
ABSTRACT
Cladribine (2CdA), a synthetic purine analog interfering with DNA synthesis, is a medication used to treat hairy cell leukemia (HCL) and B-cell chronic lymphocytic leukemia. Entinostat, a selective class I histone deacetylase (HDAC) inhibitor, shows antitumor activity in various human cancers, including hematological malignancies. The therapeutic potential of cladribine and entinostat against multiple myeloma (MM) remains unclear. Here we investigate the combinatorial effects of cladribine and entinostat within the range of their clinical achievable concentrations on MM cells. While either agent alone inhibited MM cell proliferation in a dose-dependent manner, their combinations synergistically induced anti-proliferative/anti-survival effects on all MM cell lines (RPMI8226, U266, and MM1.R) tested. Further studies showed that the combinations of cladribine and entinostat as compared to either agent alone more potently induced mitotic catastrophe in the MM cells, and resulted in a marked increase of the cells at G1 phase associated with decrease of Cyclin D1 and E2F-1 expression and upregulation of p21waf−1. Apoptotic ELISA and western blot analyses revealed that the combinations of cladribine and entinostat exerted a much more profound activity to induce apoptosis and DNA damage response, evidenced by enhanced phosphorylation of histone H2A.X and the DNA repair enzymes Chk1 and Chk2. Collectively, our data demonstrate that the combinations of cladribine and entinostat exhibit potent activity to induce anti-proliferative/anti-survival effects on MM cells via induction of cell cycle G1 arrest, apoptosis, and DNA damage response. Regimens consisting of cladribine and/or entinostat may offer a new treatment option for patients with MM.
Abbreviations: MM, multiple myeloma; HCL, hairy cell leukemia; HDAC, histone deacetylase; Ab, antibody; mAb, monoclonal Ab; FBS, fetal bovine serum; CI, combination index; PAGE, polyacrylamide gel electrophoresis; ELISA, enzyme-linked immunosorbent assay; PARP, poly(ADP-ribose) polymerase; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,inner salt
KEYWORDS:
Introduction
Multiple myeloma (MM) is a malignant hematological disease characterized by proliferation of clonal plasma cells mainly in the bone marrow. Although recent progress in understanding the biology of MM and developing novel agents and strategies, the prognosis of majority of MM patients remains poor, and resistance to chemotherapy frequently occurs [Citation1–Citation3]. Recent studies have focused on exploring the combinations of multiple agents and their mechanisms in regulation of aberrant signaling pathways [Citation4], with a hope to identify novel strategies overcoming chemotherapy resistance for the treatment of MM.
Cladribine (2-chloro-2ʹ-deoxyadenosine, 2CdA) is designed as a purine nucleoside analogue to inhibit adenosine deaminase in B cell lymphocytes. It has been tested to be safe (or tolerated) [Citation5] and effective, either by single use or in combination with other cytotoxic agents, in the treatment of several lymphocyte leukemia [Citation6–Citation10]. Cladribine functions as an inhibitor of DNA synthesis to block DNA damage repair, leading to accumulation of DNA strand breaks [Citation11]. It is believed that cladribine induces caspase-dependent apoptosis via mitochondrial and death receptor pathways [Citation6]. Recent studies indicate that cladribine in combination with other agents exerts synergistic cytotoxicity in several lymphoma cell lines [Citation12–Citation15]. We also showed that cladribine either alone or combined with a STAT3 inhibitor was able to induce growth inhibition, apoptosis, and DNA damage response in MM cells [Citation16,Citation17], suggesting that addition of cladribine into therapeutic regimens may be an effective strategy against MM.
Histone deacetylase (HDAC) was discovered to play a pivotal role in several biological processes by regulating eukaryotic gene transcription [Citation18]. It influences cell survival, proliferation, angiogenesis, inflammation and immunity [Citation19,Citation20] in a reversible manner of epigenetic regulation [Citation21]. As a pathogen, overexpression of the HDAC isoforms, especially class I HDACs, correlates with malignant tumorigenesis [Citation22] and predicts a worse outcome [Citation23]. Thus, HDACs are considered to be a trademark of human cancers [Citation24]. In order to achieve anticancer activity, HDAC inhibitors are designed as small molecules [Citation25] to target different HDACs [Citation26]. Several clinical trials have shown that HDAC inhibitors can inhibit expression of oncogenes [Citation27] to induce powerful anticancer effects via growth inhibition, differentiation, and induction of apoptosis and anti-metastasis and anti-angiogenesis activity [Citation25,Citation28,Citation29]. Mechanistic studies reveal that HDAC inhibitors interfere with DNA repair, cell cycle checkpoints, and genomic integrity in lymphoma [Citation30]. Although several HDAC inhibitors have been proved to be effective in breast cancer, lung cancer, and hematologic malignancies [Citation31], the precise mechanism of action of each HDAC inhibitor is still unclear and varies widely from each other [Citation19]. Entinostat (or SNDX-275/MS-275) is a selective class I HDAC inhibitor. In clinic testing, entinostat shows benefit in estrogen receptor-positive breast cancer [Citation32], non-small cell lung cancer [Citation33], melanoma [Citation34], and some T cell lymphoma [Citation35–Citation38]. Its combination with chemotherapy demonstrates enhanced efficacy in B cell lymphoma [Citation37,Citation39–Citation41]. In the current study, we have attempted to seek the combinatorial effects of cladribine and entinostat on cell proliferation and survival in MM cells, and explore the underlying mechanism that cladribine combined with entinostat serves as a novel therapeutic strategy against MM.
Results
Cladribine in combination with entinostat synergistically induces growth inhibition and mitotic catastrophe in MM cells
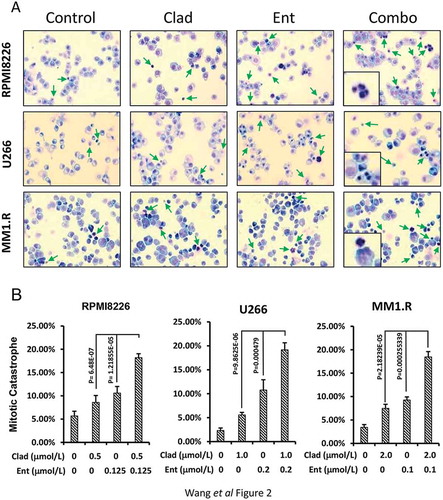
To determine the combinatorial effects of cladribine and entinostat on cell proliferation and survival in MM cells, we first examined the inhibitory effects of either cladribine or entinostat alone on RPMI8226, U266 and MM1.R cells. Pharmacokinetic analysis shows that when given as a 2-hour bolus at 0.14 mg/kg, cladribine’s mean peak plasma concentration reaches 198 nmol/L and the intracellular concentration of cladribine is several hundred-fold higher than that of cladribine in plasma [Citation42]. The clinically achievable range of entinostat in the plasma of cancer patients is less than 1 μmol/L [Citation43]. Thus, the doses of cladribine and entinostat used in the current report have been kept within the range of low micromolar levels with clinical relevance. Cell growth MTS assays showed that cladribine or entinostat significantly inhibited proliferation of RPMI8226 ( left) and U266 ( left) cells in a dose-dependent manner. MM1.R cells were less sensitive to the treatment of either cladribine or entinostat ( left). We next investigated whether the combinations of cladribine and entinostat would further inhibit proliferation of the MM cells. With a fixed ratio of each treatment group, the combinations of cladribine and entinostat as compared to either agent alone exhibited a much more potent activity to induce anti-proliferative/anti-survival effects in all three MM cell lines. These data suggested that cladribine in combination with entinostat possibly exerted a synergistic effect on growth inhibition in MM cells. Indeed, analysis of the combination index (CI) according to Chou-Talalay equation showed synergy between cladribine and entinostat in the concentrations we used for all three cell lines ( right columns). Furthermore, we performed Giemsa staining assays to test whether the combination treatment might also promote mitotic catastrophe in MM cells. By cell slides staining and morphologic observations, it appeared that single agent treatment slightly increased the mitotic catastrophe, whereas the combinations of cladribine and entinostat caused a profound increase in mitotic catastrophe in MM cells (). After calculating all the aberrant cells with mitotic catastrophe, we transferred the data into bar graphs for quantification comparison (). Statistical analysis revealed that cladribine combined with entinostat more significantly induced mitotic catastrophe in MM cells. Collectively, our data indicate that the combinations of cladribine and entinostat synergistically induce anti-proliferative/anti-survival effects on MM cells.
Figure 1. Combination of cladribine and entinostat significantly induces growth inhibition of MM cells, and is synergistic over a wide range of concentrations. Human MM cells were plated onto 96-well plates with fresh RPMI1640 medium (2.5% FBS) or same medium containing indicated concentrations of cladribine (Clad) or entinostat (Ent) or their combinations (Combo) with a fixed ratio for 48 hrs. The percentages of surviving cells as compared to controls, defined as 100% survival, were determined by reduction of MTS. Data shows the representative of three independent experiments. Bars, SD. P values vs single agent treatments. The combination index (CI) curves were calculated using Calcusyn software according to the Chou–Talalay equation. (a) RPMI8226, (b) U266, (c) MM1.R.
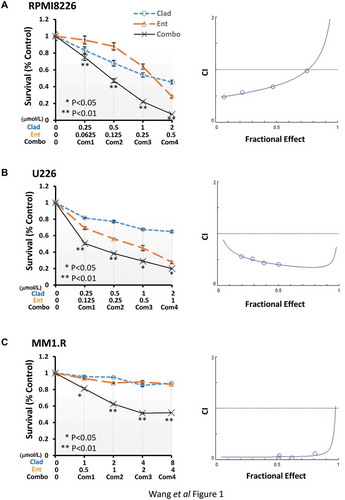
Figure 2. Combination of cladribine and entinostat causes a profound mitotic catastrophe in MM cells. (a) MM cells were cultured with RPMI1640 (2.5% FBS) in the absence or presence of entinostat (Ent), cladribine (Clad) alone or their combinations for 48 hrs. Cells were collected and subjected to cytospin onto cell slides followed by Giemsa staining and examination under a light microscope (×200 magnification). Arrows indicate the cells with mitotic catastrophe. (b) Cells that showed atypical mitotic figures, multi-nucleation, atypical chromosome clusters, and/or apoptosis were counted against normal cells, and reported in percentage. Data shows the representative of three independent experiments. Bars, SD.
The combinations of cladribine and entinostat profoundly elicit cell cycle G1 arrest in MM cells
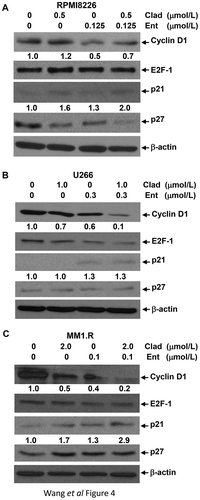
To determine the mechanisms of cladribine/entinostat-mediated inhibitory effects on MM cells, we first performed flow cytometry assays to study whether the combinations of cladribine and entinostat would impair cell cycle progression. When RPMI8226 cells were treated with either cladribine (Clad, 0.5 μmol/L) or entinostat (Ent, 0.125 μmol/L) alone, as compared to control, we did not observe significant changes of the percentage of cells at G1, S, or G2/M phase. However, the combinations of cladribine and entinostat dramatically increased the cells at G1 phase accompanied by a significant reduction of S phase ()). Similar results were also obtained in U266 and MM1.R cells untreated or treated with cladribine and/or entinostat (Supplementary Fig. S1). In general, the treatment of cladribine and/or entinostat had little effect on G2/M phase in all three MM cell lines. According to the flow cytometry results, the percentage of cells at G1, S, or G2/M phase for each cell line was further presented as bar graphs for comparison (). Thus, cladribine in combinations with entinostat significantly accumulated MM cells at G1 phase of the cell cycle, likely via blockade of the G1 phase cells entering S phase. Further studies with western blot analyses were then carried out to examine the molecular markers responsible for G1-S checkpoint of the cell cycle. We found that treatment of RPMI8226 (), U266 (), and MM1.R () cells with both cladribine and entinostat as compared to either agent alone resulted in a striking decrease in expression of Cyclin D1, a critical positive regulator of G1-S checkpoint, and a clear upregulation of p21waf−1, an inhibitor of CDK2 that promotes G1-S transition. Cladribine and/or entinostat had little effect on expression of E2F-1, another positive regulator of G1-S checkpoint, in RPMI8226 and MM1.R cells ()), whereas the combinations of cladribine and entinostat more potently than either agent alone downregulated E2F-1 in U266 cells (). Unlike p21waf−1, the expression of another CDK inhibitor p27kip−1 was not significantly induced by cladribine and/or entinostat in MM cells (). Taken together, our studies conclude that cladribine in combination with entinostat profoundly elicits G1 arrest in MM cells, correlated with alteration of the expression levels of both positive (Cyclin D1) and negative (p21waf−1) regulators of G1-S transition. Since entinostat is a class I HDAC inhibitor, we wondered if its combination with cladribine could potently induce Histone H3 acetylation, which might correlated with their effects on Cyclin D1 and p21waf−1. We found that treatment of the MM cells with entinostat alone, as expected, resulted in a striking increase in Histone H3 acetylation (Ac-Histone H3) (Supplementary Fig. S2). In U266 cells, combination of entinostat and cladribine showed additional increase of Ac-Histone H3 than either agent alone, which was correlated with the changes of Cyclin D1 and p21waf−1 we observed (). However, we found that the combo treatment even decreased Ac-Histone H3 as compared to entinostat alone in RPMI8226 and MM1.R cells. These data suggest that the acetylation status of Histone H3, in general, might not be a major indicator of the synergistic effect we observed in MM cells.
Figure 3. Entinostat and cladribine block cell cycle progression. U226 cells and MM1.R were cultured with RPMI1640 (2.5% FBS) in the absence or presence of entinostat (Ent), cladribine (Clad) alone or the combinations of entinostat and cladribine for 48 hrs. (a) Cells were harvested and subjected to flow cytometric analysis of cell cycle distribution. The percentages of RPMI8226 cells in each cell cycle phase were presented in the table underneath. (b) The bar graph reflects the percentage of cells in G1, S, or G2/M phase of the cell cycle for all three MM cell lines (RPMI8226, U266, and MM1.R). Data shows the representative of three independent experiments.
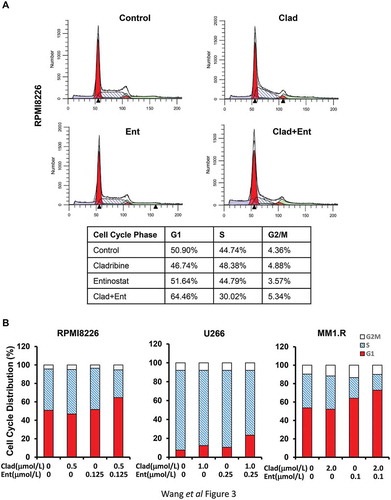
Figure 4. The combinations of cladribine and entinostat markedly alter expression of several key molecular markers critical for G1-S transition. MM cells were cultured with RPMI1640 (2.5% FBS) in the absence or presence of entinostat (Ent), cladribine (Clad) alone or their combinations for 48 hrs. Cells were collected and subjected to western blot analyses with specific antibody directed against Cyclin D1, E2F-1, p21waf−1, p27kip−1, or β-actin. The densitometry analyses of p21waf-1 and Cyclin D1 signals are shown underneath, and the arbitrary numbers indicate the intensities of each cell line relative to controls, defined as 1.0. (a) RPMI8226; (b) U226; (c) MM1.R.
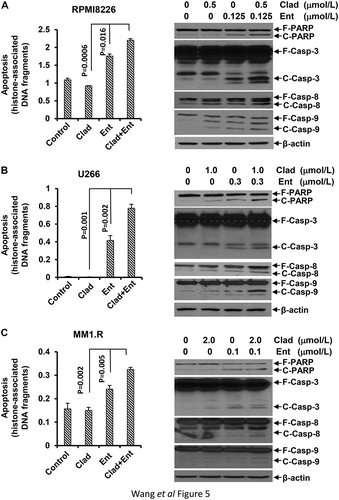
Combinations of cladribine and entinostat significantly induce apoptosis in MM cells
To further explore the combinatorial effects of cladribine and entinostat on MM cells, we next considered whether cladribine and/or entinostat would trigger apoptosis in MM cells. A specific apoptotic ELISA showed that cladribine at the concentrations we used could not induce apoptosis, whereas entinostat alone clearly promoted MM cells undergoing apoptosis. Importantly, the combinations of cladribine and entinostat exhibited a much more potent activity than entinostat alone to trigger apoptosis in all three MM cell lines ( left columns). Consistently, western blot assays revealed that the combinations of cladribine and entinostat more profoundly than entinostat alone induced PARP cleavage, a hallmark of apoptosis ( right columns). Additional studies showed that cladribine in combination with entinostat strongly activated caspases in MM cells, evidenced by increased cleaved caspase-3, caspase-8, and caspase-9 (). These data suggest that the combinations of cladribine and entinostat significantly induce apoptosis in MM cells via caspase-dependent signaling pathways.
Figure 5. Combination of cladribine and entinostat significantly promotes MM cells undergoing apoptosis. MM cells were cultured with RPMI1640 (2.5% FBS) in the absence or presence of either entinostat (Ent) or cladribine (Clad) alone, or their combinations for 48 hrs. Cells were collected and subjected to apoptotic ELISA or western blot analyses with specific antibody directed against PARP (F-PARP, full length PARP; C-PARP, cleaved PARP), caspase-3 (F-Casp-3, full length caspase-3; C-Casp-3, cleaved caspase-3), caspase-8 (F-Casp-8, full length caspase-8; C-Casp-8, cleaved caspase-8), caspase-9 (F-Casp-9, full length caspase-9; C-Casp-9, cleaved caspase-9), or β-actin. Bars, SD. (a) RPMI8226; (b) U226; (c) MM1.R.
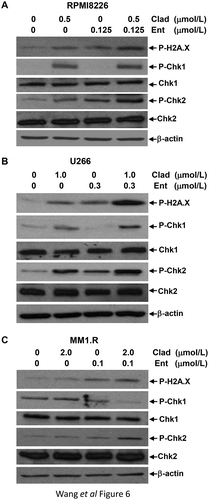
Cladribine and/or entinostat enhances DNA damage response in MM cells
To define the molecular changes of mitotic catastrophe-initiated by cladribine and/or entinostat (), we surveyed expression of the crucial markers for DNA damage response. In MM cells treated with cladribine and/or entinostat for 48 hrs, we found that the combinations of cladribine and entinostat more potently than either cladribine or entinostat alone increased the levels of phosphorylated H2A.X (P-H2A.X), a hallmark of DNA damage response and Chk2 (P-Chk2) (). Cladribine and/or entinostat displayed distinct effects on the phosphorylation levels of Chk1 (P-Chk1) in the MM cell lines. While cladribine alone increased P-Chk1 in all three cell lines, entinostat alone clearly decreased P-Chk1 levels in U266 and MM1.R cells (). However, the combinations of cladribine and entinostat did not further increase the levels of P-Chk1 in RPMI8226 cells (), slightly increased P-Chk1 in U266 cells (), and dramatically reduced P-Chk1 levels in MM1.R cells (). Thus, our data suggest that the combinations of cladribine and entinostat significantly enhance DNA damage response in MM cells accompanied by increased levels of P-H2A.X and P-Chk2.
Figure 6. The combinations of cladribine and entinostat potently influence expression of the molecular markers involving in DNA damage response. MM cells were cultured with RPMI1640 (2.5% FBS) in the absence or presence of entinostat (Ent), cladribine (Clad) alone or their combinations for 48 hrs. Cells were collected and subjected to western blot analyses with specific antibody directed against P-H2A.X, P-Chk1, Chk1, P-Chk2, Chk2, or β-actin. (a) RPMI8226; (b) U226; (c) MM1.R.
Discussion
Identifying novel therapeutic strategy against multiple myeloma (MM) has been a challenge. Purine nucleoside analogues are antimetabolites that inhibit lymphocyte proliferation by interfering DNA synthesis. As a specific one, cladribine was designed to treat hairy cell leukemia (HCL); and it has been proved to be safe and effective in some B cell originated leukemia [Citation44,Citation45]. However, its activity in MM remains elusive. HDAC inhibitors exert potent anti-tumor activity in a variety of human cancers, including lymphomas. Although the underlying mechanisms are not clear, HDAC inhibitors are wildly used in combination with chemotherapeutic agents to enhance anticancer activity [Citation46–Citation49]. As a selective class I HDAC inhibitor, entinostat has shown promising efficacy in the treatment of B cell lymphomas [Citation39,Citation40,Citation50]. Our previous studies also revealed that entinostat in combination with melphalan or bendamustine synergistically induced growth inhibition and apoptosis in MM cells [Citation51,Citation52]. Thus, the synergistic enhancement made entinostat as a potential agent in combination with other anticancer drugs for the treatment of MM. In the current report, we provide experimental evidence showing that cladribine is capable of inhibiting proliferation of MM cells. Moreover, the combinations of cladribine and entinostat exhibit a synergistic anti-MM activity.
In clinic, single drug treatment is hard to reach a satisfying efficacy for cancer patients. It is expected to have a better outcome by practicing multi-drug combination in the treatment of MM. Combined with proteasome inhibitors, some HDAC inhibitors, including panobinostat and vorinostat have been shown synergistic effects in growth inhibition of MM [Citation53,Citation54]. It is also reported that HDAC inhibitors combined with traditional chemotherapies synergistically induced apoptosis in MM cells. In most of these studies, the investigators emphasized on growth inhibition or pro-apoptosis regulation separately. Our data presented here revealed remarkable changes in cell cycle progression, DNA damage response, and apoptosis regulation simultaneously in MM cells upon combinatorial treatment of cladribine and entinostat, supporting the notion that combinations of the two agents exerted a synergistic activity to induce anti-proliferative/anti-survival effects on MM cells. Our studies also suggest that cross-talks among the multiple signaling pathways – cell cycle G1 arrest, DNA damage response, and caspase-dependent apoptosis – may exist. Further investigations are warranted to test this hypothesis.
In general, our studies on the molecular changes were in the agreement with the flow cytometry analysis showing cell cycle G1 arrest and the mitotic catastrophe revealed by Giemsa staining, yet we had two in-consistent observations of the molecular markers in the signaling pathways. First, while we found an upregulation of p27kip−1 expression in MM1.R cells upon treatment with cladribine and/or entinostat, the expression of p27kip−1 in RPMI8226 cells was dramatically decreased by the combinatorial treatment (). By screening gene type in the MM cells, we discovered that MM1.R and RPMI8226 cells contained distinct gene phenotype of p53, which could serve as the upstream signaling protein of p27kip−1 [Citation55,Citation56]. Thus, we speculated that the mutant p53 gene expression might cause the different response of p27kip−1 in the two MM cell lines. Additionally, we also found a reduction of P-Chk1 levels in MM1.R cells, which was very different from that of U266 and RPMI8226 cells (). Nonetheless, a striking induction of P-H2A.X, the hallmark of DNA damage response and a profound mitotic catastrophe were observed in all three MM cell lines by the combinatorial treatment. To the best of our knowledge, there is currently no studies to explain the discordant expression of P-Chk1 and P-H2A.X in MM1.R cells, but we cannot exclude the possible involvement of dexamethasone resistance and/or p53 gene mutation.
Based on the pharmacokinetic analysis, the concentrations of both cladribine and entinostat we used in this study have been kept in low levels – within their clinically achievable ranges [Citation42,Citation43]. Entinostat could cause strong inhibition towards HDAC1 and HDAC3 with IC50 for 0.51 μmol/L and 1.7 μmol/L, respectively. It was also tested in patients with lymphoma with healthy volunteers as comparison, and the results of continuous treatment showed that entinostat functioned significant and high in selective to lymphoma than normal leukocytes, with LC50 = 0.32 μmol/L in lymphoma [Citation57]. Additionally, the peak plasma concentration of entinostat has been calculated to be 0.34 μmol/L in clinical trials of MM patients [Citation52]. The concentrations of entinostat we used in the current report were much lower than that in those publications, and our CI analyses demonstrated that entinostat exhibited synergistic effects within such a low dose when combined with cladribine in MM cells. Taken together, our studies make entinostat a promising therapeutic agent for further evaluations in animal experiments and even clinical trials for patients with MM.
In summary, we demonstrate that the combinations of cladribine and entinostat exert a synergistic enhancement in growth inhibition by inducing cell cycle G1 arrest, DNA damage response, and caspase-dependent apoptosis in MM cells. This combination approach may be added into the treatment regimens for effective management of MM patients.
Materials and methods
Reagents and antibodies
Cladribine (Sigma Co., St. Louis, MO) and entinostat (LC Laboratories, Inc., Woburn, MA) were dissolved in dimethyl sulfoxide (DMSO) to make a stock solution at 250 mmol/L and 200 mmol/L, respectively. The stock solutions were stored at −20°C.
The sources of antibodies for western blot assays were as follows: caspase-3 rabbit mAb (8G10), caspase-8 (1C12) mouse mAb, caspase-9 (Asp353) rabbit mAb, PARP rabbit mAb, P-Histone H2A.X (Ser139) rabbit antibody, Acetyl-Histone H3 (Lys9), Histone H3, P-CHK1 (Ser345) (133D3) rabbit mAb, CHK1 rabbit antibody, P-CHK2 (Thr68) rabbit polyclonal antibody, CHK2 rabbit polyclonal antibody and p21Waf1/Cip1 (12D1) rabbit mAb (Cell Signaling Technology, Inc., Beverly, MA); Cyclin D1 rabbit mAb, E2F-1 mouse mAb (KH95), p27 (F-8) mouse mAb (Santa Cruz Biotechnology Inc., Santa Cruz, CA); β-actin mouse mAb (clone AC-75) (Sigma Co.). All other reagents were purchased from Sigma Co. unless otherwise specified.
Cells and cell culture
Human MM cell lines RPMI8226 and U266 were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Human MM cell line MM1.R was kindly provided by Dr. Steven Rosen (Robert H. Lurie Comprehensive Cancer Center, Northwestern University, Chicago, IL). All cell lines were maintained in RPMI1640 cell culture medium supplemented with 10% fetal bovine serum (FBS) at a 37°C humidified atmosphere containing 95% air and 5% CO2 and were split twice a week.
Cell proliferation assays
The CellTiter96TM AQ non-radioactive cell proliferation kit (Promega Corp., Madison, WI) was used to evaluate cell viability as we previously described [Citation17,Citation51,Citation52]. In brief, cells were plated on 96-well plates with 0.1 ml medium containing 2.5% FBS as control, or 0.1 ml of the same medium with either cladribine or entinostat alone, or their combinations, and incubated for 48 hrs in a cell culture incubator. After reading all wells at 490 nM with a microplate reader, the percentages of surviving cells from each group relative to controls, defined as 100% survival, were determined by reduction of MTS.
Morphologic evaluation of mitotic catastrophe
MM cells were cultured with RPMI1640 (2.5% FBS) in the absence or presence of entinostat (Ent), cladribine (Clad) alone or their combinations for 48 hrs. Cultured MM cells were harvested and suspended with FACS buffer (PBS + 0.5% RPMI1640 medium). A total of 150 ul each cell suspension were loaded into the bottom of SHANDON EZ Double Cytofunnel for cytospin (500 rpm, 5 min at room temperature). After the slides were air dried on bench top, they were incubated in KWIK DIFF Solution #1, 2, 3 sequentially. The slides were then washed with ddH2O 5–10 times till the slides were clear. The air-dried slides were mounted with Cytoseal 60 and examined under a light microscope. All slides were read by two independent pathologists who were blinded on each slide set regarding the treatment group. Cells whose nuclei showed abnormal mitotic figures, including chromatin condensation and DNA fragmentation were counted against normal cells and reported in percentage.
Flow cytometric analysis of cell cycle
Flow cytometric analyses were performed as described previously [Citation17,Citation51,Citation52] to define the cell cycle distribution for treated and untreated cells. In brief, cells grown in 100-mm culture dishes were harvested and fixed with 70% ethanol. Cells were then stained for total DNA content with a solution containing 50 μg/ml propidium iodide and 100 μg/ml RNase I in PBS for 30 min at 37°C. Cell cycle distribution was analyzed at the Flow Cytometry Core Facility of University of Colorado Cancer Center with a Gallios flow cytometer (Beckman Coulter Gallios, Beckman Coulter Life Sciences, Indianapolis, IN).
Quantification of apoptosis
An apoptosis ELISA kit (Roche Diagnostics Corp., Indianapolis, IN) was used to quantitatively measure cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes) as previously reported [Citation17,Citation51,Citation52].
Western blot analysis
Protein expression levels were measured as previously described [Citation17,Citation51,Citation52]. In brief, cells were lysed in a buffer containing 50 mM Tris, pH 7.4, 50 mM NaCl, 0.5% NP-40, 50 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 25 μg/ml leupeptin, and 25 μg/ml aprotinin. The protein concentrations of total cell lysates were measured by the Coomassie Plus protein assay reagent (Pierce Chemical Co., Rockford, IL). Equal amounts of cell lysates were boiled in Laemmli SDS-sample buffer, resolved by SDS-PAGE, and western blot analysis with specific antibodies as described in the figure legends.
Statistical analysis
Statistical analyses of the experimental data were performed using a two-sided Student’s t test. Significance was set at a P < 0.05. Combination index (CI) and evaluation of synergy vs. antagonism between cladribine and entinostat were performed using the Calcusyn software (Biosoft, Ferguson, MO), which was designed based on Chou-Talalay method [Citation58,Citation59]. CI values less than, equal to and more than 1 represent synergistic, additive and antagonistic effects, respectively.
Supplemental Material
Download Zip (44.7 KB)Acknowledgments
The authors are grateful to Ms. Lisa Litzenberger (Department of Pathology, University of Colorado Anschutz Medical Campus) for her excellent art preparation. This work was supported in part by the National Institute of Health (NIH) under Grant R01CA201011 (to BL) and the National Natural Science Foundation of China under Grant 81472763 (to BL). This work was also supported in part by the University of Colorado Cancer Center’s Flow Cytometry Shared Resource funded by the NIH/NCI grant P30CA046934.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary data
Supplementary data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–2520.
- Madan S, Dispenzieri A, Lacy MQ, et al. Clinical features and treatment response of light chain (AL) amyloidosis diagnosed in patients with previous diagnosis of multiple myeloma. Mayo Clin Proc. 2010;85:232–238.
- Laubach J, Garderet L, Mahindra A, et al. Management of relapsed multiple myeloma: recommendations of the International Myeloma Working Group. Leukemia. 2016;30:1005–1017.
- Ocio EM, Richardson PG, Rajkumar SV, et al. New drugs and novel mechanisms of action in multiple myeloma in 2013: a report from the International Myeloma Working Group (IMWG). Leukemia. 2014;28:525–542.
- Leist TP, Weissert R. Cladribine: mode of action and implications for treatment of multiple sclerosis. Clin Neuropharmacol. 2011;34:28–35.
- Rogalinska M, Blonski JZ, Hanausek M, et al. 2-Chlorodeoxyadenosine alone and in combination with cyclophosphamide and mitoxantrone induce apoptosis in B chronic lymphocytic leukemia cells in vivo. Cancer Detect Prev. 2004;28:433–442.
- Inwards DJ, Fishkin PA, Hillman DW, et al. Long-term results of the treatment of patients with mantle cell lymphoma with cladribine (2-CDA) alone (95-80-53) or 2-CDA and rituximab (N0189) in the North Central Cancer Treatment Group. Cancer. 2008;113:108–116.
- Tobinai K, Watanabe T, Tanimoto K, et al. Phase I/II and pharmacokinetic study of cladribine with 2-h infusion in Japanese patients with relapsed indolent B-cell lymphoma mostly pretreated with rituximab. Cancer Sci. 2009;100:1344–1350.
- Inaba H, Stewart CF, Crews KR, et al. Combination of cladribine plus topotecan for recurrent or refractory pediatric acute myeloid leukemia. Cancer. 2010;116:98–105.
- Oguri T, Ozasa H, Uemura T, et al. Preclinical rationale for synergistic interaction of pemetrexed and cytotoxic nucleoside analogues. Oncol Lett. 2012;4:571–575.
- Robak T, Robak P. Purine nucleoside analogs in the treatment of rarer chronic lymphoid leukemias. Curr Pharm Des. 2012;18:3373–3388.
- Ji J, Valdez BC, Li Y, et al. Cladribine, gemcitabine, busulfan, and SAHA combination as a potential pretransplant conditioning regimen for lymphomas: A preclinical study. Exp Hematol. 2016;44:458–465.
- Sun XS, Hasanali ZS, Chen A, et al. Suberoylanilide hydroxamic acid (SAHA) and cladribine synergistically induce apoptosis in NK-LGL leukaemia. Br J Haematol. 2015;168:371–383.
- Robak T, Smolewski P, Cebula B, et al. Rituximab combined with cladribine or with cladribine and cyclophosphamide in heavily pretreated patients with indolent lymphoproliferative disorders and mantle cell lymphoma. Cancer. 2006;107:1542–1550.
- Chow KU, Boehrer S, Napieralski S, et al. In AML cell lines Ara-C combined with purine analogues is able to exert synergistic as well as antagonistic effects on proliferation, apoptosis and disruption of mitochondrial membrane potential. Leuk Lymphoma. 2003;44:165–173.
- Ma J, Wang SL, Zhao M, et al. Therapeutic potential of cladribine in combination with STAT3 inhibitor against multiple myeloma. BMC Cancer. 2011;11:255.
- Cai B, Wang SL, Huang JC, et al. Cladribine and bendamustine exhibit inhibitory activity in dexamethasone-sensitive and -resistant multiple myeloma cells. Am J Transl Res. 2013;5:36–46.
- Witt O, Deubzer HE, Milde T, et al. HDAC family: what are the cancer relevant targets? Cancer Lett. 2009;277:8–21.
- Micelli C, Rastelli G. Histone deacetylases: structural determinants of inhibitor selectivity. Drug Discov Today. 2015;20:718–735.
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784.
- Wagner JM, Hackanson B, Lubbert M, et al. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics. 2010;1:117-136.
- Ajm DR, Van Gennip AH, Caron HN, et al. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749.
- Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280:168–176.
- Zhang H, Shang YP, Chen HY, et al. Histone deacetylases function as novel potential therapeutic targets for cancer. Hepatology Res. 2017;47:149–159.
- Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Inv Drug. 2010;19:1049–1066.
- West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–39.
- Cotto M, Cabanillas F, Tirado M, et al. Epigenetic therapy of lymphoma using histone deacetylase inhibitors. Clin Transl Oncol. 2010;12:401–409.
- Tan JH, Cang SD, Ma YH, et al. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J Hematol Oncol. 2010;3:5.
- Mottamal M, Zheng SL, Huang TL, et al. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898–3941.
- Bose P, Dai Y, Grant S. Histone deacetylase inhibitor (HDACI) mechanisms of action: emerging insights. Pharmacol Therapeut. 2014;143:323–336.
- Manal M, Chandrasekar MJN, Priya JG, et al. Inhibitors of histone deacetylase as antitumor agents: A critical review. Bioorg Chem. 2016;67:18–42.
- Yardley DA, Ismail-Khan RR, Melichar B, et al. Double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31:2128-+.
- Juergens RA, Wrangle J, Vendetti FP, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1:598–607.
- Hornig E, Heppt MV, Graf SA, et al. Inhibition of histone deacetylases in melanomaa perspective from bench to bedside. Exp Dermatol. 2016;25:831–838.
- De Souza C, Chatterji BPHDAC. Inhibitors as novel anti-cancer therapeutics. Recent Pat Anti-Canc. 2015;10:145–162.
- Zain J. Role of histone deacetylase inhibitors in the treatment of lymphomas and multiple myeloma. Hematol Oncol Clin N. 2012;26:671-+.
- Jona A, Khaskhely N, Buglio D, et al. The histone deacetylase inhibitor entinostat (SNDX-275) induces apoptosis in Hodgkin lymphoma cells and synergizes with Bcl-2 family inhibitors. Exp Hematol. 2011;39:1007–1017.
- Copeland A, Buglio D, Younes A. Histone deacetylase inhibitors in lymphoma. Curr Opin Oncol. 2010;22:431–436.
- Frys S, Simons Z, Hu Q, et al. Entinostat, a novel histone deacetylase inhibitor is active in B-cell lymphoma and enhances the anti-tumour activity of rituximab and chemotherapy agents. Br J Haematol. 2015;169:506–519.
- Simmons JK, Patel J, Michalowski A, et al. TORC1 and class I HDAC inhibitors synergize to suppress mature B cell neoplasms. Mol Oncol. 2014;8:261–272.
- Hideshima T, Cottini F, Ohguchi H, et al. Rational combination treatment with histone deacetylase inhibitors and immunomodulatory drugs in multiple myeloma. Blood Cancer J. 2015;5:e312.
- Sigal DS, Miller HJ, Schram ED, et al. Beyond hairy cell: the activity of cladribine in other hematologic malignancies. Blood. 2010;116:2884–2896.
- Gore L, Rothenberg ML, O’Bryant CL, et al. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, MS-275, in patients with refractory solid tumors and lymphomas. Clin Cancer Res. 2008;14:4517–4525.
- Wei Z, Li J, Cheng ZX, et al. A single center experience: rituximab plus cladribine is an effective and safe first-line therapy for unresectable bronchial-associated lymphoid tissue lymphoma. J Thorac Dis. 2017;9:1081–1092.
- Kiesewetter B, Dolak W, Simonitsch-Klupp I, et al. Long-term safety and activity of cladribine in patients with extranodal B-cell marginal zone lymphoma of the mucosa-associated lymphoid tissue (MALT) lymphoma. Hematol Oncol. 2017;35:177–186.
- Badros A, Burger AM, Philip S, et al. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin Cancer Res. 2009;15:5250–5257.
- Galli M, Salmoiraghi S, Golay J, et al. A phase II multiple dose clinical trial of histone deacetylase inhibitor ITF2357 in patients with relapsed or progressive multiple myeloma. Ann Hematol. 2010;89:185–190.
- Gimsing P, Hansen M, Knudsen LM, et al. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur J Haematol. 2008;81:170–176.
- Wolf JL, Siegel D, Goldschmidt H, et al. Phase II trial of the pan-deacetylase inhibitor panobinostat as a single agent in advanced relapsed/refractory multiple myeloma. Leuk Lymphoma. 2012;53:1820–1823.
- Apuri S, Sokol L. An overview of investigational Histone deacetylase inhibitors (HDACis) for the treatment of non-Hodgkin’s lymphoma. Expert Opin Inv Drug. 2016;25:687–696.
- Lee CK, Wang S, Huang X, et al. HDAC inhibition synergistically enhances alkylator-induced DNA damage responses and apoptosis in multiple myeloma cells. Cancer Lett. 2010;296:233–240.
- Cai B, Lyu H, Huang J, et al. Combination of bendamustine and entinostat synergistically inhibits proliferation of multiple myeloma cells via induction of apoptosis and DNA damage response. Cancer Lett. 2013;335:343–350.
- Santo L, Hideshima T, Kung AL, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119:2579–2589.
- Garnock-Jones KP. Panobinostat: first global approval. Drugs. 2015;75:695–704.
- Lopez-Iglesias AA, Herrero AB, Chesi M, et al. Preclinical anti-myeloma activity of EDO-S101, a new bendamustine-derived molecule with added HDACi activity, through potent DNA damage induction and impairment of DNA repair. J Hematol Oncol. 2017;10:127.
- Paino T, Sarasquete ME, Paiva B, et al. Phenotypic, genomic and functional characterization reveals no differences between CD138(++) and CD138(low) subpopulations in multiple myeloma cell lines. PLoS One. 2014;9.
- Lucas DM, Davis ME, Parthun MR, et al. The histone deacetylase inhibitor MS-275 induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia cells. Leukemia. 2004;18:1207–1214.
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55.
- Chou TC, Talaly P. A simple generalized equation for the analysis of multiple inhibitions of Michaelis-Menten kinetic systems. J Biol Chem. 1977;252:6438–6442.