
ABSTRACT
Unraveling the key mechanisms governing the retention versus loss of the cancer stem cell (CSC) state would open new therapeutic avenues to eradicate cancer. Mitochondria are increasingly recognized key drivers in the origin and development of CSC functional traits. We here propose the new term “mitostemness” to designate the mitochondria-dependent signaling functions that, evolutionary rooted in the bacterial origin of mitochondria, regulate the maintenance of CSC self-renewal and resistance to differentiation. Mitostemness traits, namely mitonuclear communication, mitoproteome components, and mitochondrial fission/fusion dynamics, can be therapeutically exploited to target the CSC state. We briefly review the pre-clinical evidence of action of investigational compounds on mitostemness traits and discuss ongoing strategies to accelerate the clinical translation of new mitostemness drugs. The recognition that the bacterial origin of present-day mitochondria can drive decision-making signaling phenomena may open up a new therapeutic dimension against life-threatening CSCs. New therapeutics aimed to target mitochondria not only as biochemical but also as biophysical and morpho-physiological hallmarks of CSC might certainly guide improvements to cancer treatment.
Keywords:
In ancient Rome, the slaves accompanying generals on victory parades whispered the words memento mori [“remember (that you have) to die”] as a reminder of their commander’s mortality, thereby preventing them from being obsessed by excessive pride and self-confidence.
Within a tumor, there are special forms of selfish and egoistic commander cells called cancer stem cells, which are endowed with unique powers including immortality, drug refractoriness, and tumor/metastasis-initiating potential.
We here propose that mitochondria would provide new means to pharmacologically whisper memento mori! to selfish and life-threatening CSC.
Table 1. Pharmacological targeting of mitochondrial dynamics reduces cancer stemness.
Cancer stem cells (CSCs) are a particularly aggressive type of malignant cell endowed with perpetuating properties such as self-renewal, refractoriness to therapy, and tumor/metastasis-initiating capacity. They account for the majority of clinical phenomena leading to cancer deaths including tumor relapse, treatment failure, and metastatic progression [Citation1–Citation3]. A detailed understanding of the underlying mechanism(s) governing the retention (self-renewal) versus loss (differentiation) of the CSC state would open new avenues to overcome therapeutic resistance and eradicate cancer.
Cellular metabolism is beginning to be recognized as a central controller of the occurrence and function of CSCs [Citation4–Citation9], and we are rapidly amassing knowledge of the metabolic features that are important for the functional traits that define CSCs. Metabolic traits such as bioenergetic/biosynthetic retuning, redox balance, and switching (activation/deactivation) of energy-sensing integrators can no longer be viewed as secondary consequences of the acquisition of cancer stemness, as they appear to elicit the CSC phenotype. In this respect, we recently coined the term metabostemness to refer to the metabolic parameters causally controlling the epigenetic reprogramming that drives and maintains CSC-like cellular states [Citation10–Citation13]. We proposed that some of the aforementioned metabolic traits would operate as pivotal molecular events rendering cells permissive for the epigenetic rewiring necessary for aberrant stemness while promoting refractoriness to differentiation. Notably, such a metabolism-centric regulation of cancer stemness incorporates mitochondria as key organelles in the maintenance of CSC properties.
Most research on the apparent dependence of CSCs on mitochondrial function has focused on the relationship between mitochondrial bioenergetics and the distinguishing metabolic profiles of CSCs. While it remains a matter of debate why and how CSCs present a highly glycolytic or a mitochondrial-dependent primary phenotype in a cancer type- and/or environment-dependent manner [Citation4–Citation8], it is clear that mitochondrial function per se takes central stage in CSC functionality irrespective of the metabolic features of the primary tumor. This mitochondrial dependence of stemness and cell fate specification compels us to reconsider mitochondria as (the) key signaling organelles that can be therapeutically exploited to eliminate the dynamic pool of CSCs within heterogeneous cancer populations.
We here propose the new term “mitostemness” to designate the mitochondria-dependent signaling functions that, anciently rooted in the engulfment of free-living α-proteobacteria (present-day mitochondria) by eukaryotic host cells approximately 1.5 billion years ago [Citation14,Citation15], drive CSC self-renewal and fate specification (). We briefly discuss how the mitostemness traits, namely mitonuclear communication, mitoproteome components, and mitochondrial fission/fusion dynamics, offer new possibilities to directly target the CSC state.
Metabolic symbiosis-related mitonuclear communication
Although the nature and benefit of the endosymbiotic relationship between α-proteobacteria and the host archaeon is still a matter of deliberation [Citation16,Citation17], a metabolic symbiosis would have been originated between the waste products of α-proteobacteria (H2, CO2, and acetate) and their use by the archaeon host (e.g., methanogenesis or biomethanation). As an evolutionary consequence, the signaling function of mitochondria in the first eukaryotic cell likely involved the exploitation of original waste products such as acetate–which is readily converted to acetyl-CoA for protein acetylation–as nuclear-sensed signals of correct mitochondrial functionality (, top). Accordingly, while acetate-derived acetyl-CoA is known to function as a key regulator in the yeast cell cycle via histone acetylation [Citation18], modern metazoans regulate this process via generation of acetyl-CoA from the mitochondrial tricarboxylic acid cycle intermediates acetate and citrate. From an evolutionary perspective, it is not surprising that the biosynthetic role of present-day mitochondria is to provide the most common intermediate metabolites (e.g., acetyl-CoA, NAD+, α-ketoglutarate, succinate, FAD, ATP and S-adenosylmethionine, among others) to generate and modify epigenetic marks in the nucleus (, top). The usage of these metabolites as donor substrates and regulating cofactors of chromatin-modifying enzymes provides a direct link between the metabolic state and epigenetics [Citation19–Citation21]. Correspondingly, alterations in their production would have profound effects on CSC identity and fate decisions by dysregulating nuclear transcriptional programs.
Figure 1. Mitostemness: A new therapeutic dimension in cancer stem cells. The new term mitostemness designates the mitochondria-dependent signaling functions that, anciently rooted in the engulfment of free-living α-proteobacteria by eukaryotic host cells approximately 1.5 billion years ago, now drives CSC self-renewal and fate specification. The mitostemness traits, namely mitonuclear communication [Citation1], mitoproteome components [Citation2], and mitochondrial fission/fusion dynamics [Citation3], could be therapeutically exploited to target the CSC state in tumors.
![Figure 1. Mitostemness: A new therapeutic dimension in cancer stem cells. The new term mitostemness designates the mitochondria-dependent signaling functions that, anciently rooted in the engulfment of free-living α-proteobacteria by eukaryotic host cells approximately 1.5 billion years ago, now drives CSC self-renewal and fate specification. The mitostemness traits, namely mitonuclear communication [Citation1], mitoproteome components [Citation2], and mitochondrial fission/fusion dynamics [Citation3], could be therapeutically exploited to target the CSC state in tumors.](/cms/asset/fb06a697-3517-47e1-89da-8ca6000fe62f/kccy_a_1467679_f0001_oc.jpg)
Figure 2. Mitostemness traits: New therapeutic opportunities against cancer stem cells. The bacterial origin of present-day mitochondria can drive decision-making signaling phenomena such as those governing the retention versus loss of cancer stemness. Accordingly, metabolic symbiosis-related mitonuclear communication (top panels), bacteria-related mitoproteome (middle panels), and ancient host-related mitochondrial fission/fusion dynamics (bottom panels) might introduce new molecular avenues to therapeutically target self-renewal and maintenance of the CSC phenotype. (SAM: S-adenosylmethionine; SAH: S-adenosylhomocysteine; NAD+: nicotinamide adenine dinucleotide; α-KG: alpha-ketoglutarate; FAD: flavin adenine dinucleotide; 2-HG: 2-hydroxyglutarate; Ac: acetylation; Me: methylation; DNMT: DNA methyltransferase; HDAC: histone deacetylase; HDM: histone demethylase; Drp1: dynamin-related protein 1; Mfn: mitofusin).
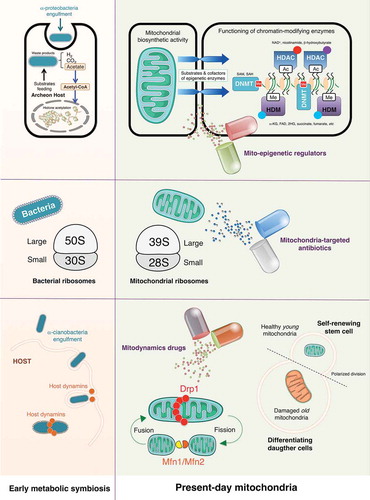
Figure 3. A.) Cell2Sphere™ assays (StemTek Therapeutics) using MDA-MB-436 breast cancer cells were performed as per the manufacturer’s instructions. Graded concentrations of mdivi-1 (M0119, Sigma-Aldrich) or mitochondrial fusion promoter M1 hydrazone (SML0629, Sigma-Aldrich) were added to quintuplicate sets of wells on days 1 and 4 without replenishing the medium. ImageJ (NIH) was used to quantify the size (left; central lines indicate mean values ± SD) and number (right; representative photomicrographs) of 6 day-old mammospheres. Comparisons of means were performed by ANOVA. B.) Monolayers of double-thymidine block synchronized SUM-159 and HMLERshEcad cells were pre-treated with graded concentrations of mdivi-1 for 6 hours, trypsinized and re-plated for mammosphere assays in the absence of mdivi-1. Mammosphere-forming efficiency (MSFE) was calculated as described previously [Citation95]. MSFE of vehicle-alone control cells was normalized to one; three technical replicates per n; n = 3 experimental replicates. (Drp1: dynamin-related protein 1; Mfn: mitofusins; DMSO: dimethyl sulfoxide). The results are presented as the mean (columns) ± SD (bars) of three independent experiments performed in triplicate. Comparisons of means were performed by ANOVA. SUM-159 and HMLERshEcad cells were routinely grown in Ham’s F12 and Clonetics™ MEGM™ (Mammary Epithelial Cell Growth Medium), respectively [Citation95].
![Figure 3. A.) Cell2Sphere™ assays (StemTek Therapeutics) using MDA-MB-436 breast cancer cells were performed as per the manufacturer’s instructions. Graded concentrations of mdivi-1 (M0119, Sigma-Aldrich) or mitochondrial fusion promoter M1 hydrazone (SML0629, Sigma-Aldrich) were added to quintuplicate sets of wells on days 1 and 4 without replenishing the medium. ImageJ (NIH) was used to quantify the size (left; central lines indicate mean values ± SD) and number (right; representative photomicrographs) of 6 day-old mammospheres. Comparisons of means were performed by ANOVA. B.) Monolayers of double-thymidine block synchronized SUM-159 and HMLERshEcad cells were pre-treated with graded concentrations of mdivi-1 for 6 hours, trypsinized and re-plated for mammosphere assays in the absence of mdivi-1. Mammosphere-forming efficiency (MSFE) was calculated as described previously [Citation95]. MSFE of vehicle-alone control cells was normalized to one; three technical replicates per n; n = 3 experimental replicates. (Drp1: dynamin-related protein 1; Mfn: mitofusins; DMSO: dimethyl sulfoxide). The results are presented as the mean (columns) ± SD (bars) of three independent experiments performed in triplicate. Comparisons of means were performed by ANOVA. SUM-159 and HMLERshEcad cells were routinely grown in Ham’s F12 and Clonetics™ MEGM™ (Mammary Epithelial Cell Growth Medium), respectively [Citation95].](/cms/asset/a5a461ff-82e2-4223-ac1e-144ba185c43f/kccy_a_1467679_f0003_oc.jpg)
We recently presented a first-in-class computational model of the causative relationship between the abundance of mitochondrial intermediate metabolites, epigenetic landscapes, and cell state transitions [Citation22]. This mathematical approach predicted that regulating the stochastic translation of mitochondrial-driven metabolic inputs into resilient versus plastic cell states via epigenetics could promote the establishment of epigenomes refractory to (CSC-related) loss of cell fate and de-differentiation phenomena. In this context, apparently simple drugs such as the anti-diabetic biguanide metformin, which operates as a metabolo-epigenetic reprogrammer of the molecular conduits linking mitochondrial-driven synthesis of epigenetic metabolites with the structure and functioning of the epigenome [Citation23–Citation26], are now recognized as molecules that selectively target CSCs [Citation27,Citation28]. Metformin has been shown to potently and specifically suppress the undifferentiated stem cell compartment responsible for malignant carcinoma-like growth within tumor masses generated upon injection of induced pluripotent stem cells (iPSCs) into mice [Citation29–Citation30]. At the same time, however, metformin treatment fully preserves the competency of iPSCs to differentiate into derivatives of all three germ layers in vivo [Citation29,Citation30], thereby providing a good example of the control of the tumorigenic fate of CSCs that would be expected from the pharmacological fine-tuning of mitochondria-derived metabolic epigenetic substrates and cofactors [Citation31].
Bacteria-related mitoproteome
Beyond the function of mitochondria in the regulation of epigenetics and mitonuclear communication [Citation32], we have recently learned that the adverse effects of anti-bacterial antibiotics on human cells likely reflect the above-mentioned evolutionary relationship between proteobacteria and eukaryotic mitochondria. Accordingly, whereas clinically relevant doses of bactericidal antibiotics including quinolones, aminoglycosides, and β-lactams have been shown to cause mitochondrial dysfunction in mammalian cells [Citation33,Citation34], mutations that cause human mitochondrial ribosomes to more closely resemble bacterial ribosomes enable stronger interactions with some anti-bacterial antibiotics [Citation35]. Tetracyclines, such as doxycycline, and chloramphenicol were described also as mitochondrial inhibitors more than 50 years ago [Citation36]; recent mechanistic studies have definitively confirmed that both types of antibiotics lead to a state of so-called mitonuclear protein imbalance (i.e., protein synthesis from mitochondrial DNA is not matched by protein synthesis from nuclear DNA) through their capacity to inhibit both bacterial and mitochondrial translation [Citation37]. The ability of the tetracyclines to globally repress mitochondrial protein synthesis, which is accompanied by severely impaired mitochondrial activity, leads to changes in nuclear gene expression [Citation38], further supporting the notion that mitochondrial proteostasis and function ultimately communicate with the mammalian symbiotic partner regarding whether or not the mitochondrial bioenergetic and/or biosynthetic activity status would allow any decision-making activity.
Consistent with the concept that CSCs are critically dependent on mitochondrial biogenesis and function [Citation39,Citation40], several recent preclinical studies have exploited the fact that many FDA-approved antibiotics actually target the mitochondrial proteome (a priori undesirable but mild off-target effect), to clinically repurpose them as CSC-targeted therapies [Citation41–Citation44]. Because of conserved similarities between the 70S bacterial and the 55S mitochondrial ribosomes, the erythromycins (e.g., azithromycin) and chloramphenicol can selectively bind to the 39S large subunit of the mitochondrial ribosome and prevent the translation of mitochondrial proteins mainly related to the mitochondrial OXPHOS complexes. Moreover, the tetracyclines (e.g., doxycycline) and glycylcyclines (e.g., tigecycline) can both bind with high affinity to the 28S small subunit of the mitochondrial ribosome to inhibit mitochondrial protein translation (, middle). All of these compounds have been found to significantly reduce the anchorage-independent survival and propagation of CSC-like cells in mammosphere assays, a widely-used approach to evaluate the potential of cancer cells to exhibit CSC-like tumor-initiating and self-renewal activities [Citation45], even when employed at low, clinically-achievable concentrations [Citation41]. In our hands, breast cancer subtypes enriched for stem cell-like properties exhibit an exacerbated sensitivity to microbicides targeting active sites of the ribosome (e.g., emetine, puromycin, and cycloheximide), inhibitors of ribosome biogenesis (e.g., dactinomycin), and ribotoxic stress agents such as daunorubicin [Citation42]. Other bacterial antibiotics blocking either mitochondrial OXPHOS by targeting Complex II & III of the electron transport chain (e.g., pyrvinium pamoate, atovaquone), or ATP synthesis by targeting mitochondrial ATP-synthase/Complex V (e.g., bedaquiline) [Citation41,Citation43] have also been shown to inhibit propagation and expansion of CSC-like cells in mammosphere assays. A virtual high-throughput screening and computational chemistry approach coupled to phenotypic drug screening has recently identified four classes of a new family of mitochondrial-based antibiotics, termed mitoriboscins, which effectively inhibit the growth of bacteria and pathogenic yeast as well as CSC-like cells in vitro [Citation44].
Ancient host-related mitochondrial fission/fusion dynamics
Faithful to their bacterial origin, mitochondria constantly move, merge, and divide to form extensive tubular networks that ensure a uniform distribution of energy throughout the cell and the delivery of appropriate signals (e.g., stemness) to the correct sub-cellular location. This dynamic network is maintained by two opposing processes–fission and fusion–that are largely under control of large GTP hydrolyzing enzymes of the dynamin superfamily, namely Drp1, and mitofusins (Mfn1/Mfn2), respectively [Citation46–Citation48]. Viewed through an evolutionary lens, endosymbiotic bacteria likely employed the host cell dynamin-related GTPases during the endocytosis-like engulfment through the plasma membrane, and then once again during mitochondrial division, before the emergence of modern-day mitochondria [Citation49,Citation50]. Such co-option of host dynamins could therefore be considered part of the symbiotic relationship in which the mammalian cell endowed the mitochondria with a division apparatus (, bottom).
Whereas mitochondria in healthy cells tend to be elongated and fuse together into filamentous structures, both the initial steps of malignant transformation [Citation51,Citation52] and also self-renewal and resistance to differentiation in some types of stem cells [Citation53] have been associated with high levels of mitochondrial fission activity. In the latter regard, fusion/fission cycles coupled to mitophagy–a quality control process that prevents dysfunctional mitochondria from fusing with healthy mitochondria [Citation54,Citation55]–begin to be recognized as critical, Drp1-related mechanisms through which stem cells asymmetrically segregate their old mitochondria to the differentiating daughter cells, while the self-renewing daughter cell receives only young mitochondria [Citation56]. Crucially, the failure to asymmetrically apportion, in a Drp1-dependent manner, old versus young mitochondria in a single division appears to be sufficient to cause a persistent loss of stemness properties in the progeny cell [Citation56]. A recent study has confirmed that mitochondrial fission appears to drive a distinct mitochondrial profile in brain tumor-initiating CSC-like cells when compared with non-initiating tumor cells [Citation57]. Targeting Drp1 activity with mdivi-1, an inhibitor of mitochondrial division originally described by its ability to impair yeast Dnm1 GTPase activity [Citation58], has been shown to inhibit tumor-forming capacity and stemness-related signaling in brain [Citation57] and breast cancer [Citation59] cell populations (). Because mitochondrial fission might be coupled to the nuclear transcriptional events that drive the acquisition and maintenance of stemness in iPSCs and CSCs [Citation60,Citation61], it is reasonable to suggest that Drp1-driven mitochondrial fission could be a novel target for the therapeutic elimination of CSCs. Importantly, the opposing force of mitofusins-regulated mitochondrial fusion might also impact cancer stemness. If we consider that CSC-related mitochondrial features of iPSCs provide a framework in which the infrastructure and functioning of mitochondria might control the CSC state, it is noteworthy that Drp1 activation and Mfn1/2 ablation both facilitate the acquisition and maintenance of stem cell pluripotency through the restructuring of mitochondrial dynamics [Citation62,Citation63]. Accordingly, compounds such as (E)-4-Chloro-2-(1-(2-(2,4,6-trichlorophenyl) hydrazono) ethyl) phenol (small molecule M1), which are capable of specifically shifting the mitochondrial dynamic balance toward fusion [Citation64] might cause depletion of the tumor-initiating CSC-like subpopulation ().
Mitostemness-targeting drugs as anti-csc therapies: the challenges ahead
Targeting mitochondrial reprogramming as an essential regulator of CSC identity might deactivate a core mechanism of drug resistance and metastatic progression irrespective of cancer mutational landscapes. However, despite the ever-growing evidence for the central involvement of mitochondria in CSC maintenance and fate [Citation53], the clinical translation of such experimental therapies remains an elusive challenge.
A strong body of epidemiological [Citation65–Citation67] and pre-clinical [Citation28] evidence has motivated numerous studies to probe the anti-cancer effects of metformin through clinical trials. However, the majority of ongoing and completed trials have focused on the impact of metformin on various proliferative and apoptotic surrogate markers in cancer cells, and very little clinical effort has been directed to evaluate metformin as a CSC targeting agent for prevention of cancer relapse. Nevertheless, a recently conducted prospective study of metformin in epithelial ovarian cancer noted a significant reduction in the number of CSCs at baseline in metformin-treated tumors [Citation68,Citation69]. Upcoming trials may therefore need to shift focus and explore the ability of metformin to direct stem-like plastic states to re-enter into epigenetic resilience. In this respect, studies ongoing in our laboratory have been designed to develop a new generation of pharmacophoric mimetics of metformin aimed to redirect cellular identity and cell fate transitions via normalization of the aberrant mitonuclear communication existing in CSC states.
The clinical use of either generic, FDA-approved antibiotics with manageable side effects, or new compounds capable of binding to the mitoribosome and inhibiting mitochondrial function, is therapeutically appealing and might significantly reduce cancer cost. Nevertheless, future clinical studies should determine whether these mitoproteome-targeting antibiotics are solely investigational tool-bench compounds to assess the required mitochondria function for CSCs in vitro, or could also be bedside, clinically-valuable adjuvant oncology drugs providing a more effective means to eliminate CSCs when combined with current cancer therapies. Moreover, although ATP production is commonly employed as an end-point screening marker of dysfunctional mitochondria in response to mitochondria-targeted antibiotics, one should acknowledge that mitochondria can maintain their biosynthetic (and therefore their mitonuclear communication) capabilities even in the absence of ATP generation [Citation15]. Noteworthy, the polyether ionophore antibiotic salinomycin, the first anti-CSC molecule identified in a chemical screen designed to discover compounds capable of specifically inhibiting CSC proliferation through the induction of differentiation [Citation70], alters mitochondrial bioenergetic performance at the concentration range used in studies showing selective toxicity for breast CSCs [Citation71,Citation72]. Although salinomycin is apparently being investigated in clinical trials [Citation73], none of them have been reported under the clinicaltrials.gov website [Citation74]. Because machine-learning methods capable of predicting the lifespan–promoting capability of chemicals have identified compounds affecting mitochondria among the top hit compounds [Citation75], such databases of aging-related drugs [Citation76] would be an interesting resource for helping in the discovery of new classes of mitostemness-targeting drugs.
The classical view of regulated mitochondrial dynamics as a crucial requirement for energy production should be broadened to consider changes in mitochondrial morphology as a sufficient means to elicit self-renewal capacity in CSCs. However, the current Drp1- and Mfn-targeting regulators of mitochondrial division have not been tested in patients because of limitations imparted by their expected pharmacological properties or side-effect profiles. Thus, there is an urgent need to develop selective compounds that control mitochondrial dynamics without affecting basal mitochondrial functions such as ATP production or generation of reactive oxygen species (ROS). Indeed, the current assumption that mitodynamics drugs such as mdivi-1 can cause a loss of the functional properties possessed by CSCs should be reconsidered in light of the recently reported finding that mdivi-1 is a poor inhibitor of recombinant Drp1 GTPase activity, but modifies mitochondrial ROS production via reversible inhibition of mitochondrial complex I without affecting mitochondrial elongation [Citation77]. New pharmacological strategies are being developed in our laboratory to design negative allosteric modulators of Drp1 activity based on the key structural features recently described in more potent and specific mdivi-1 derivatives [Citation78]. Such deconstruction of Drp1 druggability aims also to generate new Drp1-targeted small molecules capable of mimicking the chemical behavior of the so-called P110 peptide, which has been shown to selectively prevent the GTPase activity of Drp1 and recruitment to mitochondria via blocking the interaction between Drp1 and one of its receptors (Fis1) at the mitochondrial outer membrane only under pathological conditions [Citation79].
Mitostemness: echoes from the past?
The recognition that the bacterial origin of present-day mitochondria can drive decision-making signaling phenomena such as those governing the retention versus loss of cancer stemness would be viewed as a continuation of the atavism hypothesis of cancer, which states that tumor development and metastasis relies on the functional disruption of genes that sustain multicellularity upon reactivation of primitive transcriptional programs that evolved in the earliest single-celled organisms [Citation80–Citation82]. We acknowledge that an ever-growing number of studies begin to support the notion that the high degree of robustness and plasticity occurring via activation of unicellular networks might confer CSC-like tumor adaptability to drug exposure [Citation83–Citation85]. However, our view merely proposes that the usage of mitochondria-dependent signaling functions evolutionary rooted in the bacterial origin of mitochondria might be considered a primary cause of non-genetic distortions in the manner of Waddingtonian non-CSC versus CSC cellular states (attractors), which might be amenable to broad-spectrum cancer therapies capable of mitochondrially tackling CSC-driven inter- and intra-tumor heterogeneity [Citation9]. Our mitostemness proposal is consistent with a number of descriptions in which the determination of cell fate involves the asymmetric distribution of certain mitochondrial controllers into CSC-like cell entities [Citation84–Citation89]. Indeed, our proposal complements the view that changes in intracellular, intercellular, and extracellular mitochondrial traffic [Citation90–Citation94] appear to ensure the functional endurance of mitochondria in the tumor-initiating and drug-resistant subpopulation of CSC. New therapeutics aimed to target mitochondria not only as biochemical but also as biophysical and morpho-physiological hallmarks of CSC might certainly guide improvements to cancer treatment.
Acknowledgments
This work was supported by grants from the Ministerio de Ciencia e Innovación (Grant SAF2016-80639-P to J. A. Menendez), Plan Nacional de I+D+I, Spain, and the Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR) (Grant 2014 SGR229 to J. A. Menendez). This work was supported also by a grant of the Obra Social La Caixa Foundation on Collaborative Mathematics awarded to the Centre de Recerca Matemàtica. This study was supported also by unrestricted research grants from Roche Pharma (Spain) and Astellas Pharma (Spain) to the Program Against Cancer Therapeutic Resistance (ProCURE, Catalan Institute of Oncology). Elisabet Cuyàs is supported by the Sara Borrell post-doctoral contract (CD15/00033) from the Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria (FIS), Spain. Núria Folguera-Blasco and Tomás Alarcón acknowledge MINECO for funding under grant MTM2015-71509-C2-1-R and AGAUR for funding under grant 2014 SGR1307. The authors would like to thank Dr. Kenneth McCreath for editorial support.
Disclosure statement
Stock ownership: Ángel G. Martin, StemTek Therapeutics (CEO). All other authors have no competing interests to declare.
Additional information
Funding
Related Research Data
References
- Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014;13:497–512.
- Chaffer CL, Weinberg RA. How does multistep tumorigenesis really proceed? Cancer Discov. 2015;5:22–24.
- Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–629.
- Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. 2016;114:1305–1312.
- Menendez JA. Metabolic control of cancer cell stemness: lessons from iPS cells. Cell Cycle. 2015;14:3801–3811.
- Song IS, Jeong JY, Jeong SH, et al. Mitochondria as therapeutic targets for cancer stem cells. World J Stem Cells. 2015;7:418–427.
- Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, et al. Cancer stem cell metabolism. Breast Cancer Res. 2016;18:55.
- Loureiro R, Mesquita KA, Magalhães-Novais S, et al. Mitochondrial biology in cancer stem cells. Semin Cancer Biol. 2017;47:18–28.
- Cuyàs E, Verdura S, Fernández-Arroyo S, et al. Metabolomic mapping of cancer stem cells for reducing and exploiting tumor heterogeneity. Oncotarget. 2017;8:99223–99236.
- Menendez JA, Alarcón T. Metabostemness: a new cancer hallmark. Front Oncol. 2014 Sep;29(4):262.
- Menendez JA, Corominas-Faja B, Cuyàs E, et al. Metabostemness: metaboloepigenetic reprogramming of cancer stem-cell functions. Oncoscience. 2014;1:803–806.
- Menendez JA. The metaboloepigenetic dimension of cancer stem cells: evaluating the market potential for new metabostemness-targeting oncology drugs. Curr Pharm Des. 2015;21:3644–3653.
- Menendez JA, Corominas-Faja B, Cuyàs E, et al. Oncometabolic nuclear reprogramming of cancer stemness. Stem Cell Reports. 2016;6:273–283.
- Chandel NS. Mitochondria as signaling organelles. BMC Biol. 2014;12:34.
- Chandel NS. Evolution of mitochondria as signaling organelles. Cell Metab. 2015;22:204–206.
- Martin W, Müller M. The hydrogen hypothesis for the first eukaryote. Nature. 1998;392:37–41.
- Gray MW. Mitochondrial evolution. Cold Spring Harb Perspect Biol. 2012;4:a011403.
- Shi L, Tu BP. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol. 2015;33:125–131.
- Johnson C, Warmoes MO, Shen X, et al. Epigenetics and cancer metabolism. Cancer Lett. 2015;356:309–314.
- Mentch SJ, Locasale JW. One-carbon metabolism and epigenetics: understanding the specificity. Ann N Y Acad Sci. 2016;1363:91–98.
- Reid MA, Dai Z, Locasale JW. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol. 2017;19:1298–1306.
- Folguera-Blasco N, Cuyàs E, Menendez JA, et al. Epigenetic regulation of cell fate reprogramming in aging and disease: A predictive computational model. PLoS Comput Biol. 2018;14:e1006052.
- Corominas-Faja B, Quirantes-Piné R, Oliveras-Ferraros C, et al. Metabolomic fingerprint reveals that metformin impairs one-carbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging (Albany NY). 2012;4:480–498.
- Janzer A, German NJ, Gonzalez-Herrera KN, et al. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc Natl Acad Sci U S A. 2014;111:10574–10579.
- Cuyàs E, Fernández-Arroyo S, Joven J, et al. Metformin targets histone acetylation in cancer-prone epithelial cells. Cell Cycle. 2016;15:3355–3361.
- Cuyàs E, Fernández-Arroyo S, Verdura S, et al. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene. 2017 Oct 23. DOI:10.1038/onc.2017.367.
- Hirsch HA, Iliopoulos D, Tsichlis PN, et al. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–7511.
- Del Barco S, Vazquez-Martin A, Cufí S, et al. Metformin: multi-faceted protection against cancer. Oncotarget. 2011;2:896–917.
- Vazquez-Martin A, Cufi S, Lopez-Bonet E, et al. Metformin limits the tumourigenicity of iPS cells without affecting their pluripotency. Sci Rep. 2012;2:964.
- Vazquez-Martin A, Vellon L, Quirós PM, et al. Activation of AMP-activated protein kinase (AMPK) provides a metabolic barrier to reprogramming somàtic cells into stem cells. Cell Cycle. 2012;11:974–989.
- Menendez JA, Alarcón T. Senescence-inflammatory regulation of reparative cellular reprogramming in aging and cancer. Front Cell Dev Biol. 2017;5:49.
- Matilainen O, Quirós PM, Auwerx J. Mitochondria and epigenetics - crosstalk in homeostasis and stress. Trends Cell Biol. 2017;27:453–463.
- Kalghatgi Lowes DA, Wallace C, Murphy MP, et al. The mitochondria targeted antioxidant MitoQ protects against fluoroquinolone-induced oxidative stress and mitochondrial membrane damage in human Achilles tendon cells. Free Radic Res. 2009;43:323–328.
- Kalhatgi S, Spina CS, Costello JC, et al. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in mammalian cells. Sci Transl Med. 2013;5:192ra85.
- Gao Z, Chen Y, Guan M-X. Mitochondrial DNA mutations with aminoglycoside induced ototoxicity. J Otol. 2017;12:1–8.
- Clark-Walker GD, Linnane AW. In vivo differentiation of yeast cytoplasmic and mitochondrial protein synthesis with antibiotics. Biochem Biophys Res Commun. 1966;25:8–13.
- Houtkooper RH, Mouchiroud L, Ryu D, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457.
- Moullan N, Mouchiroud L, Wang X, et al. Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep. 2015;10:1681–1691.
- Lamb R, Harrison H, Hulit J, et al. Mitochondria as new therapeutic targets for eradicating cancer stem cells: quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget. 2014;5:11029–11037.
- De Luca A, Fiorillo M, Peiris-Pagès M, et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget. 2015;6:14777–14795.
- Lamb R, Ozsvari B, Lisanti CL, et al. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. 2015;6:4569–4584.
- Cuyàs E, Martin-Castillo B, Corominas-Faja B, et al. Anti-protozoal and anti-bacterial antibiotics that inhibit protein synthesis kill cancer subtypes enriched for stem cell-like properties. Cell Cycle. 2015;14:3527–3532.
- Fiorillo M, Lamb R, Tanowitz HB, et al. Bedaquiline, an FDA-approved antibiotic, inhibits mitochondrial function and potently blocks the proliferative expansion of stem-like cancer cells (CSCs). Aging (Albany NY). 2016;8:1593–1607.
- Ozsvari B, Fiorillo M, Bonuccelli G, et al. Mitoriboscins: mitochondrial-based therapeutics targeting cancer stem cells (CSCs), bacteria and pathogenic yeast. Oncotarget. 2017;8:67457–67472.
- Manuel Iglesias J, Beloqui I, Garcia-Garcia F, et al. Mammosphere formation in breast carcinoma cell lines depends upon expression of E-cadherin. PLoS One. 2013;8:e77281.
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884.
- Archer SL. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236–2251.
- Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. 2014;15:634–646.
- Kuroiwa T, Nishida K, Yoshida Y, et al. Structure, function and evolution of the mitochondrial division apparatus. Biochim Biophys Acta. 2006;1763:510–521.
- van der Bliek AM. A mitochondrial division apparatus takes shape. J Cell Biol. 2000;151:F1–F4.
- Serasinghe MN, Wieder SY, Renault TT, et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell. 2015;57:521–536.
- Kashatus JA, Nascimento A, Myers LJ, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. 2015;57:537–551.
- Chen H, Chan DC. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 2017;26:39–48.
- Guan JL, Simon AK, Prescott M, et al. Autophagy in stem cells. Autophagy. 2013;9:830–849.
- Vazquez-Martin A, Van den Haute C, Cufí S, et al. Mitophagy-driven mitochondrial rejuvenation regulates stem cell fate. Aging (Albany NY). 2016;8:1330–1352.
- Katajisto P, Döhla J, Chaffer CL, et al. Stem cells. Asymmetric Apportioning of Aged Mitochondria between Daughter Cells Is Required for Stemness. Science. 2015;348:340–343.
- Xie Q, Wu Q, Horbinski CM, et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. 2015;18:501–510.
- Cassidy-Stone A, Je C, Ingerman E, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204.
- Peiris-Pagès M, Bonuccelli G, Sotgia F, et al. Mitochondrial fission as a driver of stemness in tumor cells: mDIVI1 inhibits mitochondrial function, cell migration and cancer stem (CSC) signaling. Oncotarget. 2018;9:13254–13275.
- Vazquez-Martin A, Cufi S, Corominas-Faja B, et al. Mitochondrial fusion by pharmacological manipulation impedes somatic cell reprogramming to pluripotency: new insight into the role of mitophagy in cell stemness. Aging (Albany NY). 2012;4:393–401.
- Khacho M, Clark A, Svoboda DS, et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell. 2016;19:232–247.
- Son MY, Choi H, Han YM, et al. Unveiling the critical role of REX1 in the regulation of human stem cell pluripotency. Stem Cells. 2013;31:2374–2387.
- Son MJ, Kwon Y, Son MY, et al. Mitofusins deficiency elicits mitochondrial metabolic reprogramming to pluripotency. Cell Death Differ. 2015;22:1957–1969.
- Wang D, Wang J, Bonamy GM, et al. A small molecule promotes mitochondrial fusion in mammalian cells. Angew Chem Int Ed Engl. 2012;51:9302–9305.
- Libby G, Donnelly LA, Donnan PT, et al. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–1625.
- Bodmer M, Meier C, Krähenbühl S, et al. Long-term metformin use is associated with decreased risk of breast cancer. Diabetes Care. 2010;33:1304–1308.
- Gong Z, Aragaki AK, Chlebowski RT, et al. Diabetes, metformin and incidence of and death from invasive cancer in postmenopausal women: results from the women’s health initiative. Int J Cancer. 2016;138:1915–1927.
- Shank JJ, Yang K, Ghannam J, et al. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecol Oncol. 2012;127:390–397.
- Buckanovich RJ, Brown J, Shank J, et al. A phase II clinical trial of metformin as a cancer stem cell targeting agent in stage IIc/III/IV ovarian, fallopian tube, and primary peritoneal cancer. J Clin Oncol. 2017;35 (suppl; abstr 5556).
- Gupta PB, Onder TT, Jiang G, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659.
- Mitani M, Yamanishi T, Miyazaki Y, et al. Salinomycin effects on mitochondrial ion translocation and respiration. Antimicrob Agents Chemother. 1976;9:655–660.
- Managò A, Leanza L, Carraretto L, et al. Early effects of the antineoplastic agent salinomycin on mitochondrial function. Cell Death Dis. 2015;6:e1930.
- Naujokat C, Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. J Biomed Biotechnol. 2012;2012:950658.
- Marcucci F, Rumio C, Lefoulon F. Anti-cancer stem-like cell compounds in clinical development - an overview and critical appraisal. Front Oncol. 2016;6:115.
- Barardo DG, Newby D, Thornton D, et al. Machine learning for predicting lifespan-extending chemical compounds. Aging (Albany NY). 2017;9:1721–1737.
- Barardo D, Thornton D, Thoppil H, et al. The DrugAge database of aging-related drugs. Aging Cell. 2017;16:594–597.
- Bordt EA, Clerc P, Roelofs BA, et al. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex i inhibitor that modulates reactive oxygen species. Dev Cell. 2017;40:583–594.
- Numadate A, Mita Y, Matsumoto Y, et al. Development of 2-thioxoquinazoline-4-one derivatives as dual and selective inhibitors of dynamin-related protein 1 (Drp1) and puromycin-sensitive aminopeptidase (PSA). Chem Pharm Bull (Tokyo). 2014;62:979–988.
- Qi X, Qvit N, Su YC, et al. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. 2013;126:789–802.
- Davies PCW, Lineweaver CH. Cancer tumors as Metazoa 1.0: tapping genes of ancient ancestors. Phys Biol. 2011;8:1–7.
- Lineweaver CH, Davies PC, Vincent MD. Targeting cancer’s weaknesses (not its strengths): therapeutic strategies suggested by the atavistic model. Bioessays. 2014;36:827–835.
- Vincent M. Cancer: a de-repression of a default survival program common to all cells?: a life-history perspective on the nature of cancer. Bioessays. 2012;34:72–82.
- As T, Rb P, At P, et al. Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors. Proc Natl Acad Sci U S A. 2017;114:6406–6411.
- Bussey KJ, Cisneros LH, Lineweaver CH, et al. Ancestral gene regulatory networks drive cancer. Proc Natl Acad Sci U S A. 2017;114:6160–6162.
- Trigos AS, Pearson RB, Papenfuss AT, et al. How the evolution of multicellularity set the stage for cancer. Br J Cancer. 2018;118:145–152.
- Erenpreisa J, Cragg MS. Cancer: a matter of life cycle? Cell Biol Int. 2007;31:1507–1510.
- Erenpreisa J, Cragg MS. Three steps to the immortality of cancer cells: senescence, polyploidy and self-renewal. Cancer Cell Int. 2013;13:92.
- Erenpreisa J, Salmina K, Huna A, et al. The “virgin birth”, polyploidy, and the origin of cancer. Oncoscience. 2014;2:3–14.
- Erenpreisa J, Salmiņa K, Belyayev A, et al. Survival at the brink: chromatin autophagy of tumour cells in response to genotoxic challenge. In: Editor Hayat MA. Autophagy: cancer, other pathologies, inflammation, immunity, infection, and aging. 2017. Vol. 12. Elsevier Chapter 12, p. 275–294.
- Díaz-Carballo D, Gustmann S, Jastrow H, et al. Atypical cell populations associated with acquired resistance to cytostatics and cancer stem cell features: the role of mitochondria in nuclear encapsulation. DNA Cell Biol. 2014;33:749–774.
- Diaz-Carballo D, Saka S, Klein J, et al. A distinct oncogenerative multinucleated cancer cell serves as a source of stemness and tumor heterogeneity. Cancer Res. 2018 Feb 12. pii: canres.1861.2017. . Epub ahead of print. DOI:10.1158/0008-5472.CAN-17-1861
- Tan AS, Baty JW, Dong LF, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015;21:81–94.
- Berridge MV, Dong L, Neuzil J. Mitochondrial DNA in tumor initiation, progression, and metastasis: role of horizontal mtDNA transfer. Cancer Res. 2015;75:3203–3208.
- Berridge MV, Neuzil J. The mobility of mitochondria: intercellular trafficking in health and disease. Clin Exp Pharmacol Physiol. 2017;44(Suppl 1):15–20.
- Corominas-Faja B, Cuyàs E, Lozano-Sánchez J, et al. Extra-virgin olive oil contains a metabolo-epigenetic inhibitor of cancer stem cells. Carcinogenesis. 2018 Feb 14. Epub ahead of print. DOI:10.1093/carcin/bgy023