
ABSTRACT
Truncating de novo mutations in ADNP have been identified in patients with the Helsmoortel-Van der Aa syndrome. However correlations between the distinct mutations and their impact on the protein have not been studied before. Here we report the effect of mutations in ADNP by examining the expression and subcellular localization of GFP-tagged mutant transcripts in transfected HEK293T cells. ADNP encloses a bipartite nuclear localization signal and we found mutations therein to stall the mutant protein within the cytoplasm. Using immunocytochemistry, we could demonstrate colocalization of wild-type ADNP with heterochromatin. We found mutations presenting a pattern based on the genetic position. For certain mutant proteins enrichment at pericentromeric heterochromatin seems partially lost. Finally, N-terminal truncated ADNP mutants are routed towards cytosolic proteasomal degradation and rescued with the proteasome inhibitor MG132. Our results suggest a correlation between the position of the mutations across the protein, its stability and subcellular localization.
Introduction
Brain functioning is affected in patients with various neurodevelopmental disorders (NDDs), including intellectual disabilities (ID), autism spectrum disorders (ASD) and epilepsy [Citation1]. According to the DSM-V, ASDs are characterized by a combination of limited social skills together with stereotyped restricted and repetitive behavior and interests [Citation2]. ASDs have seen a rising incidence over the past decades, with a prevalence currently estimated at 1–1.2% of the adult population [Citation3,Citation4]. However, due to the high heterogeneity of the clinical features and etiologies, the biological basis of ASD still remains poorly understood. While a role of genetics in the occurrence of autism has been well-established, the exact cause remains to be identified in many forms of autism and a mechanistic model remains elusive for the majority of identified ASD genes.
We have identified de novo mutations in the Activity-Dependent Neuroprotective Protein (ADNP) gene as a frequent cause of syndromic autism in a cohort of patients with ASD and ID [Citation5,Citation6]. Since then, we have collected almost 100 patients with mutations in ADNP, classified as Helsmoortel-Van der Aa syndrome (OMIM 615,873) [Citation7]. The ADNP gene maps to chr20q13.13 and contains five exons of which the last three are translated. It encodes a transcription factor that is described to interact with certain components of the SWI/SNF chromatin remodeling complex, including BRG1, BAF250a and BAF170 [Citation8]. The gene was first described as vasoactive intestinal peptide (VIP)-responsive. VIP is a neuroprotective peptide that is active during embryonic development, protecting damaged nerve cells from cell death by inducing glia-derived, survival-promoting substances. Being VIP-responsive, ADNP presents a similar neuroprotective function with increased expression after VIP treatment [Citation9,Citation10]. The neuroprotective function of ADNP is attributed to an octapeptide, NAP (NAPVSIPQ = Asn–Ala–Pro–Val–Ser–Ile–Pro–Gln) [Citation9,Citation11–Citation13]. Predicted from its sequence, the ADNP protein contains 9 zinc fingers and a DNA-binding homeobox domain, features indicating a role in embryonic development. It also comprises a HP1α-binding PxVxL motif [Citation14,Citation15]. In line with the predicted roles of ADNP, literature indicates the presence of a nuclear localization signal (NLS; )), suggesting a role for ADNP in the nucleus [Citation16].
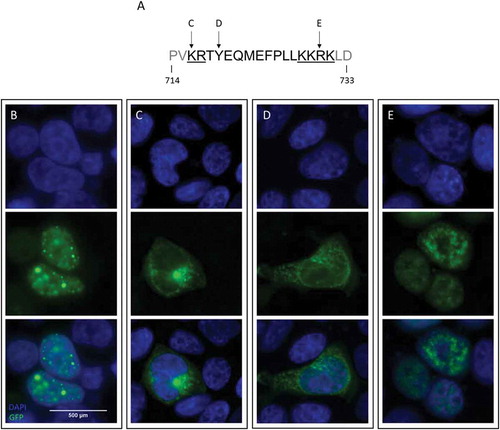
Figure 1. Nuclear import of ADNP is impaired upon mutagenesis of its NLS sequence.
Cell localization studies in HEK293T cells using an expression vector of human ADNP, carrying a N-terminal eGFP-tag. The GFP signal (green) is indicative of ADNP expression and localization. The cell nucleus was visualized using DAPI (blue). (a) The modeled mutations positioned on the ADNP bipartite NLS sequence. The NLS basic amino acid regions are underlined and separated by the central spacer sequence. (b) Expression of wild-type ADNP is restricted to the nucleus. (c) Mutation p.Lys716Glu, affecting the first basic region of the ADNP NLS, causes the mutant ADNP protein to stall in the cytoplasm. (d) Introduction of the common p.Tyr719* mutation presents cytoplasmic mislocalization like the previous mutation. (e) p.Arg730* transcripts are imported in the nucleus.
Essentially all of our patients carry nonsense or frameshift mutations throughout the protein, introducing a premature stop in the transcript. Most of the de novo premature stop mutations are located in the last exon of ADNP, which encodes 94% of the protein. As we demonstrated previously, Helsmoortel-Van der Aa patients still generate a mutated form of the mRNA [Citation5], consistent with the predicted escape from nonsense-mediated decay (NMD) in the absence of a downstream exon-exon junction [Citation17,Citation18]. We therefore hypothesized the syndrome to be the consequence of a dominant negative mechanism, where mutant ADNP competes with the wild-type protein to interact with its targets (Helsmoortel et al., 2014).
The current study aims to examine the effect of mutations in various domains of ADNP, reflecting our patient cohort, on the subcellular level in order to increase insights into genotype-phenotype correlations of the disorder.
Results
Firstly, we validated the subcellular distribution of the wild-type (WT) hADNP-eGFP construct in HEK293T cells. As indicated by the fluorescent signal of the eGFP-tag (), we found WT ADNP to locate in defined DAPI-positive foci in the nucleus. This nuclear localization is in line with the presence of a bipartite NLS from residue 714 to 733, as the cNLS mapper tool predicts. The ADNP NLS consists of the classic bipartite NLS of 2 basic amino acid regions, separated by a central spacer of 9 to 12 residues ()).
Preservation of the NLS is necessary to shuttle ADNP to the nucleus
To validate the function of the predicted NLS, we selectively mutated both basic regions and the spacer region, introducing variants predicted by cNLS mapper to disrupt the NLS function. Side-directed mutagenesis of Lysine716, in the first basic region, into a Glutamate caused the mutant protein to stall within the cytoplasm ()). Likewise, the p.Tyr719* mutation in the spacer sequence restricted the mutant ADNP exclusively to the cytoplasm ()). In contrast, the p.Arg730* mutated protein, with a nonsense mutation in the second basic region of the NLS, was still imported into the nucleus ()). Interestingly, the p.Tyr719* and p.Arg730* nonsense mutations make up the most frequent Helsmoortel-Van der Aa syndrome-related mutations. Both introduce a premature stop in the protein at slightly different positions within the NLS sequence, but apparently display a distinct subcellular expression pattern.
Mutations in ADNP present in a pattern of mislocalization
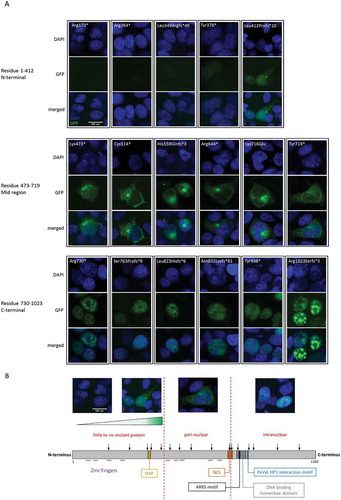
In addition to variants affecting the NLS, we wanted to evaluate the effect on subcellular localization of variants throughout the remainder of the protein. shows the position of all the modeled mutations and their effect on ADNP expression and subcellular localization. Depending on the position of the mutation, our results defined three groups of mutant ADNP with distinct cellular presentation. Mutations at the C-terminus, from residue 730 on, presented nuclear import of the mutant ADNP forms in accordance with the presence of an intact NLS. Cytoplasmic localization was observed for a group of mutations spanning residue 473 up to 719, likely due to absence of a functional NLS sequence. Interestingly, mutations grouped at the N-terminus of the protein, before the fifth zinc finger (up to residue 447), presented little to no GFP-signal. We observed a gradient in signal ranging from no detectable signal for the shortest transcripts to a fluorescent signal of undefined subcellular location for longer N-terminal mutants. ()). Thus, mutant ADNP present a specific pattern of expression and (mis)localization according to the position of the mutation across the protein domains ()).
Figure 2. Mutations in ADNP present in a pattern of protein expression and localization.
(a) HEK293T cells were transiently transfected with a vector expressing mutated forms of GFP-tagged hADNP and analyzed using ICC. Cell nuclei are stained with DAPI (blue), ADNP is visualized using GFP (green). Mutations at the N-terminus at residue 1–412 are shown to negatively impact ADNP expression. Mutations in residue 473–719, the mid region of the protein, cause mislocalization of the mutant ADNP transcripts in the cytoplasm. C-terminal mutations from residue 730 on leave the first basic region of the NLS unaffected and are imported into the nucleus. (b) Schematic representation of the modeled mutations positioned across the ADNP protein. Based on the ICC results, the mutations can be categorized into 3 groups according to their effect on ADNP expression and localization. The first group is made up by the most N-terminal mutations where the GFP-signal for ADNP expression could be observed in a gradient of absent to undefined ADNP expression. Mutations in the mid region span zinc finger 5 up to the middle of the NLS sequence and make up the second group of mutations, where ADNP is mislocalized in the cytoplasm. Nuclear localization of ADNP is seen for mutations downstream of the second part of the NLS, the third group of mutations.
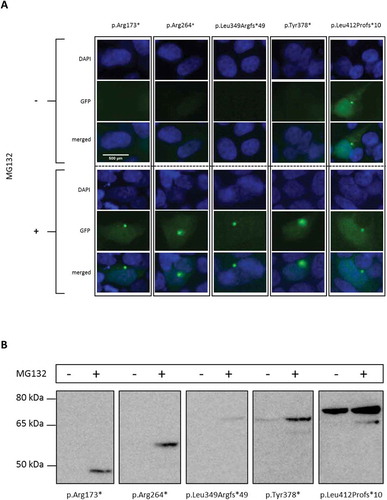
N-terminal mutations shift truncated ADNP towards proteasomal degradation
Since we previously demonstrated escape of mRNA from NMD [Citation5], the absence of protein due to N-terminal mutations was remarkable. To assess whether truncated ADNP transcripts of N-terminal mutations fail translation or are degraded after synthesis, we inhibited proteasomal degradation with MG132 in HEK293T cells. In accordance with the ICC experiments ()), constructs p.Arg173*, p.Arg264* and p.Leu349Argfs*49 were undetectable by western blot assessment, while p.Tyr378* and p.Leu412Profs*10 constructs showed lower expression in absence of a proteasome inhibitor ()). After treatment with proteasome inhibitor MG132 all these mutant forms of ADNP could be detected in confined GFP-positive spots in the cytoplasm () and by western blot analysis (). The western blot data confirm a gradual reduction of ADNP protein levels as mutations approach the N-terminus (as concluded in , ). Treatment with MG132 rescued the lower expression levels of these shorter proteins from proteasomal degradation. Overall, these results indicate that N-terminal mutations shift the mutated protein towards cytosolic proteasomal degradation.
Figure 3. Degradation of N-terminal mutated ADNP.
HEK293T cells were transiently transfected with expression vector encoding GFP-tagged hADNP and where indicated treated with the proteasome inhibitor MG132 (200 nM). (a) ICC of HEK293T cells transfected with a vector expressing distinct N-terminal mutant hADNP transcripts. ADNP is shown in green (GFP), the cell nucleus in blue (DAPI). A gradient of no to little GFP-signal, indicative for ADNP expression is observed in untreated conditions. After treatment of the transfected cells with proteasome inhibitor MG132, ADNP expression is visible in confined spots in the cytoplasm. (b) Western blot analysis of the N-terminal truncated ADNP proteins. Cells were lysed in RIPA buffer and analyzed by immunoblotting for ADNP with anti-GFP antibody. Total protein normalization by means of Ponceau S was used for cell loading control. The predicted molecular mass for the transcripts are the following: p.Arg173* at 47 kDa, pArg264* at 57 kDa, p.Leu349Argfs*49 at 72 kDa, p.Tyr378* at 69 kDa and p.Leu412Profs*10 at 74 kDa. Only after treatment with MG132 are transcripts detectable or increased in expression. One representative experiment out of 2 is shown here.
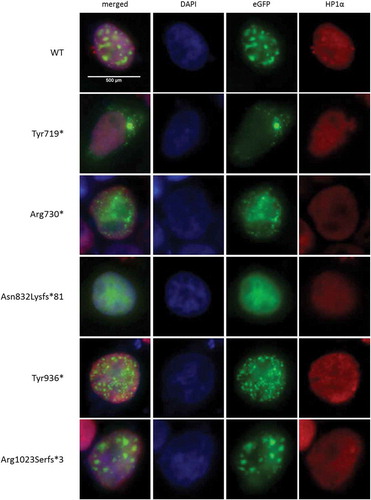
(Mis)localization of mutant ADNP in the nucleus
ADNP has previously been described as a binding partner of HP1α and HP1β [Citation19]. Since we initially observed overlap of the WT GFP-ADNP signal with DAPI-positive foci, representing mainly pericentromeric heterochromatin (), we opted to perform immunolabeling of the well-established heterochromatin marker HP1α to verify co-localization of ADNP with heterochromatin. In line with available data, we observed the GFP-signal for WT ADNP expression co-localized with the heterochromatin marker and enriched at pericentromeric heterochromatin foci (). Modeling the Tyr936* and Arg1023Serfs*3 mutants including an intact NLS sequence and PxVxL HP1 interaction motif (residues 819–823) showed results comparable to the WT construct. The protein lacking the NLS (p.Tyr719*) was used as a negative control to envision distinction between the cytoplasmic enriched ADNP signal and the heterochromatin signals from DAPI and HP1α stainings. Interestingly, the mutant proteins p.Arg730* and p.Asn832Lysfs*81 showed less prounounced punctuate distribution as seen for the WT construct ( and ). While the p.Arg730* mutation renders a protein lacking the HP1α interaction motif, we still observed co-localization with HP1α, however specific enrichment at pericentromeric heterochromatin seems partially lost. Strikingly, the p.Asn832Lysfs*81 mutant, although containing the PxVxL motif, presents a distribution similar to the p.Arg730* mutated protein.
Figure 4. Mutant ADNP (mis)localization in the nucleus.
HEK293T cells were transiently transfected with GFP-tagged hADNP. Cells were fixed and co-localization of ADNP with heterochromatin was analyzed by immunolabeling for HP1α, a well-established heterochromatin marker. Nucleic DNA with percentromeric heterochromatin foci was stained with DAPI (blue), ADNP was visualized using the GFP-tag (green) inherent to the construct and HP1α (red) was stained using anti-HP1α antibody. Co-localization of ADNP with HP1α can be observed as a yellow signal in the composite image. The signal for WT ADNP is clearly enriched at pericentromeric heterochromatin. Similar data are obtained for ADNP mutants Tyr936* and Arg1023Serfs*3. The cytoplasm-stalling p.Tyr719* mutation was used as a negative control. While the protein lacking the HP1α interaction motif (p.Arg730*) and the mutant p.Asn832Lysfs*81 still co-localize with HP1, specific enrichment at pericentromeric heterochromatin seems partially lost.
Discussion
In this study we have characterized mutations in ADNP. This is to our knowledge the first study of the mutational effect upon ADNP expression and subcellular localization.
Here, we modeled the effect of mutations across ADNP on expression and localization of the protein within the cell. We were able to classifiy the mutations into three groups of distinct subcellular localization depending on the position in the gene. We found a nuclear localization for products of the most C-terminal mutations, similar to the WT protein. In contrast, we observed mislocalization in the cytoplasm due to mutations in the central region of the protein, truncating the conserved bipartite NLS sequence. Even more striking is the observation of absent or undefined ADNP expression as mutations approach the N-terminus. With proteasomal arrest experiments, we found that these shortest mutant ADNP transcripts are degraded shortly after their synthesis, likely by ubiquitination and targeting to the proteasome for clearance [Citation20]. The reduction in ADNP expression and stability seems proportional to the length of the truncated protein, but nonetheless have all these short-lived transcripts lost their nuclear localization and function.
Thus, there might be multiple mechanisms by which ADNP mutations result in disease. Our previous data demonstrating escape of mutant ADNP mRNA from NMD in our patient cohort, at that time point including primarily C-terminal mutations, led us to speculate a competition between wild-type ADNP and its mutant counterpart. The observation of intact shuttling of C-terminal mutated ADNP to the nucleus is compatible with this hypothesis. Mutations affecting the central part of ADNP show mislocalization in the cytoplasm. It is unlikely that these mutant proteins in the cytoplasm compete with the wild-type protein in the nucleus, but it cannot be excluded that these mutated forms of ADNP have gained novel interactions within their newly acquired subcellular location (dominant negative effect). The N-terminal mutations most likely result in de facto haploinsufficiency.
An independent report on the expression of exogenous ADNP in transfected HEK293T cells also described a nuclear localization [Citation8]. Like in our study, these authors showed co-localization of wild-type ADNP signals with defined DAPI-positive foci in the nucleus. Nuclear DAPI foci have been described to correlate with heterochromatin densities [Citation21]. Overlap between ADNP and such foci is in line with a suggested function in chromatin remodeling. Moreover, ADNP has been identified as a binding partner of HP1 [Citation19]. Yet, the mentioned study as well as our own was carried out in HEK293T cells and it remains to be determined how mutations affect subcellular distribution in other cell types. ADNP was found to be differentially localized in either the nucleus and/or the cytoplasm, depending on the cell type studied. In studies on other non-neuronal cell types, including mouse heart, placenta and P19 differentiated cardiovascular-like cells, ADNP was constraint to the nucleus [Citation22]. However, in rat neurons, ADNP was predominantly allocated to in the cytoplasm, with occasional weak nuclear signals [Citation23]. Immunolabeling of P19 neuronal- and glia-differentiated cells showed ADNP localization in the nucleus, cytoplasm and neuronal extensions [Citation22]. Experiments on rat astrocytes presented discordant observations. A study by Furman et al. (2004) indicated both nuclear as well as cytoplasmic expression of ADNP, with higher concentrations found in the nucleus [Citation24]. In contrast, very weak to no cytoplasmic signal in rat astrocytes was detected by Gennet et al. (2008). Additionally, gene expression maps show broad differential expression of ADNP with levels depending on the tissue and cell type (Expression Atlas, EMBL-EBI, available from: https://www.ebi.ac.uk).
Similar to the variability in ADNP expression and localization by tissue and cell type, are ADNP levels dependent on the developmental stage. ADNP levels are highest during embryonic development and decrease with age progression [Citation10,Citation25–Citation27]. Throughout development and aging, the focus of ADNP synthesis shifts from forebrain to mid- and hindbrain (Allen Mouse Brain Atlas 2004, Allen Developing Mouse Brain Atlas 2008, Allen Human Brain Atlas 2010, BrainSpan Atlas of the Developing Human Brain 2010, available through the Allen Brain Atlas Data Portal: http://www.brain-map.org/). ADNP levels fluctuate in the placenta and decidua during pregnancy and in the female arcuate nucleus of the hypothalamus within the estrus cycle [Citation25,Citation28]. Differential expression in these key regulators of the reproductive system further emphasize the importance of timing of ADNP expression. Furthermore is ADNP regulation subjected to sexual dimorphism, with differences in several brain region between male and the estrus cycle of the female [Citation28–Citation30]. The combination of these findings on ADNP expression support a developmental role and point towards a multimodal aspect, where the protein’s cellular function depends on the cell type. The results of our study should be viewed in light of this information. Ascribing an underlying mechanistic model to specific ADNP mutation should rather be considered per cell type throughout development.
In summary, we classified mutations in ADNP into three groups of distinct subcellular presentation, depending on their position. These results bring insights into the molecular basis underlying the Helsmoortel-Van Der Aa syndrome. However, given the developmental, cell-specific and sex-dependent role of ADNP, most mutations seem to display a combination of mutational effects and thus ascribing one general pathogenic mechanism (either loss-of-function or dominant negative) per mutational group would be incomplete. Further studies on the subcellular function of mutant ADNP transcripts might explain the individual patients’ symptoms and praise hope for targeted treatment.
Materials and methods
Plasmid constructs
The pEZ-M29 expression vector encoding WT hADNP fused to a N-terminal eGFP-tag was purchased from GeneCopoeia (Rockville, EX-T0346-M29, http://www.genecopoeia.com/product/search/detail.php?prt=1&cid=&key=T0346). Mutations were inserted into this wild-type vector by PCR mutagenesis using Q5 Site-Directed Mutagenesis Kit (New England Biolabs, E0554S, https://www.neb.com/products/e0554-q5-site-directed-mutagenesis-kit#Product%20Information) following the manufacturer’s protocol. Mutagenesis primers were designed using the NEBaseChanger tool (http://nebasechanger.neb.com/) and ordered through IDT (https://eu.idtdna.com). The modeled mutations with used primer sequences are listed in Supplementary Table 1. DNA was purified from E. coli using the PureYield Plasmid Midiprep System (Promega, A2492, https://be.promega.com/products/dna-purification-quantitation/plasmid-purification/pureyield-plasmid-midiprep-system/?catNum=A2492) according to the manual. Mutations were verified using Sanger sequencing following standard protocols.
Cell culture and cellular assays
HEK293T cells were cultured in DMEM, supplemented with 10% FBS and 1% penicillin/streptomycin in a humidified CO2 incubator at 37°C. Transient transfections in the HEK293T cells were performed with Lipofectamine 3000 transfection reagent (Invitrogen, L3000008, https://www.thermofisher.com/order/catalog/product/L3000008) in accordance with the manufacturer’s instructions. Transfection efficiency was about 70%, in line with the manufacturer’s tested performance. For the experiments regarding ADNP protein degradation, the growth medium was removed 6 hours after transfection and replaced with fresh medium containing 200 nM of proteasome inhibitor MG132 (Sigma-Aldrich, M7449-200UL, https://www.sigmaaldrich.com/catalog/product/sigma/m7449).
Immunocytochemistry
For immunocytochemical experiments, HEK293T cells were seeded to a concentration of 2 × 105 cells per coverslip. 24 hours after transfection, the transfected HEK293T cells were washed with PBS and fixed in 3,7% PFA. Fixed cells were permeabilized with PBS containing 1% BSA and 1% Triton X-100 for 30 minutes at room temperature. For the heterochromatin co-localization experiments, the coverslips were incubated in anti-HP1α (clone EPR5777, Abcam, ab109028, http://www.abcam.com/hp1-alpha-antibody-epr5777-heterochromatin-marker-ab109028.html) diluted 1:500 in permeabilization buffer (PBS supplemented with 1% BSA and 1% Triton X-100) for 1 h, washed three times for 5 minutes with wash buffer (PBS supplemented with 0,1% Triton X-100), incubated with goat anti-rabbit Alexa Fluor 647 (Abcam, ab150075, http://www.abcam.com/donkey-rabbit-igg-hl-alexa-fluor-647-ab150075.html) diluted 1:5000 in permeabilization buffer for 1 h and then washed three times. Antibody incubation and wash steps were conducted at room temperature. After immunolabeling, coverslips were mounted using Vectashield Mounting Medium with DAPI (Vector Laboratories, H-1200, https://vectorlabs.com/vectashield-mounting-medium-with-dapi.html) and imaged using a Leica DM5500 microscope (Leica Microsystems, Wetzlar, Germany). Given the transfection efficiency of 70%, all cells emiting GFP signal were presumed to be succesfully transfected with the ADNP construct. As the observed effect was equal for all transfected cells, 40 cells per condition were randomly selected and imaged for examination of localization.
Western blot
Cell harvesting, lysis and immunoblotting were performed in accordance with standard protocols. Cells were lysed in a RIPA buffer (Thermo Scientific, Waltham, USA), supplemented with protease and phosphatase inhibitor (Roche, Basel, Switzerland). Cell lysates were collected and quantified using the Pierce BCA Protein Assay Kit (Thermo Scientific, 23,227, https://www.thermofisher.com/order/catalog/product/23227). A total protein concentration of 5 μg of cell lysate was loaded for separation using 4–12% SDS PAGE (Invitrogen Novex, NP0322BOX, https://www.thermofisher.com/order/catalog/product/NP0322BOX), followed by trans-blotting onto a nitrocellulose membrane (GE Healthcare Life Sciences). Mouse monoclonal anti-GFP HRP-tagged antibody (Miltenyi Biotec, 130–091-833, http://www.miltenyibiotec.com/en/products-and-services/macsmolecular/reagents/protein-research/epitope-tagged-protein-isolation-and-detection/anti-gfp-antibodies.aspx) was used for detection. The signal was detected using ECL Femto chemiluminescent reagent (Thermo Scientific, 34,096, https://www.thermofisher.com/order/catalog/product/34096).
NLS prediction
The amino acid sequence of ADNP and its mutant transcripts was queried in the cNLS Mapper tool (http://nls-mapper.iab.keio.ac.jp based on Kosugi et al., 2009 [Citation31]) for prediction of the presence of classic NLS sequences.
Supplemental Material
Download MS Word (14.1 KB)Acknowledgments
The authors thank Ellen Elinck, Katja Michielsen and Stefaan Schreers for technical support and Anke Van Dijck for providing information of the patient-specific mutations.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data can be accessed here.
Additional information
Funding
Related Research Data
References
- Hu WF, Chahrour MH, Walsh CA. The diverse genetic landscape of neurodevelopmental disorders. Annu Rev Genomics Hum Genet. 2014;15:195–213. PubMed PMID: 25184530; eng.
- APA. American psychiatric association: diagnostic and statistical manual of mental disorders DSM-V, Fifth edition, Text revision. Washington DC: American Psychiatric Association; 2013.
- McPartland J, Volkmar FR. Autism and related disorders. Handb Clin Neurol. 2012;106:407–418. PubMed PMID: 22608634; PubMed Central PMCID: PMCPmc3848246. eng.
- Surveillance DDMN. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. Morbidity mortality weekly report surveillance summaries. 2014 Mar 28;63(2):1–21. PubMed PMID: 24670961.
- Helsmoortel C, Vulto-Van Silfhout AT, Coe BP, et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat Genet. 2014 Apr;46(4):380–384. PubMed PMID: 24531329; PubMed Central PMCID: PMC3990853.
- Vandeweyer G, Helsmoortel C, Van Dijck A, et al. The transcriptional regulator ADNP links the BAF (SWI/SNF) complexes with autism. Am J Med Genet C: Semin Med Genet. 2014;166C(3):315–326.
- Van Dijck A, Vulto-Van Silfhout AT, Cappuyns E, et al. Clinical presentation of a complex neurodevelopmental disorder caused by mutations in ADNP. Biol Psychiatry. 2018. DOI:10.1016/j.biopsych.2018.02.1173
- Mandel S, Gozes I. Activity-dependent neuroprotective protein constitutes a novel element in the SWI/SNF chromatin remodeling complex. J Biol Chem. 2007 Nov 23;282(47):34448–34456. PubMed PMID: 17878164; eng.
- Bassan M, Zamostiano R, Davidson A, et al. Complete sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J Neurochem. 1999 Mar;72(3):1283–1293. PubMed PMID: 10037502; eng.
- Pinhasov A, Mandel S, Torchinsky A, et al. Activity-dependent neuroprotective protein: a novel gene essential for brain formation. Brain Res Dev Brain Res. 2003 Aug 12;144(1):83–90. PubMed PMID: 12888219; eng.
- Gozes I. Microtubules, schizophrenia and cognitive behavior: preclinical development of davunetide (NAP) as a peptide-drug candidate. Peptides. 2011 Feb;32(2):428–431. PubMed PMID: 21050875; eng.
- Magen I, Davunetide: GI. Peptide therapeutic in neurological disorders. Curr Med Chem. 2014 Feb 17;21(23):2591–2598. PubMed PMID: 24533805.
- D’Amico AG, Maugeri G, Rasa DM, et al. NAP counteracts hyperglycemia/hypoxia induced retinal pigment epithelial barrier breakdown through modulation of HIFs and VEGF expression. J Cell Physiol. 2017 Apr 24. PubMed PMID: 28436035; eng. DOI:10.1002/jcp.25971
- Mandel S, Rechavi G, Gozes I. Activity-dependent neuroprotective protein (ADNP) differentially interacts with chromatin to regulate genes essential for embryogenesis. Dev Biol. 2007 Mar 15;303(2):814–824. PubMed PMID: 17222401; eng.
- Mosch K, Franz H, Soeroes S, et al. HP1 recruits activity-dependent neuroprotective protein to H3K9me3 marked pericentromeric heterochromatin for silencing of major satellite repeats [Research Support, Non-U.S. Gov’t]. PLoS One. 2011;6(1):e15894. PubMed PMID: 21267468; PubMed Central PMCID: PMC3022755. eng.
- Zamostiano R, Pinhasov A, Gelber E, et al. Cloning and characterization of the human activity-dependent neuroprotective protein [Research Support, Non-U.S. Gov’t]. J Biol Chem. 2001 Jan 5;276(1):708–714. PubMed PMID: 11013255; eng.
- Maquat LE, Tarn WY, Isken O. The pioneer round of translation: features and functions. Cell. 2010 Aug 06;142(3):368–374. PubMed PMID: 20691898; PubMed Central PMCID: PMCPMC2950652. eng.
- Schoenberg DR, Maquat LE. Regulation of cytoplasmic mRNA decay. Nat Reviews. 2012 Mar 6;13(4):246–259. PubMed PMID: 22392217; PubMed Central PMCID: PMCPMC3351101. eng.
- Rosnoblet C, Vandamme J, Volkel P, et al. Analysis of the human HP1 interactome reveals novel binding partners. Biochem Biophys Res Commun. 2011 Sep 23;413(2):206–211. PubMed PMID: 21888893; eng.
- Varshavsky A. The ubiquitin system, autophagy, and regulated protein degradation. Annu Rev Biochem. 2017 Jun 20;86:123–128.
- Zhang R, Poustovoitov MV, Ye X, et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005 Jan;8(1):19–30. PubMed PMID: 15621527; eng.
- Mandel S, Spivak-Pohis I, Gozes I. ADNP differential nucleus/cytoplasm localization in neurons suggests multiple roles in neuronal differentiation and maintenance [Research Support, Non-U.S. Gov’t]. J Mol Neurosci: MN. 2008 Jun;35(2):127–141. PubMed PMID: 18286385; eng.
- Gennet N, Herden C, Bubb VJ, et al. Expression of activity-dependent neuroprotective protein in the brain of adult rats. Histol Histopathol. 2008 Mar;23(3):309–317. PubMed PMID: 18072088.
- Furman S, Steingart RA, Mandel S, et al. Subcellular localization and secretion of activity-dependent neuroprotective protein in astrocytes. Neuron Glia Biol. 2004 Aug;1(3):193–199. PubMed PMID: 16845437; PubMed Central PMCID: PMC1502393.
- Poggi SH, Vink J, Goodwin K, et al. Differential expression of embryonic and maternal activity-dependent neuroprotective protein during mouse development. Am J Obstet Gynecol. 2002 Oct;187(4):973–976. PubMed PMID: 12388989; eng.
- Zusev M, Gozes I. Differential regulation of activity-dependent neuroprotective protein in rat astrocytes by VIP and PACAP. Regul Pept. 2004 Dec 15;123(1–3):33–41. PubMed PMID: 15518891; eng.
- Schirer Y, Malishkevich A, Ophir Y, et al. Novel marker for the onset of frontotemporal dementia: early increase in activity-dependent neuroprotective protein (ADNP) in the face of Tau mutation. PLoS One. 2014;9(1):e87383. PubMed PMID: 24489906; PubMed Central PMCID: PMC3906161.
- Furman S, Hill JM, Vulih I, et al. Sexual dimorphism of activity-dependent neuroprotective protein in the mouse arcuate nucleus. Neurosci Lett. 2005 Jan 3;373(1):73–78. PubMed PMID: 15555780; eng.
- Malishkevich A, Amram N, Hacohen-Kleiman G, et al. Activity-dependent neuroprotective protein (ADNP) exhibits striking sexual dichotomy impacting on autistic and Alzheimer’s pathologies. Transl Psychiatry. 2015;5:e501. PubMed PMID: 25646590.
- Gozes I. Sexual divergence in activity-dependent neuroprotective protein impacting autism, schizophrenia, and Alzheimer’s disease. J Neurosci Res. 2017 Jan 2;95(1–2):652–660. PubMed PMID: 27870441; eng.
- Kosugi S, Hasebe M, Tomita M, et al. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A. 2009 Jun 23;106(25):10171–10176. PubMed PMID: 19520826; PubMed Central PMCID: PMCPMC2695404. eng.