongest hotspots, GAT1 and CCT6, the neighboring weaker hotspots become stronger when Tel1 is missing [Citation23]. This phenomenon may also be observed in mouse (), although further analysis of mouse SPO11-oligo maps will be required to reveal similar behavior.
Importantly, Tel1 (and its paralog Mec1) in S. cerevisiae also suppresses clustered DSB formation in trans, which is the formation of DSBs at the same position on the sister chromatid or homologous chromosome [Citation20,Citation25]. Because multiple cutting may remove available repair templates or lead to formation of potentially harmful double recombination products, this form of suppression may be important to prevent aberrant meiotic recombination. It has been speculated that the establishment of trans-interference is also locally regulated and distance-dependent, both between sister chromatids and between non-sister chromatids undergoing homologous pairing, although these hypotheses are largely unexplored (discussed in ref [Citation25,Citation31].).
Large-scale DSB patterning
At larger size scales, the nonrandom distribution of DSBs defines large chromosomal domains in yeast, mice, and humans [Citation13,Citation16,Citation17,Citation23,Citation56,Citation64,Citation66,Citation67,Citation90,Citation106,Citation107]. Strikingly, the loss of ATM in spermatocytes leads to substantial large-scale alterations in DSB distribution [Citation17]. Specifically, large, Mb-scale domains that are normally DSB-poor (cold) appear to be more sensitive to ATM loss than those that are DSB-rich (hot). Consequently, although all SPO11-oligo hotspots become stronger, weaker hotspots that populate DSB-poor domains experience a greater increase in DSB formation. Large-scale ATM-dependent DSB suppression occurs in domains of correlated behavior (measured as the correlation in fold change in SPO11-oligo densities when comparing neighboring regions) and is estimated to decay by ~ 15–20 Mb. In addition, in the absence of ATM a substantial number of normally weak SPO11-oligo clusters rises above the arbitrary threshold used to call hotspots, which leads to an overall increase in the number of detectable hotspots [Citation17] (see, e.g., ). This behavior predicts that more new hotspots will be detected within normally DSB-poor domains than in DSB-rich domains (). Recent findings demonstrate that ATM and PRDM9 contribute independently to hotspot strength [Citation63]. However, it remains to be elucidated why DSB-poor domains respond more strongly to the absence of ATM and what molecular events within these domains are regulated by ATM to impose this stronger suppression of DSB formation.
In budding yeast, Tel1 provides a more subtle contribution to genome-wide DSB patterning [Citation23]. Nevertheless, Tel1 absence causes changes in DSB distributions, as distinct subchromosomal domains (e.g., hotspots in subtelomeric or pericentric regions) accumulate fewer DSBs than the genome average early in meiosis. These alterations are not permanent, as later in meiosis DSB frequencies in the regions less sensitive to Tel1 loss level off at the genome average. Remarkably, this homeostatic compensatory behavior is implemented despite the increase in DSB numbers in tel1, and so it must be mediated by other regulatory systems, with this behavior showing the ability of meiotic cells to adjust to variations in DSB numbers.
Whole chromosomes provide an even larger size scale to evaluate DSB distribution. Smaller chromosomes typically experience more DSBs, which is attributed to a negative feedback mechanism tied to some feature of the engagement of homologous chromosomes [Citation56]. This evolutionarily conserved pathway negatively regulates DSB numbers by inhibiting further DSB formation on successfully synapsed chromosome segments [Citation10,Citation56,Citation108,Citation109]. Perhaps smaller chromosomes take longer on average to engage with one another, allowing them to continue to undergo DSB formation for longer periods. Thus, in budding yeast and mouse, this behavior can be observed as a negative correlation of SPO11-oligo density with chromosome size [Citation16,Citation17,Citation56]. In S. cerevisiae, this anti-correlation is lost in mutants missing components of the homolog engagement pathway (“ZMM” mutants defective in homologous synapsis and recombination [Citation110]). Similarly to tel1, these mutants produce more DSBs and display changes in DSB distributions [Citation56]. However, recent work in S. cerevisiae and mouse has reinforced the idea that homolog engagement and ATM/Tel1 represent distinct pathways for negative control of DSB formation [Citation10]: In Atm–/– and tel1 mutants, SPO11-oligo density remains negatively correlated with chromosome size despite the increase in DSBs, although Tel is required for timely establishment of this relationship. Thus, chromosome-scale DSB patterns are not observably affected by ATM/Tel1.
ATM has an even more pronounced role in suppressing DSB formation on the non-homologous parts of the X and Y chromosomes. In wild-type spermatocytes, SPO11-oligo density in these regions is much lower than on autosomes, but in Atm–/–, these regions experience a greater increase in SPO11-oligo density than genome average [Citation17]. Although the molecular details remain to be elucidated, it is plausible that this suppression is needed to retain low numbers of DSBs in non-homologous sex-chromosome segments, as they are not competent for synapsis and meiotic recombination.
A role for ATR in DSB control?
The more profound DSB distribution phenotype in Atm–/– mice than tel1 yeast may reflect the quantitatively more severe DSB number phenotype of Atm–/– or the acquisition of different functions for the two proteins during meiosis. Redundancy with other factors must also be considered, especially in budding yeast, where redundancy between Tel1 and the Mec1 kinase (related to mammalian ATR) has been observed in other meiotic contexts. Both proteins phosphorylate Rec114, a Spo11 partner required for DSB formation, and Sae2 [Citation26,Citation97]. Moreover, both Tel1 and Mec1 play roles in DSB resection and directing DSB repair towards the homolog by inhibiting intersister recombination [Citation53,Citation104,Citation105]. While Tel1 controls the repair of low abundance DSBs occurring early in meiosis, it appears to become redundant with Mec1 for the repair of high abundance DSBs later in meiosis [Citation105].
If redundancy between Tel1 and Mec1 exists in the control of DSB distribution, timing may be a key factor. For example, this could explain why the observed alterations in DSB patterning in the tel1 mutant change over time in meiosis [Citation23]. We emphasize, however, that even though Mec1 may substitute for Tel1 to some extent or at specific times, Tel1 plays a dominant role in this process. This is supported by the fact that in the presence of Tel1, the Mec1 pathway is dispensable for the local control of DSB distribution [Citation20].
In mice, ATR does not appear to play a major role in regulating DSB numbers. SPO11-oligo complexes formed in spermatocytes with conditionally-deleted Atr are similar in number to those in wild type [Citation111], while they are slightly increased in spermatocytes carrying a hypomorphic Atr allele [Citation112]. As in yeast, epistasis studies could bring new perspectives on the relationships of these two kinases. Redundancy is suggested in another context in that ATR partially substitutes for ATM in response to DSBs to phosphorylate H2AX in meiosis [Citation27,Citation30]. ATR has an essential function in executing DSB repair in mouse meiosis, in addition to its previously described role in meiotic silencing of unsynapsed chromatin [Citation113,Citation114].
Mechanism of ATM/Tel1-dependent regulation of DSB formation
How ATM/Tel1 negatively regulates DSB formation is an active area of investigation. It is envisioned that ATM/Tel1 is activated by DSBs to phosphorylate proteins that regulate DSB formation. Kinase activity is critical, as a kinase-dead tel1 mutation increases levels of Spo11-oligo complexes similarly to tel1 null mutation [Citation23]. Similar experiments have not been performed in mice, however, because kinase-dead Atm mutations lead to embryonic lethality [Citation115,Citation116].
Relevant phosphotarget(s) are not yet known, although REC114 and HORMAD1 are good candidates. REC114 is a conserved component of the DSB-promoting complex (REC114-MEI4-IHO1 in mouse, Rec114-Mei4-Mer2 in S. cerevisiae; see also ref [Citation117].), whereas HORMAD1 and its yeast ortholog, Hop1, are structural elements of meiotic chromosome axes that are required for normal DSB levels [Citation4,Citation26,Citation53,Citation109,Citation118–Citation122]. Yeast Rec114 and Hop1 are known Tel1 and Mec1 targets, and they are phosphorylated in a DSB-dependent manner [Citation26,Citation53,Citation123]. Also, mouse HORMAD1 contains putative ATM/ATR phosphorylation sites [Citation109,Citation124] and DSB formation promotes HORMAD1 phosphorylation [Citation124].
In yeast, experiments testing whether Tel1-mediated DSB control operates via Rec114 phosphorylation have given ambiguous results. A rec114 mutant lacking potential Tel1 and Mec1 target SQ/TQ sites (rec114-8A) shows evidence of precocious and elevated DSB formation, and a putative phosphomimetic mutant (rec114-8D) shows reduced DSB levels [Citation26]. However, neither rec114-8A alone or in combination with a phosphomutant hop1 phenocopies the elevated Spo11-oligo complex formation caused by Tel1 deficiency [Citation23]. Moreover, formation of multiple nearby DSBs on the same chromatid is not observed with non-phosphorylatable rec114 and hop1 alleles (V. Garcia and M. Neale, personal communication), unlike what is observed with the absence of Tel1 [Citation20]. These findings can be reconciled if there are multiple Tel1 targets (including Rec114 and Hop1), any of which is sufficient to inhibit DSB formation when phosphorylated [Citation23]. Alternatively, as proposed by Carballo et al. [Citation26], the modest phenotype of rec114-8A may be explained if this allele mainly causes repeated cleavage by the same activated DSB machinery at the same location, which would hardly increase the DSB signals measured by Southern.
In mouse, so far, only one of the several HORMAD1 phosphorylation sites, Ser375, has been investigated [Citation113,Citation124]. Ser375-phosphorylated HORMAD1 appears early in meiosis and it is associated with unsynapsed chromosomes throughout meiotic prophase. However, whether phosphorylation at this site depends on ATM has only been examined at late meiotic stages, not the crucial early meiotic stages when negative feedback circuits are expected to regulate DSB formation.
It has been proposed that DSB-promoting complexes assemble on chromosome axes to which the chromatin loop is tethered and a DSB is formed [Citation92,Citation93,Citation125,Citation126] (). Molecular details of this model integrating DSB formation with the loop-axis structure of meiotic chromosomes derive principally from data in yeast, but it has long been envisioned as an evolutionarily conserved process [Citation127]. Recent studies in mouse support this view [Citation95,Citation118]. If correct, the tethering of chromatin loops at DSB sites would provide a structural framework within which ATM could phosphorylate members of the DSB-promoting complex and/or HORMAD1 to negatively regulate DSB formation and thus locally shape DSB distribution and hotspot usage ().
Figure 3. Large-scale DSB patterning
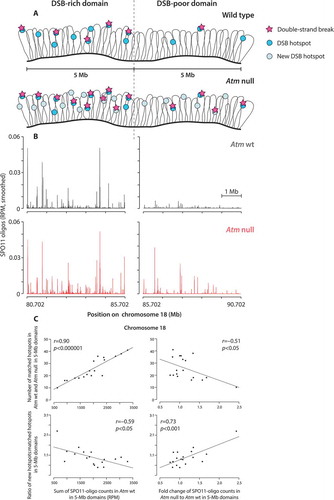
Figure 4. Hypothetical model for ATM-mediated DSB control. (top) Chromatin loops containing DSB hotspots are anchored to the chromosome axis. Before DSB formation, the IHO1-REC114-MEI4 complex and the axis-structural component HORMAD1 assemble along the chromosome axis. The IHO1-REC114-MEI4 complex together with HORMAD1 promotes DSB formation by SPO11. At present, physical interactions between IHO1, REC114, and MEI4 are inferred only from yeast two-hybrid analyzes [Citation118,Citation120], but further support for these interactions comes from extensive two-hybrid and coimmunoprecipitation studies of the orthologous proteins in budding and fission yeasts: Mer2-Rec114-Mei4 [Citation142–Citation145] and Rec15-Rec7-Rec24 [Citation146,Citation147], respectively. (bottom) A DSB is formed preferentially at a DSB hotspot in a chromatin loop tethered to the axis. In response to the DSB, ATM is activated and phosphorylates REC114, HORMAD1, and/or other proteins. Phosphorylation events separately or in combination inhibit additional DSB formation at the same chromatin loop (1) or at the adjacent loop (2) which is pre-activated for DSB formation, by inhibiting or destabilizing of the IHO1-REC114-MEI4 complex. HORMAD1 phosphorylation in the vicinity of the DSB prevents the assembly of the IHO1-REC114-MEI4 complexes and thus DSB formation at other nearby chromatin loop (3)
![Figure 4. Hypothetical model for ATM-mediated DSB control. (top) Chromatin loops containing DSB hotspots are anchored to the chromosome axis. Before DSB formation, the IHO1-REC114-MEI4 complex and the axis-structural component HORMAD1 assemble along the chromosome axis. The IHO1-REC114-MEI4 complex together with HORMAD1 promotes DSB formation by SPO11. At present, physical interactions between IHO1, REC114, and MEI4 are inferred only from yeast two-hybrid analyzes [Citation118,Citation120], but further support for these interactions comes from extensive two-hybrid and coimmunoprecipitation studies of the orthologous proteins in budding and fission yeasts: Mer2-Rec114-Mei4 [Citation142–Citation145] and Rec15-Rec7-Rec24 [Citation146,Citation147], respectively. (bottom) A DSB is formed preferentially at a DSB hotspot in a chromatin loop tethered to the axis. In response to the DSB, ATM is activated and phosphorylates REC114, HORMAD1, and/or other proteins. Phosphorylation events separately or in combination inhibit additional DSB formation at the same chromatin loop (1) or at the adjacent loop (2) which is pre-activated for DSB formation, by inhibiting or destabilizing of the IHO1-REC114-MEI4 complex. HORMAD1 phosphorylation in the vicinity of the DSB prevents the assembly of the IHO1-REC114-MEI4 complexes and thus DSB formation at other nearby chromatin loop (3)](/cms/asset/16f835dd-3a6b-47f4-a37b-4c49d6e88f94/kccy_a_1464847_f0004_oc.jpg)
If tethering of chromatin loops explains local effects of ATM, how might ATM influence DSB distributions at larger size scales? On the assumption that the relevant ATM targets are the same for both local and large-scale DSB control, it has been proposed that the differential susceptibility of Mb-sized chromosome domains to ATM-mediated DSB suppression reflects variability in the amount of ATM substrates (e.g., REC114 and/or HORMAD1) along the chromosome axis [Citation17]. In this view, the underlying density of these factors may determine domains of differing intrinsic DSB-forming potential as well as of differential sensitivity to ATM. In budding yeast, DSB-rich genomic regions are enriched for Hop1 and Rec114 [Citation126]. These domains of enrichment also tend to be more responsive to the Tel1-independent negative feedback regulation of DSBs attributed to homolog engagement [Citation56].
In mice, there is an inverse correlation for Mb-sized domains between the SPO11-oligo density in wild type and the fold-increase in SPO11-oligo density in the absence of ATM [Citation17]. That is, relatively DSB-poor domains in wild type also tend to be the domains that are more highly suppressed by ATM (also see ). Thus, studies in mice of correlations between the density of potential ATM substrates and the strength of ATM-dependent regulation are likely to be informative. Intuitively, domains enriched for substrates might be more responsive to ATM (positive correlation), but the opposite can be also imagined (negative correlation). In particular, domains occupied by fewer substrates might be more responsive if the ATM proficiency is measured by how “good” ATM is in phosphorylating all of its available targets. In this case, fewer targets would simply mean less “work” for ATM.
As mentioned above, histone H2AX is also a target for ATM-mediated phosphorylation at residue 139 (γH2AX) [Citation128]. Because γH2AX is not spatially restricted to DSB sites but spreads from DSBs to surrounding chromatin [Citation129], ATM-dependent γH2AX spreading could theoretically function to suppress DSB formation in the vicinity of the initial break. The extent of γH2AX-positive domains in meiosis has been not assessed, but in mitotically cycling cells, γH2AX extends for up to 1–2 Mb in mammals [Citation130] and for up to 50 kb in S. cerevisiae [Citation131]. However, although this idea is appealing, H2ax-null mice do not phenocopy Atm–/ – [Citation132,Citation133]. Also, in S. cerevisiae, h2a mutants do not show DSB clustering like tel1 (V. Garcia and M. Neale, personal communication, and ref [Citation20].). Thus, H2AX is not critical for ATM-regulated negative feedback. Possible redundancy should also be considered in this case, however, since at least one other Tel1/Mec1-dependent histone phosphorylation (Thr 129 phosphorylation on H2B) has been identified in vegetative yeast [Citation134].
Consequences of the lack of ATM
While the grossly elevated number of DSBs in Atm-null meiosis is presumably a major source of meiotic defects [Citation22], alteration in DSB distribution is likely to be as critical [Citation17]. As discussed above, loss of local DSB suppression in Atm–/ – may provoke SPO11 to cut multiple chromatids at the same time, which would damage the repair template, or to cut multiple times at adjacent genomic positions, which may be problematic to repair. It is interesting to consider how loss of control of local DSB patterning contributes to the overall increase in DSB numbers. Atm–/ – spermatocytes have a relatively small increase in cytological foci of DMC1 (the meiosis-specific paralog of the RAD51 strand-exchange protein that assembles on resected strands) compared with the increase in SPO11-oligo complexes;(ref [Citation22]. and A.L., J.L., S.K., M.J., unpublished observations) and similar observations have been reported previously for RAD51 foci in Spo11+/–Atm–/ – spermatocytes [Citation51] suggesting that fewer foci than expected may mark multiple, closely spaced DSBs. An alternative is that fewer DMC1/RAD51 arise than expected because of the involvement of ATM in other functions, such as resection, as seen in mitotic cells [Citation135–Citation137] and in yeast meiosis [Citation104,Citation105], and/or interhomolog bias [Citation53], which may cause more rapid intersister DSB repair. If ATM has these functions in meiosis, like Tel1 in budding yeast, their loss might exacerbate the consequences of dysregulated DSB formation.
In this regard, it is interesting to consider the synapsis phenotypes of Atm–/ – mutants. Whereas synapsis is gravely affected in Atm–/ – spermatocytes [Citation27,Citation28], synapsis in Spo11+/–Atm–/ – is nearly normal and cells progress substantially further [Citation30,Citation51]. Thus, the synapsis defects in Atm–/ – mutants are probably secondary to the altered DSB formation, most likely the increased levels of DSBs but possibly also their altered distribution. Chromosome fragments that are sometimes observed in Spo11+/–Atm–/ – spermatocytes [Citation30,Citation51] could be an indication of inappropriate DSB formation at the same location on sister chromatids (and, possibly, the homolog).
Other consequences of the lack of ATM should also be considered that were not previously anticipated. For example, extra DSBs formed in DSB-cold domains may present issues for repair if the chromatin composition or chromosome structure of cold domains limits access of the DSB repair machinery. Although rather speculative at this point, chromatin context is known to affect DSB repair pathway choice in mitotic cells [Citation138]. Enrichment for meiosis-specific factors in different domains could also affect recombination pathway choice, for example, as proposed for differential Zip3 or Hop1 occupancy in yeast [Citation139,Citation140]. It will be interesting to determine whether changes in recombination outcomes observed in the absence of ATM/Tel1 [Citation24,Citation51] are a direct consequence of large-scale changes in DSB distributions.
Concluding remarks
The number of DSBs and their distribution in meiosis is highly regulated and the ATM kinase critically contributes to this complex process. While clearly important for a successful meiosis, ATM control of DSB formation also has the power to shape genome evolution. Understanding the underlying mechanisms is a key future priority. Deciphering relevant ATM targets and mapping their positions in the genome relative to SPO11-oligo maps will shed light onto how ATM molds the DSB landscape. Moreover, correlating SPO11 activity across the genome with other cellular processes (e.g., transcription), chromatin modifications, and meiotic chromosome architecture (loop-axis organization) may reveal interesting relationships. Conversely, it could be fruitful to explore changes in chromatin dynamics in the absence of ATM. A detailed investigation of DSB repair in the absence of ATM at distinct DSB sites and genome-wide will be crucial for understanding the effect of ATM-mediated DSB control in the suppression of aberrant meiotic events.
Acknowledgments
We thank members of the Jasin and Keeney labs for helpful discussions.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Hunter N. Meiotic recombination: the essence of heredity. Cold Spring Harb Perspect Biol. 2015;7. PMID:26511629. DOI:10.1101/cshperspect.a016618
- Szekvolgyi L, Ohta K, Nicolas A. Initiation of meiotic homologous recombination: flexibility, impact of histone modifications, and chromatin remodeling. Cold Spring Harb Perspect Biol. 2015;7:a016527. PMID:25934010.
- Thacker D, Keeney S. Homologous recombination during meiosis. DNA replication, recombination, and repair: molecular mechanisms and pathology. Tokyo: Springer; 2016. p. 131–151.
- Lam I, Keeney S. Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb Perspect Biol. 2014;7:a016634. PMID:25324213.
- Robert T, Vrielynck N, Mezard C, et al. A new light on the meiotic DSB catalytic complex. Semin Cell Dev Biol. 2016;54:165–176. PMID:26995551.
- de Massy B. Initiation of meiotic recombination: how and where? Conservation and specificities among eukaryotes. Annu Rev Genet. 2013;47:563–599. PMID:24050176.
- Kim S, Peterson SE, Jasin M, et al. Mechanisms of germ line genome instability. Semin Cell Dev Biol. 2016;54:177–187. PMID:26880205.
- Sasaki M, Lange J, Keeney S. Genome destabilization by homologous recombination in the germ line. Nat Rev Mol Cell Biol. 2010;11:182–195. PMID:20164840.
- Hochwagen A, Amon A. Checking your breaks: surveillance mechanisms of meiotic recombination. Curr Biol. 2006;16:R217–28. PMID:16546077.
- Keeney S, Lange J, Mohibullah N. Self-organization of meiotic recombination initiation: general principles and molecular pathways. Annu Rev Genet. 2014;48:187–214. PMID:25421598.
- Murakami H, Keeney S. Temporospatial coordination of meiotic DNA replication and recombination via DDK recruitment to replisomes. Cell. 2014;158:861–873. PMID:25126790.
- Cooper TJ, Garcia V, Neale MJ. Meiotic DSB patterning: A multifaceted process. Cell Cycle. 2016;15:13–21. PMID:26730703.
- Kauppi L, Jeffreys AJ, Keeney S. Where the crossovers are: recombination distributions in mammals. Nat Rev Genet. 2004;5:413–424. PMID:15153994.
- Petes TD. Meiotic recombination hot spots and cold spots. Nat Rev Genet. 2001;2:360–369. PMID:11331902.
- Baudat F, Imai Y, de Massy B. Meiotic recombination in mammals: localization and regulation. Nat Rev Genet. 2013;14:794–806. PMID:24136506.
- Pan J, Sasaki M, Kniewel R, et al. A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell. 2011;144:719–731. PMID:21376234.
- Lange J, Yamada S, Tischfield SE, et al. The landscape of mouse meiotic double-strand break formation, processing, and repair. Cell. 2016;167:695–708e16. PMID:27745971.
- Tischfield SE, Keeney S. Scale matters: the spatial correlation of yeast meiotic DNA breaks with histone H3 trimethylation is driven largely by independent colocalization at promoters. Cell Cycle. 2012;11:1496–1503. PMID:22433953.
- Nambiar M, Smith GR. Repression of harmful meiotic recombination in centromeric regions. Semin Cell Dev Biol. 2016;54:188–197. PMID:26849908.
- Garcia V, Gray S, Allison RM, et al. Tel1(ATM)-mediated interference suppresses clustered meiotic double-strand-break formation. Nature. 2015;520:114–118. PMID:25539084.
- Joyce EF, Pedersen M, Tiong S, et al. Drosophila ATM and ATR have distinct activities in the regulation of meiotic DNA damage and repair. J Cell Biol. 2011;195:359–367. PMID:22024169.
- Lange J, Pan J, Cole F, et al. ATM controls meiotic double-strand-break formation. Nature. 2011;479:237–240. PMID:22002603.
- Mohibullah N, Keeney S. Numerical and spatial patterning of yeast meiotic DNA breaks by Tel1. Genome Res. 2017;27:278–288. PMID:27923845.
- Anderson CM, Oke A, Yam P, et al. Reduced crossover interference and increased ZMM-independent recombination in the absence of Tel1/ATM. PLoS Genet. 2015;11:e1005478. PMID:26305689.
- Zhang L, Kim KP, Kleckner NE, et al. Meiotic double-strand breaks occur once per pair of (sister) chromatids and, via Mec1/ATR and Tel1/ATM, once per quartet of chromatids. Proc Natl Acad Sci USA. 2011;108:20036–20041. PMID:22123968.
- Carballo JA, Panizza S, Serrentino ME, et al. Budding yeast ATM/ATR control meiotic double-strand break (DSB) levels by down-regulating Rec114, an essential component of the DSB-machinery. PLoS Genet. 2013;9:e1003545. PMID:23825959.
- Barchi M, Mahadevaiah S, Di Giacomo M, et al. Surveillance of different recombination defects in mouse spermatocytes yields distinct responses despite elimination at an identical developmental stage. Mol Cell Biol. 2005;25:7203–7215. PMID:16055729.
- Barlow C, Liyanage M, Moens PB, et al. Atm deficiency results in severe meiotic disruption as early as leptonema of prophase I. Development. 1998;125:4007–4017. PMID:9735362.
- Xu Y, Ashley T, Brainerd EE, et al. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996;10:2411–2422. PMID:8843194.
- Bellani MA, Romanienko PJ, Cairatti DA, et al. SPO11 is required for sex-body formation, and Spo11 heterozygosity rescues the prophase arrest of Atm-/- spermatocytes. J Cell Sci. 2005;118:3233–3245. PMID:15998665.
- Cooper TJ, Wardell K, Garcia V, et al. Homeostatic regulation of meiotic DSB formation by ATM/ATR. Exp Cell Res. 2014;329:124–131. PMID:25116420.
- Cole F, Kauppi L, Lange J, et al. Homeostatic control of recombination is implemented progressively in mouse meiosis. Nat Cell Biol. 2012;14:424–430. PMID:22388890.
- Baudat F, de Massy B. Regulating double-stranded DNA break repair towards crossover or non-crossover during mammalian meiosis. Chromosome Res. 2007;15:565–577. PMID:17674146.
- Buhler C, Borde V, Lichten M. Mapping meiotic single-strand DNA reveals a new landscape of DNA double-strand breaks in Saccharomyces cerevisiae. PLoS Biol. 2007;5:e324. PMID:18076285.
- Loidl J, Lorenz A. DNA double-strand break formation and repair in Tetrahymena meiosis. Semin Cell Dev Biol. 2016;54:126–134. PMID:26899715.
- Edlinger B, Schlogelhofer P. Have a break: determinants of meiotic DNA double strand break (DSB) formation and processing in plants. J Exp Bot. 2011;62:1545–1563. PMID:21220780.
- Kauppi L, Jasin M, Keeney S. How much is enough? Control of DNA double-strand break numbers in mouse meiosis. Cell Cycle. 2013;12:2719–2720. PMID:23966150.
- Chen SY, Tsubouchi T, Rockmill B, et al. Global analysis of the meiotic crossover landscape. Dev Cell. 2008;15:401–415. PMID:18691940.
- Savitsky K, Bar-Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. PMID:7792600.
- Paull TT. Mechanisms of ATM Activation. Annu Rev Biochem. 2015;84:711–738. PMID:25580527.
- Derheimer FA, Kastan MB. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett. 2010;584:3675–3681. PMID:20580718.
- Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6:931–942. PMID:17457059.
- Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31:402–410. PMID:16774833.
- Di Domenico EG, Romano E, Del Porto P, et al. Multifunctional role of ATM/Tel1 kinase in genome stability: from the DNA damage response to telomere maintenance. Biomed Res Int. 2014;2014:787404. PMID:25247188.
- Barlow C, Hirotsune S, Paylor R, et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. PMID:8689683.
- Elson A, Wang Y, Daugherty CJ, et al. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci USA. 1996;93:13084–13089. PMID:8917548.
- Sedgewick R, Boder E. Ataxia-telangiectasia. Handbook of clinical neurology. Amsterdam: Elsevier Scientific Publishers; 1991. p. 347–423.
- Di Giacomo M, Barchi M, Baudat F, et al. Distinct DNA-damage-dependent and -independent responses drive the loss of oocytes in recombination-defective mouse mutants. Proc Natl Acad Sci USA. 2005;102:737–742. PMID:15640358.
- Pacheco S, Marcet-Ortega M, Lange J, et al. The ATM signaling cascade promotes recombination-dependent pachytene arrest in mouse spermatocytes. PLoS Genet. 2015;11:e1005017. PMID:25768017.
- Bellani MA, Boateng KA, McLeod D, et al. The expression profile of the major mouse SPO11 isoforms indicates that SPO11beta introduces double strand breaks and suggests that SPO11alpha has an additional role in prophase in both spermatocytes and oocytes. Mol Cell Biol. 2010;30:4391–4403. PMID:20647542.
- Barchi M, Roig I, Di Giacomo M, et al. ATM promotes the obligate XY crossover and both crossover control and chromosome axis integrity on autosomes. PLoS Genet. 2008;4:e1000076. PMID:18497861.
- Madigan JP, Chotkowski HL, Glaser RL. DNA double-strand break-induced phosphorylation of Drosophila histone variant H2Av helps prevent radiation-induced apoptosis. Nucleic Acids Res. 2002;30:3698–3705. PMID:12202754.
- Carballo JA, Johnson AL, Sedgwick SG, et al. Phosphorylation of the axial element protein Hop1 by Mec1/Tel1 ensures meiotic interhomolog recombination. Cell. 2008;132:758–770. PMID:18329363.
- Argunhan B, Farmer S, Leung WK, et al. Direct and indirect control of the initiation of meiotic recombination by DNA damage checkpoint mechanisms in budding yeast. PLoS One. 2013;8:e65875. PMID:23762445.
- Blitzblau HG, Hochwagen A. ATR/Mec1 prevents lethal meiotic recombination initiation on partially replicated chromosomes in budding yeast. Elife. 2013;2:e00844. PMID:24137535.
- Thacker D, Mohibullah N, Zhu X, et al. Homologue engagement controls meiotic DNA break number and distribution. Nature. 2014;510:241–246. PMID:24717437.
- Silva E, Tiong S, Pedersen M, et al. ATM is required for telomere maintenance and chromosome stability during Drosophila development. Curr Biol. 2004;14:1341–1347. PMID:15296750.
- McKim KS, Green-Marroquin BL, Sekelsky JJ, et al. Meiotic synapsis in the absence of recombination. Science. 1998;279:876–878. PMID:9452390.
- Mehrotra S, McKim KS. Temporal analysis of meiotic DNA double-strand break formation and repair in Drosophila females. PLoS Genet. 2006;2:e200. PMID:17166055.
- Gerton JL, Hawley RS. Homologous chromosome interactions in meiosis: diversity amidst conservation. Nat Rev Genet. 2005;6:477–487. PMID:15931171.
- Wu TC, Lichten M. Factors that affect the location and frequency of meiosis-induced double-strand breaks in Saccharomyces cerevisiae. Genetics. 1995;140:55–66. PMID:7635308.
- Baker CL, Walker M, Kajita S, et al. PRDM9 binding organizes hotspot nucleosomes and limits Holliday junction migration. Genome Res. 2014;24:724–732. PMID:24604780.
- Yamada S, Kim S, Tischfield SE, et al. Genomic and chromatin features shaping meiotic double-strand break formation and repair in mice. Cell Cycle. 2017;1–15. PMID:28820351. DOI:10.1080/15384101.2017.1361065
- Fowler KR, Sasaki M, Milman N, et al. Evolutionarily diverse determinants of meiotic DNA break and recombination landscapes across the genome. Genome Res. 2014;24:1650–1664. PMID:25024163.
- Lichten M, de Massy B. The impressionistic landscape of meiotic recombination. Cell. 2011;147:267–270. PMID:22000007.
- Pratto F, Brick K, Khil P, et al. DNA recombination. Recombination initiation maps of individual human genomes. Science. 2014;346:1256442. PMID:25395542.
- Smagulova F, Gregoretti IV, Brick K, et al. Genome-wide analysis reveals novel molecular features of mouse recombination hotspots. Nature. 2011;472:375–378. PMID:21460839.
- Blitzblau HG, Bell GW, Rodriguez J, et al. Mapping of meiotic single-stranded DNA reveals double-stranded-break hotspots near centromeres and telomeres. Curr Biol. 2007;17:2003–2012. PMID:18060788.
- Brick K, Smagulova F, Khil P, et al. Genetic recombination is directed away from functional genomic elements in mice. Nature. 2012;485:642–645. PMID:22660327.
- Grey C, Clement JA, Buard J, et al. In vivo binding of PRDM9 reveals interactions with noncanonical genomic sites. Genome Res. 2017;27:580–590. PMID:28336543.
- Gerton JL, DeRisi J, Shroff R, et al. Global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2000;97:11383–11390. PMID:11027339.
- Hayashi K, Yoshida K, Matsui Y. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature. 2005;438:374–378. PMID:16292313.
- Mihola O, Trachtulec Z, Vlcek C, et al. A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science. 2009;323:373–375. PMID:19074312.
- Myers S, Bowden R, Tumian A, et al. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science. 2010;327:876–879. PMID:20044541.
- Parvanov ED, Petkov PM, Paigen K. Prdm9 controls activation of mammalian recombination hotspots. Science. 2010;327:835. PMID:20044538.
- Baudat F, Buard J, Grey C, et al. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2010;327:836–840. PMID:20044539.
- Buard J, Barthes P, Grey C, et al. Distinct histone modifications define initiation and repair of meiotic recombination in the mouse. EMBO J. 2009;28:2616–2624. PMID:19644444.
- Grey C, Barthes P, Chauveau-Le Friec G, et al. Mouse PRDM9 DNA-binding specificity determines sites of histone H3 lysine 4 trimethylation for initiation of meiotic recombination. PLoS Biol. 2011;9:e1001176. PMID:22028627.
- Powers NR, Parvanov ED, Baker CL, et al. The Meiotic Recombination Activator PRDM9 Trimethylates Both H3K36 and H3K4 at Recombination Hotspots In Vivo. PLoS Genet. 2016;12:e1006146. PMID:27362481.
- Wu H, Mathioudakis N, Diagouraga B, et al. Molecular basis for the regulation of the H3K4 methyltransferase activity of PRDM9. Cell Rep. 2013;5:13–20. PMID:24095733.
- Eram MS, Bustos SP, Lima-Fernandes E, et al. Trimethylation of histone H3 lysine 36 by human methyltransferase PRDM9 protein. J Biol Chem. 2014;289:12177–12188. PMID:24634223.
- Howe FS, Fischl H, Murray SC, et al. Is H3K4me3 instructive for transcription activation? Bioessays. 2017;39:1–12. PMID:28004446.
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. PMID:17218268.
- Axelsson E, Webster MT, Ratnakumar A, et al. Death of PRDM9 coincides with stabilization of the recombination landscape in the dog genome. Genome Res. 2012;22:51–63. PMID:22006216.
- Oliver PL, Goodstadt L, Bayes JJ, et al. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet. 2009;5:e1000753. PMID:19997497.
- Auton A, Rui Li Y, Kidd J, et al. Genetic recombination is targeted towards gene promoter regions in dogs. PLoS Genet. 2013;9:e1003984. PMID:24348265.
- Campbell CL, Bherer C, Morrow BE, et al. A pedigree-based map of recombination in the domestic dog genome. G3 (Bethesda). 2016. PMID:27591755. DOI:10.1534/g3.116.034678
- Munoz-Fuentes V, Di Rienzo A, Vila C. Prdm9, a major determinant of meiotic recombination hotspots, is not functional in dogs and their wild relatives, wolves and coyotes. PLoS One. 2011;6:e25498. PMID:22102853.
- Singhal S, Leffler EM, Sannareddy K, et al. Stable recombination hotspots in birds. Science. 2015;350:928–932. PMID:26586757.
- Baudat F, Nicolas A. Clustering of meiotic double-strand breaks on yeast chromosome III. Proc Natl Acad Sci USA. 1997;94:5213–5218. PMID:9144217.
- Borde V, Robine N, Lin W, et al. Histone H3 lysine 4 trimethylation marks meiotic recombination initiation sites. EMBO J. 2009;28:99–111. PMID:19078966.
- Acquaviva L, Szekvolgyi L, Dichtl B, et al. The COMPASS subunit Spp1 links histone methylation to initiation of meiotic recombination. Science. 2013;339:215–218. PMID:23160953.
- Sommermeyer V, Beneut C, Chaplais E, et al. Spp1, a member of the Set1 Complex, promotes meiotic DSB formation in promoters by tethering histone H3K4 methylation sites to chromosome axes. Mol Cell. 2013;49:43–54. PMID:23246437.
- Parvanov ED, Tian H, Billings T, et al. PRDM9 interactions with other proteins provide a link between recombination hotspots and the chromosomal axis in meiosis. Mol Biol Cell. 2017;28:488–499. PMID:27932493.
- Imai Y, Baudat F, Taillepierre M, et al. The PRDM9 KRAB domain is required for meiosis and involved in protein interactions. Chromosoma. 2017;126:681–695. PMID:28527011.
- Zhu X, Keeney S. High-resolution global analysis of the influences of Bas1 and Ino4 transcription factors on meiotic DNA break distributions in saccharomyces cerevisiae. Genetics. 2015;201:525–542. PMID:26245832.
- Cartagena-Lirola H, Guerini I, Viscardi V, et al. Budding yeast Sae2 is an in vivo target of the Mec1 and Tel1 checkpoint kinases during meiosis. Cell Cycle. 2006;5:1549–1559. PMID:16861895.
- Terasawa M, Ogawa T, Tsukamoto Y, et al. Sae2p phosphorylation is crucial for cooperation with Mre11p for resection of DNA double-strand break ends during meiotic recombination in Saccharomyces cerevisiae. Genes Genet Syst. 2008;83:209–217. PMID:18670132.
- Garcia V, Phelps SE, Gray S, et al. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature. 2011;479:241–244. PMID:22002605.
- Neale MJ, Pan J, Keeney S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature. 2005;436:1053–1057. PMID:16107854.
- Cannavo E, Cejka P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature. 2014;514:122–125. PMID:25231868.
- Moreau S, Ferguson JR, Symington LS. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol Cell Biol. 1999;19:556–566. PMID:9858579.
- Keeney S, Giroux CN, Kleckner N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell. 1997;88:375–384. PMID:9039264.
- Mimitou EP, Yamada S, Keeney S. A global view of meiotic double-strand break end resection. Science. 2017;355:40–45. PMID:28059759.
- Joshi N, Brown MS, Bishop DK, et al. Gradual implementation of the meiotic recombination program via checkpoint pathways controlled by global DSB levels. Mol Cell. 2015;57:797–811. PMID:25661491.
- Smagulova F, Brick K, Pu Y, et al. The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes Dev. 2016;30:266–280. PMID:26833728.
- Borde V, Wu TC, Lichten M. Use of a recombination reporter insert to define meiotic recombination domains on chromosome III of Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:4832–4842. PMID:10373533.
- Kauppi L, Barchi M, Lange J, et al. Numerical constraints and feedback control of double-strand breaks in mouse meiosis. Genes Dev. 2013;27:873–886. PMID:23599345.
- Wojtasz L, Daniel K, Roig I, et al. Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal proteins, are depleted from synapsed chromosome axes with the help of TRIP13 AAA-ATPase. PLoS Genet. 2009;5:e1000702. PMID:19851446.
- Lynn A, Soucek R, Borner GV. ZMM proteins during meiosis: crossover artists at work. Chromosome Res. 2007;15:591–605. PMID:17674148.
- Widger A, Mahadevaiah SK, Lange J, et al. ATR is a multifunctional regulator of male mouse meiosis. bioRxiv. 2017. DOI:10.1101/172106
- Pacheco S, Maldonado-Linares A, Marcet-Ortega M, et al. ATR is required to complete meiotic recombination in mice. bioRxiv. 2017. DOI:10.1101/133744
- Royo H, Prosser H, Ruzankina Y, et al. ATR acts stage specifically to regulate multiple aspects of mammalian meiotic silencing. Genes Dev. 2013;27:1484–1494. PMID:23824539.
- Turner JM. Meiotic sex chromosome inactivation. Development. 2007;134:1823–1831. PMID:17329371.
- Yamamoto K, Wang Y, Jiang W, et al. Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice. J Cell Biol. 2012;198:305–313. PMID:22869596.
- Daniel JA, Pellegrini M, Lee BS, et al. Loss of ATM kinase activity leads to embryonic lethality in mice. J Cell Biol. 2012;198:295–304. PMID:22869595.
- Tesse S, Bourbon HM, Debuchy R, et al. Asy2/Mer2: an evolutionarily conserved mediator of meiotic recombination, pairing, and global chromosome compaction. Genes Dev. 2017;31:1880–1893. PMID:29021238.
- Stanzione M, Baumann M, Papanikos F, et al. Meiotic DNA break formation requires the unsynapsed chromosome axis-binding protein IHO1 (CCDC36) in mice. Nat Cell Biol. 2016;18:1208–1220. PMID:27723721.
- Daniel K, Lange J, Hached K, et al. Meiotic homologue alignment and its quality surveillance are controlled by mouse HORMAD1. Nat Cell Biol. 2011;13:599–610. PMID:21478856.
- Kumar R, Bourbon HM, de Massy B. Functional conservation of Mei4 for meiotic DNA double-strand break formation from yeasts to mice. Genes Dev. 2010;24:1266–1280. PMID:20551173.
- Kumar R, Ghyselinck N, Ishiguro K, et al. MEI4 - a central player in the regulation of meiotic DNA double-strand break formation in the mouse. J Cell Sci. 2015;128:1800–1811. PMID:25795304.
- Shin YH, Choi Y, Erdin SU, et al. Hormad1 mutation disrupts synaptonemal complex formation, recombination, and chromosome segregation in mammalian meiosis. PLoS Genet. 2010;6:e1001190. PMID:21079677.
- Niu H, Wan L, Baumgartner B, et al. Partner choice during meiosis is regulated by Hop1-promoted dimerization of Mek1. Mol Biol Cell. 2005;16:5804–5818. PMID:16221890.
- Fukuda T, Pratto F, Schimenti JC, et al. Phosphorylation of chromosome core components may serve as axis marks for the status of chromosomal events during mammalian meiosis. PLoS Genet. 2012;8:e1002485. PMID:22346761.
- Blat Y, Protacio RU, Hunter N, et al. Physical and functional interactions among basic chromosome organizational features govern early steps of meiotic chiasma formation. Cell. 2002;111:791–802. PMID:12526806.
- Panizza S, Mendoza MA, Berlinger M, et al. Spo11-accessory proteins link double-strand break sites to the chromosome axis in early meiotic recombination. Cell. 2011;146:372–383. PMID:21816273.
- Kleckner N. Chiasma formation: chromatin/axis interplay and the role(s) of the synaptonemal complex. Chromosoma. 2006;115:175–194. PMID:16555016.
- Rogakou EP, Pilch DR, Orr AH, et al. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. PMID:9488723.
- Mahadevaiah SK, Turner JM, Baudat F, et al. Recombinational DNA double-strand breaks in mice precede synapsis. Nat Genet. 2001;27:271–276. PMID:11242108.
- Rogakou EP, Boon C, Redon C, et al. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. PMID:10477747.
- Shroff R, Arbel-Eden A, Pilch D, et al. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol. 2004;14:1703–1711. PMID:15458641.
- Fernandez-Capetillo O, Mahadevaiah SK, Celeste A, et al. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell. 2003;4:497–508. PMID:12689589.
- Celeste A, Petersen S, Romanienko PJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. PMID:11934988.
- Lee CS, Lee K, Legube G, et al. Dynamics of yeast histone H2A and H2B phosphorylation in response to a double-strand break. Nat Struct Mol Biol. 2014;21:103–109. PMID:24336221.
- Wang H, Shi LZ, Wong CC, et al. The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet. 2013;9:e1003277. PMID:23468639.
- Jazayeri A, Falck J, Lukas C, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. PMID:16327781.
- Chen CC, Kass EM, Yen WF, et al. ATM loss leads to synthetic lethality in BRCA1 BRCT mutant mice associated with exacerbated defects in homology-directed repair. Proc Natl Acad Sci USA. 2017;114:7665–7670. PMID:28659469.
- Clouaire T, Legube G. DNA double strand break repair pathway choice: a chromatin based decision? Nucleus. 2015;6:107–113. PMID:25675367.
- Medhi D, Goldman AS, Lichten M. Local chromosome context is a major determinant of crossover pathway biochemistry during budding yeast meiosis. Elife. 2016;5. PMID:27855779. DOI:10.7554/eLife.19669
- Serrentino ME, Chaplais E, Sommermeyer V, et al. Differential association of the conserved SUMO ligase Zip3 with meiotic double-strand break sites reveals regional variations in the outcome of meiotic recombination. PLoS Genet. 2013;9:e1003416. PMID:23593021.
- Froenicke L, Anderson LK, Wienberg J, et al. Male mouse recombination maps for each autosome identified by chromosome painting. Am J Hum Genet. 2002;71:1353–1368. PMID:12432495.
- Arora C, Kee K, Maleki S, et al. Antiviral protein Ski8 is a direct partner of Spo11 in meiotic DNA break formation, independent of its cytoplasmic role in RNA metabolism. Mol Cell. 2004;13:549–559. PMID:14992724.
- Kee K, Protacio RU, Arora C, et al. Spatial organization and dynamics of the association of Rec102 and Rec104 with meiotic chromosomes. EMBO J. 2004;23:1815–1824. PMID:15044957.
- Li J, Hooker GW, Roeder GS. Saccharomyces cerevisiae Mer2, Mei4 and Rec114 form a complex required for meiotic double-strand break formation. Genetics. 2006;173:1969–1981. PMID:16783010.
- Maleki S, Neale MJ, Arora C, et al. Interactions between Mei4, Rec114, and other proteins required for meiotic DNA double-strand break formation in Saccharomyces cerevisiae. Chromosoma. 2007;116:471–486. PMID:17558514.
- Steiner S, Kohli J, Ludin K. Functional interactions among members of the meiotic initiation complex in fission yeast. Curr Genet. 2010;56:237–249. PMID:20364342.
- Miyoshi T, Ito M, Kugou K, et al. A central coupler for recombination initiation linking chromosome architecture to S phase checkpoint. Mol Cell. 2012;47:722–733. PMID:22841486.