
ABSTRACT
Aging is characterized by progressive decay of biological systems and although it is not considered a disease, it is one of the main risk factors for chronic diseases and many types of cancers. The accumulation of senescent cells in various tissues is thought to be a major factor contributing to aging and age-related diseases. Removal of senescent cells during aging by either genetic or therapeutic methods have led to an improvement of several age related disease in mice. In this preview, we highlight the significance of developing senotherapeutic approaches to specifically kill senescent cells (senolytics) or suppress the senescence-associated secretory phenotype (SASP) that drives sterile inflammation (senomorphics) associated with aging to extend healthspan and potentially lifespan. Also, we provide an overview of the senotherapeutic drugs identified to date. In particular, we discuss and expand upon the recent identification of inhibitors of the HSP90 co-chaperone as a new class of senolytics.
As we age, certain biological changes occur that increase the risk of diseases and disability. Aging is defined by molecular and cellular “hallmarks of aging”, each of which has shown to contribute of overall aging of organisms [Citation1]. One of these hall marks is cell senescence, a cell state defined by loss of proliferative capacity, increased metabolic activity and, importantly, resistance to apoptosis [Citation2]. Cellular senescence is an example of evolutionary antagonistic pleiotropy, being beneficial at young ages, with respect to development, wound healing and cancer prevention, but being detrimental at older ages as the damaging, senescent cell burden increases [Citation3]. Senescent cells can release pro-inflammatory cytokines/chemokines, proteases, and other factors, termed the senescence-associated secretory phenotype (SASP) that can adversely affect not only neighboring cells, but also likely contribute to driving systemic aging [Citation4]. The role of senescent cells in driving aging and age-related decline was demonstrated by the therapeutic effects elicited by clearance of senescent cells from progeroid mice carrying a p16-INK-ATTAC transgene where a suicide gene is expressed specifically in p16INK4a-positive senescent cells [Citation5,Citation6]. In contrast, local injection of senescent cells drives age-related diseases such as osteoarthritis and frailty [Citation7]. Thus, development of senotherapeutic agents able to specifically kill or suppress senescent cells would have significant therapeutic effects to slow aging, treat age-related diseases and improve resiliency. Indeed, certain senotherapeutics are already in clinical trials and others will be entering the clinic shortly [Citation8,Citation9].
Senotherapeutics: senolytic and senomorphic drugs
Recently, several small molecules have been demonstrated to function as senotherapeutics to reduce the senescent cell burden that accumulates with age by either killing senescent cell specifically (senolytics) or by suppressing some or all features of senescent cells (senomorphics) [Citation10] (). The first senolytic compounds were developed using a bioinformatics approach to identify Senescent-Cell Anti-Apoptotic Pathways (SCAPs), including inter-related Bcl-2/Bcl-XL, p53/p21, PI3K/AKT, and serpine anti-apoptotic pathways, which are upregulated in senescent cells [Citation11]. Importantly, this first study demonstrated that senescent cells upregulate some of the same anti-apoptotic pathways as tumor cells, suggesting that certain anti-cancer drugs also target senescent cells. Initially, the combination of dasatinib and quercetin (D + Q), which target several of these pro-survival pathways, was shown to induce death specifically in senescent murine and certain human cells in vitro, as well as to enhance cardiovascular function in aged mice, treadmill endurance in radiation-exposed mice, and decrease frailty, neurologic dysfunction, and bone loss in progeroid mice [Citation11]. Furthermore, treatment with (D + Q) reduced the senescent cell burden and aortic calcification in the aortae of atherosclerotic apoE−/- mice, improved lung function in the mouse model of idiopathic pulmonary fibrosis, and reduced murine hepatic steatosis and osteoporosis[Citation12]. The combination of D + Q is now being tested in humans for diabetes-associated chronic kidney disease, idiopathic pulmonary fibrosis and hematopoietic stem cell transplant survivors. Such clinical trials likely will be the first to document a therapeutic benefit of reducing the senescent cell burden.
Table 1. Senotherapeutic Compounds.
Subsequently, it was demonstrated by several groups that inhibitors of Bcl-2 family members like Navitoclax (ABT263), ABT737, A1331852, and A1155463 are senolytic in some, but not all human and murine cell types [Citation13–Citation17]. Navitoclax, a drug that targets several BCL-2 family members, was shown to reduce senescent cell burden, alleviate radiation-induced hematopoietic stem cell dysfunction and improve osteoarthritis[Citation13]. However, the systemic toxicity associated with some, if not all, Bcl-2 family inhibitors may preclude their chronic use systemically for extending healthspan. A FOXO4-interacting peptide that blocks the association of FOXO4 with p53 also induced apoptosis of senescent mouse and human cells in vitro, and reduces senescence and improves fitness, hair growth, and renal function in progeroid and aged wild-type mice [Citation18]. Interestingly, the natural compounds fisetin, a quercetin-related flavonoid, and piperlongumine, have senolytic/senomorphic activity in certain cell types in vitro [Citation14,Citation19]. More recently, multiple clinically-used compounds targeting the chaperone protein HSP90 were identified in a screen as a novel class of senolytics, able to induce apoptosis in senescent murine and human cells [Citation10]. Intermittent treatment of the Ercc1-/∆ murine progeria model of accelerated aging with the HSP90 inhibitor 17-DMAG results in an extension of healthspan and a reduction in markers of senescence.
Importantly, it is likely that the treatment regimen with senolytics would be intermittent, serving to reduce the senescent cell burden by acute treatment every few months or possibly even years [Citation8,Citation9]. This is critical for minimizing the side effects associated with chronic drug treatments. However, not all senescent cells are the same, expressing different SASP factors, different senescence markers, and more importantly, using different mechanisms to resist apoptosis [Citation20]. To eliminate senescent cells from different tissues or even within a single tissue likely will require the development of multiple classes of senotherapeutic drugs and approaches that are ultimately used in combination [Citation11]. For example, drugs targeting specific isoform and homologs of Hsp90, either alone or in combination with other senotherapeutic agents, could provide greater specificity.
HSP90 inhibitors as senolytics
The highly conserved heat shock proteins (HSPs) were originally discovered in Drosophila as genes whose expression could be induced by heat shock [Citation21]. HSPs are considered as overall stress response proteins that stabilize unfolded or misfolded proteins, giving the cell time to repair damaged proteins under stressful conditions such as heat shock, pH shift, heavy metals or hypoxia [Citation22–Citation24] . The increase in the level of HSPs is regulated at the transcriptional level by HSF1 (heat shock factor 1), one of the main transcription factors induced in response to proteotoxic cell stress [Citation25,Citation26]. HSPs are conveniently named after their molecular size (HSP60, HSP70, HSP90) in kDa and are generally ATP dependent [Citation27,Citation28]. There also is a set of small heat shock proteins including HSP27, αA-crystallin and αB-crystallin that function to prevent protein aggregation, but are ATP independent [Citation29]. Several HSPs are constitutively expressed and function as molecular chaperones helping the cell with the synthesis and folding of proteins [Citation28]. They often interact with their client proteins in multi-protein co-chaperone complexes, such as HSP70/HSP40, which mostly acts as a shuttle complex for nascent proteins [Citation30]. They also participate in protein assembly, export, turn-over and regulation with HSP90 serving as critical player in the stabilization and activation of key signaling proteins [Citation31].
Hsp90 plays a role in various cellular functions and pathophysiological processes from helping its client proteins to fold properly and stabilizing them against heat stress, to chaperoning them for protein degradation. A number of these stabilized proteins are required for tumor growth in several types of cancer cells and therefore HSP90 has become an attractive drug target [Citation31,Citation32]. There have been and currently are clinical trials using Hsp90 inhibitors for cancer, either alone or in combination with chemotherapeutics or irradiation [Citation33]. Tumor-specific increase in Hsp90 protein levels also is being tested as a prognostic biomarker for certain types of cancer [Citation34]. However, there still are no FDA-approved HSP90 inhibitors available for cancer treatments. This lack of clear clinical efficacy could be due to the fact that Hsp90 plays an important role in such a wide variety of cellular processes or that there are multiple isoforms and homologs of Hsp90 with distinct functions [Citation35]. Recently, organelle-specific inhibitors of Hsp90 are being developed [Citation36]. Extracellular, mitochondrial and endoplasmic reticulum directed Hsp90 inhibitors can inhibit Hsp90 functions efficiently in an organelle-specific way, resulting in less toxicity to normal cells [Citation37–Citation39]. These specific HSP90 inhibitory strategies could also be useful in senolytic drug discovery as several organelle-specific HSP90 homologs are affected by aging and/or implicated in age-related diseases [Citation40,Citation41]. These inhibitors could help to overcome the suboptimal pan-Hsp90 inhibitor toxicity versus efficacy profile and lead to more potent, directed senolytics.
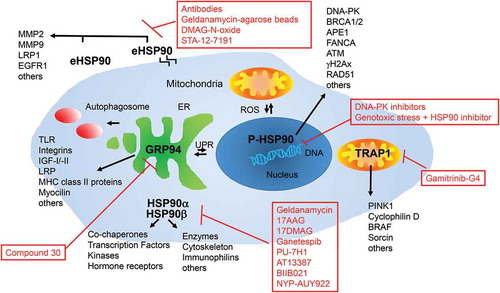
In mammals, the Hsp90 chaperone family comprises four homologs: HSP90α, HSP90β, Grp94 and Trap1 [Citation31,Citation35,Citation42]. Inducible Hsp90α and constitutively expressed Hsp90β are the major homologs, sharing approximately 90% of their sequences. They are ubiquitously expressed in all nucleated cells and are mostly found in the cytoplasm and the nucleus. Due to their importance in cellular stress response they are very closely regulated on transcriptional as well as post-translational levels [Citation43]. In addition to cytosolic HSP90, there are two organelle-specific isoforms, the endoplasmatic reticulum (ER) localized 94-kDa glucose-regulated protein (Grp94) and the mitochondrial tumor necrosis factor (TNF) receptor-associated protein 1 (Trap1) that are both upregulated under cell stress. There is also an extracellular Hsp90 (eHsp90) isoform that can be either cell-surface-bound or secreted from the cytoplasm. Each homolog or isoform has a unique biological function and participates in various physiological and pathological processes including aging related diseases [Citation35,Citation40,Citation41,Citation44]. Testing drugs that specifically target one of these homologs or isoforms for senolytic activity in different cell types should provide more insight as to how HSP90 prevents apoptosis of senescent cells. On the side, all senolytic HSP90 inhibitors identified to date bind to the N-terminal ATP binding pocket of HSP90.
Cytoplasmic HSP90s have a variety of client proteins and co-chaperones that could be influenced by inhibition of either the inducible or the constitutively expressed form of HSP90 (α or β) [Citation45]. A detailed description of cytoplasmic HSP90 and its interactors would exceed the scope of this preview but can be found in several excellent reviews and on the website of D. Picard (http://www.picard.ch)[Citation46–Citation48]. The role of HSP90 in cell senescence has received attention, recently, but is still poorly defined. Upregulation of phosphorylated AKT, a client protein of HSP90, has been shown to stabilize senescent cells from apoptosis whereas inhibition of cytoplasmic HSP90 leads to AKT de-phosphorylation, making senescent cells susceptible to apoptosis, similar to what is seen in cancer cells [Citation10,Citation49]. HSP90 is also known to inhibit upregulation of senecence in non-small cell lung cancer cells by inducing p14ARF degradation [Citation50]. However, if the cancer cells were already p14ARF positive, indicating oncogene-induced senescence, increased cell death was observed when HSP90 was inhibited [Citation50]. Recent studies also showed that inhibition of HSP90 altered the release of several cytokines in cancer cells from a pro-tumorigenic secretome to a pro-apoptotic senescence status, thus resulting in chemosensitizing effects [Citation51].
In addition to the widely discussed cytosolic function of HSP90, several cancer studies have shown that Hsp90 and its associated co-chaperones also play an essential role in the nucleus [Citation52]. Phosphorylation of HSP90 by DNA-PK as well as its interaction with several DNA maintenance and DNA repair proteins have shown to be vital for several DNA damage repair (DDR) processes from base excision repair (BER) to homologues recombination (HR) and non-homologues end-joining (NHEJ) processes [Citation53]. A combination of HSP90 inhibitors together with γ-Irradiation or other DNA damaging agents is therefore a promising approach to treat some types of cancers [Citation52,Citation54]. During aging, different extrinsic and intrinsic stressors like exposure to chemicals, radiation and reactive oxygen species (ROS) can lead to an accumulative amount of damage to different cellular components and specifically to the DNA, described as the “DNA damage theory of aging”[Citation55]. Increased DNA damage as well as a decreased DNA damage repair (DDR) both play a crucial role in the deterioration of cell function. Stabilization of nuclear HSP90 client proteins like BRCA 1, BRCA 2, FANCA, APE1 and subsequently Pol-β, ATM followed by NF-γB, γH2Ax and RAD51 might not only be important in HSP90-addicted cancer cells, but might also present a survival mechanism for senescent cells [Citation52,Citation53,Citation56–Citation58]. Thus, for senescent cells induced by irradiation or other extrinsic and intrinsic DNA damaging factors HSP90 inhibitors should have strong senolytic activity.
In contrast to nuclear HSP90, secreted or membrane bound HSP90 (eHSP90) influences cancer cells by increasing cell migration and cancer cell invasion [Citation59–Citation62]. eHsp90 client proteins include the matrix metalloproteinases MMP2 and MMP9, LDL receptor-like protein (LRP1) and EGFR2/Her/ErbB2 receptors, all known to be critical for cancer cell invasion and metastasis but also for being found in the secretome of senescent cells [Citation63–Citation65]. Specific inhibitors of eHSP90 such as anti-HSP90 antibodies, geldanamycin coupled agarose beads, DMAG-N-oxide, or STA-12–7191, a biotinylated analog of ganetespib reduce cancer cell migration and invasion. [Citation59,Citation66,Citation67,Citation68] However, whether eHSP90 regulates senescence is still unclear.
Trap1 (TNF receptor associated protein 1) is a molecular chaperone with high sequence homology to HSP90, found in the mitochondrial matrix and the inner membrane space [Citation69–Citation73]. Trap1 has anti-apoptotic and antioxidant properties and plays a critical role in mitochondrial reactive oxygen species (ROS)-mediated oxidative stress [Citation74,Citation75]. Oxidative stress leads to mitochondrial dysfunction and apoptosis and is associated with diverse human diseases, such as heart attack, stroke, neurodegenerative diseases and certain types of cancer. Trap1 is upregulated in several human carcinomas, such as prostate, colorectal, breast, and ovarian cancers [Citation76–Citation80]. In mitochondria, Trap1 is a chaperone for denatured proteins and regulates ROS metabolism by interaction with CypD, a protein in the mitochondrial permeability transition pore (MTPT)[Citation38,Citation73]. Excessive ROS production activates CypD to switch the permeability pore open, release cytochrome c, and ultimately lead to cell death [Citation81–Citation83]. Direct interaction of CypD with Trap1 can inhibit MTPT opening triggered by activated CypD and represents an essential survival mechanism of various cancer cells and possibly senescent cells. The Trap1-specific inhibitor, gamitrinib-G4, promotes the activation of CypD, MTPT opening, releasing cytochrome c, and inducing cancer cell death [Citation84,Citation85] and might also be useful as a senolytic. Other Trap1 client proteins include the kinase PINK1 [Citation75] which protects against oxidative stress, the Ca2+ binding protein sorcin [Citation86], and the cytoprotective protein kinase BRAF [Citation87]. In addition, Trap1−/− mice are not only viable, but also show reduced occurrence of age-associated pathologies like inflammatory tissue degeneration, dysplasia, spontaneous tumor formation and obesity [Citation88]. The mice show a global upregulation of oxidative phosphorylation and glycolysis transcriptomes probably caused by deregulated mitochondrial respiration, and oxidative stress [Citation88]. Cells from the Trap1−/− mice also have impaired cell proliferation and a switch to glycolytic metabolism, which might reduce the viability of not only cancer, but also highly metabolically active senescent cells.
The endoplasmic reticulum is a multifunctional organelle where protein folding, lipid biosynthesis, and calcium storage occurs. Perturbations of this processes lead to the misfolding of proteins, ER stress and up-regulation of several signaling pathways collectively called the unfolded protein response (UPR) [Citation89–Citation91]. The UPR is characterized by the induction of chaperones like the HSP90 homolog Grp94 (Glucose regulated protein 94), degradation of misfolded proteins and attenuation of protein translation [Citation92,Citation93]. During aging, a decline in the expression and activity of key ER molecular chaperones compromise proper protein folding and the adaptive response of the UPR [Citation94,Citation95]. Grp94, glucose regulated protein 78 (Grp78; also known as Immunoglobulin Binding protein – BiP), the lectins, calnexin and calreticulin, and the thiol-disulfide oxidoreductases, protein disulfide isomerase (PDI) and ERp57 are some of the major folding sensors and chaperones in the ER that diminish during the aging process [Citation95–Citation98]. In addition to their reduced expression levels, those chaperones are progressively oxidized with age. Increased oxidation significantly correlate with reductions in enzymatic activity of several chaperones [Citation97], which may contribute to their functional decline.
One mechanism used by the UPR to remove misfolded proteins from cells is to activate autophagy [Citation99]. If this process is disturbed, proteins convert from their normally soluble forms to insoluble aggregates and accumulate in several organs including the liver, brain or intestine [Citation100,Citation101]. These protein plaques can lead to certain age related diseases including Alzheimer’s, Parkinson’s, amyotrophic leteral sclerosis (ALS), Huntington disease and others [Citation102–Citation105]. The UPR is implicated in many of these protein folding diseases as well as a variety of cancers and a host of inflammatory diseases including diabetes, atherosclerosis, inflammatory bowel disease and arthritis [Citation94,Citation96,Citation106,Citation107]. Therapeutics that either reduce ER stress by promoting correct protein folding, or that improve detection and removal of misfolded proteins may not only help in delaying or preventing some of the age-related diseases, but could also be used for an early detection of a number of age-related pathologies. In regard to GRP94, several client proteins have been identified including the immune-related MHC class II proteins and Toll-like receptors, several integrins, insulin-like growth factors I and II, LRP6, and mutant myocilin that changes with aging [Citation92,Citation101,Citation108,Citation109]. GRP94 is essential during embryogenesis, but it is non-essential in developed organisms [Citation110]. Some diseases like multiple myeloma and hepatocellular carcinoma, however, rely upon a functional ER chaperone system and deletion of Grp94 decreases the viability of these cancer cells [Citation111,Citation112]. In a wound healing scratch assay with several aggressive and metastatic cancer cells, a specific GRP94 inhibitor decreased migratory capabilities of the cells and induced degradation of the Grp94 client protein integrin α2, a cell adhesion protein. These data suggest that inhibition of Grp94 represents a new therapeutic strategy for cancer treatments [Citation113]. A similar pro-survival function could be hypothesized for Grp94 in senescent cells where the use of Grp94 inhibitors on senescent cells could lead to a sensitization of those cells to ER stress and increased cell death.
The ability to target Hsp90 inhibitors to particular cellular compartments should offer exciting new approaches to selectively inhibit subcellular localized Hsp90 isoforms in senescent cells with reduced toxicity to normal cells (). However, besides Hsp90 and its homologs, there are additional pathways that regulate senescent cell survival and thus it is highly likely that additional classes of senotherapeutics will be identified through bioinformatic analyses and drug screening approaches.
Figure 1. HSP90 and its homologs. HSP90 and its homologs are localized in different cellular compartments and interact with different co-chaperones and client proteins. HSP90a and b are mainly localized in the cytoplasm or the nucleus phosphorylated by DNA-PK (P-HSP90). HSP90 can also be found secreted to the extracellular matrix or membrane associated (eHSP90). It can also be found in mitochondria together with its homolog TRAP1. The endoplasmic reticulum homolog of HSP90 is GRP94. Interaction of different co-chaperones and client proteins makes it a versatile platform engaged in a range of important cellular signaling pathways. Red boxes list inhibitors of the indicated proteins tested in senescent and/or cancer cells.
In addition to senolytics, several classes of senomorphics have been identified, which suppress markers of senescence or the secretory phenotype without cytotoxicity. These include inhibitors of IγB Kinase (IKK)/NF-γB, free radical scavengers, and Jak pathway inhibitors. Even rapamycin acts as a senomorphic by reducing the expression of the SASP [Citation10,Citation114,Citation115]. Other compounds, for example fisetin, have senomorphic effects on certain cell types while having senolytic activity on others, at least in vitro.
Taken together, there is reason to be tremendously excited about the health and economic impact of this new class of drugs, termed senotherapeutics. The development of approaches to reduce age-related senescent cell burden has the ability to revolutionize how age-related diseases and even ultimately aging itself can be treated. Multiple studies with small molecule senotherapeutics, nano-particle based gene therapy and drug delivery and antibodies have shown promise in preclinical studies [Citation116]. Clinical trials with D + Q have been initiated in several age-related diseases with clinical trials with Bcl2 family inibitors and MDM2-p53 inhibitors to be initiated soon. However, there is still much to learn about how best to identify, characterize, and apply these senotherapeutic drugs to improve human health. Also, understanding their mechanisms of action should lead to safer and more effective senotherapeutic and, in particular, senolytic drugs.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Lopez-Otin C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013 Jun 6;153(6):1194–1217. PubMed PMID: 23746838; PubMed Central PMCID: PMC3836174.
- Wang E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995 Jun 1;55(11):2284–2292. PubMed PMID: 7757977.
- Campisi J. Cancer and ageing: rival demons? Nat Rev Cancer. 2003 May;3(5):339–349. PubMed PMID: 12724732.
- Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008 Dec 02;6(12):2853–2868. PubMed PMID: 19053174; PubMed Central PMCID: PMCPMC2592359.
- Baker DJ, Childs BG, Durik M, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016 Feb 11;530(7589):184–189. PubMed PMID: 26840489; PubMed Central PMCID: PMCPMC4845101.
- Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011 Nov 02;479(7372):232–236. PubMed PMID: 22048312; PubMed Central PMCID: PMC3468323.
- Xu M, Bradley EW, Weivoda MM, et al. Transplanted senescent cells induce an osteoarthritis-like condition in mice. J Gerontol A Biol Sci Med Sci. 2017 Jun 01;72(6):780–785. PubMed PMID: 27516624.
- Kirkland JL, Tchkonia T, Zhu Y, et al. The clinical potential of senolytic drugs. J Am Geriatr Soc. 2017 Sep 04 PubMed PMID: 28869295. DOI:10.1111/jgs.14969.
- Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine. 2017 Apr 12. PubMed PMID: 28416161. DOI:10.1016/j.ebiom.2017.04.013
- Fuhrmann-Stroissnigg H, Ling YY, Zhao J, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun. 2017 Sep 04;8(1):422. PubMed PMID: 28871086; PubMed Central PMCID: PMC5583353.
- Zhu Y, Tchkonia T, Pirtskhalava T, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015 Aug;14(4):644–658. PubMed PMID: 25754370; PubMed Central PMCID: PMC4531078.
- Farr JN, Xu M, Weivoda MM, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med. 2017 Sep;23(9):1072–1079. PubMed PMID: 28825716.
- Chang J, Wang Y, Shao L, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016 Jan;22(1):78–83. PubMed PMID: 26657143; PubMed Central PMCID: PMCPMC4762215.
- Zhu Y, Doornebal EJ, Pirtskhalava T, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY). 2017 Mar 08;9(3):955–963. PubMed PMID: 28273655; PubMed Central PMCID: PMC5391241.
- Moncsek A, Al-Suraih MS, Trussoni CE, et al. Targeting senescent cholangiocytes and activated fibroblasts with Bcl-xL inhibitors ameliorates fibrosis in Mdr2-/- mice. Hepatology. 2017 Aug 12 PubMed PMID: 28802066. DOI:10.1002/hep.29464.
- Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016 Jun;15(3):428–435. PubMed PMID: 26711051; PubMed Central PMCID: PMCPMC4854923.
- Yosef R, Pilpel N, Tokarsky-Amiel R, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. 2016 Apr 06;7:11190. PubMed PMID: 27048913; PubMed Central PMCID: PMCPMC4823827.
- Baar MP, Brandt RM, Putavet DA, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017 Mar 23;169(1):132–147 e16. PubMed PMID: 28340339.
- Wang Y, Chang J, Liu X, et al. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging (Albany NY). 2016 Nov 19;8(11):2915–2926. PubMed PMID: 27913811; PubMed Central PMCID: PMC5191878.
- van Deursen JM. The role of senescent cells in ageing. Nature. 2014 May 22;509(7501):439–446. PubMed PMID: 24848057; PubMed Central PMCID: PMC4214092.
- Ritossa FM. A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia. 1962;18(12): 571. PubMed PMID: 14163779.
- Courgeon AM, Maisonhaute C, Best-Belpomme M. Heat shock proteins are induced by cadmium in Drosophila cells. Exp Cell Res. 1984 Aug;153(2):515–521. PubMed PMID: 6734754.
- Trott A, West JD, Klaic L, et al. Activation of heat shock and antioxidant responses by the natural product celastrol: transcriptional signatures of a thiol-targeted molecule. Mol Biol Cell. 2008 Mar;19(3):1104–1112. PubMed PMID: 18199679; PubMed Central PMCID: PMC2262981.
- Kregel KC. Heat shock proteins: modifying factors in physiological stress responses and acquired thermotolerance. J Appl Physiol (1985). 2002 May;92(5):2177–2186. PubMed PMID: 11960972.
- Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998 Dec 15;12(24):3788–3796. PubMed PMID: 9869631.
- Dai C, Whitesell L, Rogers AB, et al. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007 Sep 21;130(6):1005–1018. PubMed PMID: 17889646; PubMed Central PMCID: PMC2586609.
- Jee H. Size dependent classification of heat shock proteins: a mini-review. J Exerc Rehabil. 2016 Aug;12(4):255–259. PubMed PMID: 27656620; PubMed Central PMCID: PMC5031383.
- Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell. 2010 Oct 22;40(2):253–266. PubMed PMID: 20965420.
- Arrigo AP, Simon S, Gibert B, et al. Hsp27 (HspB1) and alphaB-crystallin (HspB5) as therapeutic targets. FEBS Lett. 2007 Jul 31;581(19):3665–3674. PubMed PMID: 17467701.
- Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996 Jun 13;381(6583):571–579. PubMed PMID: 8637592.
- Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010 Jul;11(7):515–528. PubMed PMID: 20531426.
- Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. PubMed PMID: 2853609.
- Trepel J, Mollapour M, Giaccone G, et al. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010 Aug;10(8):537–549. PubMed PMID: 20651736.
- Barrott JJ, Haystead TA. Hsp90, an unlikely ally in the war on cancer. FEBS J. 2013 Mar;280(6):1381–1396. PubMed PMID: 23356585; PubMed Central PMCID: PMC3815692.
- Sreedhar AS, Kalmar E, Csermely P, et al. Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 2004 Mar 26;562(1–3):11–15. PubMed PMID: 15069952.
- Seo YH. Organelle-specific Hsp90 inhibitors. Arch Pharm Res. 2015 Sep;38(9):1582–1590. 10.1007/s12272-015-0636-1. PubMed PMID: 26195286.
- Plescia J, Salz W, Xia F, et al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell. 2005 May;7(5):457–468. PubMed PMID: 15894266.
- Kang BH, Plescia J, Dohi T, et al. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007 Oct 19;131(2):257–270. PubMed PMID: 17956728.
- Patel PD, Yan P, Seidler PM, et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol. 2013 Nov;9(11):677–684. PubMed PMID: 23995768; PubMed Central PMCID: PMC3982621.
- Blair LJ, Sabbagh JJ, Dickey CA. Targeting Hsp90 and its co-chaperones to treat Alzheimer’s disease. Expert Opin Ther Targets. 2014 Oct;18(10):1219–1232. PubMed PMID: 25069659; PubMed Central PMCID: PMC4625388.
- Falsone SF, Kungl AJ, Rek A, et al. The molecular chaperone Hsp90 modulates intermediate steps of amyloid assembly of the Parkinson-related protein alpha-synuclein. J Biol Chem. 2009 Nov 06;284(45):31190–31199. PubMed PMID: 19759002; PubMed Central PMCID: PMC2781518.
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005 Oct;5(10):761–772. PubMed PMID: 16175177.
- Prodromou C. Mechanisms of Hsp90 regulation. Biochem J. 2016 Aug 15;473(16):2439–2452. PubMed PMID: 27515256; PubMed Central PMCID: PMC4980810.
- Dasuri K, Zhang L, Ebenezer P, et al. Aging and dietary restriction alter proteasome biogenesis and composition in the brain and liver. Mech Ageing Dev. 2009 Nov-Dec;130(11–12):777–783. PubMed PMID: 19896962; PubMed Central PMCID: PMC2942759.
- Echeverria PC, Bernthaler A, Dupuis P, et al. An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. PLoS One. 2011;6(10):e26044. PubMed PMID: 22022502; PubMed Central PMCID: PMC3195953.
- Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol. 2017 Jun;18(6):345–360. PubMed PMID: 28429788.
- Zuehlke A, Johnson JL. Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers. 2010 Mar;93(3):211–217. PubMed PMID: 19697319; PubMed Central PMCID: PMC2810645.
- Li J, Soroka J, Buchner J. The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim Biophys Acta. 2012 Mar;1823(3):624–635. PubMed PMID: 21951723..
- Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci U S A. 2000 Sep 26;97(20):10832–10837. PubMed PMID: 10995457; PubMed Central PMCID: PMC27109.
- Han SY, Ko A, Kitano H, et al. Molecular chaperone HSP90 is necessary to prevent cellular senescence via lysosomal degradation of p14ARF. Cancer Res. 2017 Jan 15;77(2):343–354. PubMed PMID: 27793846.
- di Martino S, Amoreo CA, Nuvoli B, et al. HSP90 inhibition alters the chemotherapy-driven rearrangement of the oncogenic secretome. Oncogene. 2018 Mar;37(10):1369–1385. 10.1038/s41388-017-0044-8. PubMed PMID: 29311642.
- Pennisi R, Ascenzi P, di Masi A. Hsp90: a new player in DNA repair? Biomolecules. 2015 Oct 16;5(4):2589–2618. PubMed PMID: 26501335; PubMed Central PMCID: PMC4693249.
- Stecklein SR, Kumaraswamy E, Behbod F, et al. BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation. Proc Natl Acad Sci U S A. 2012 Aug 21;109(34):13650–13655. PubMed PMID: 22869732; PubMed Central PMCID: PMC3427093.
- Spiegelberg D, Dascalu A, Mortensen AC, et al. The novel HSP90 inhibitor AT13387 potentiates radiation effects in squamous cell carcinoma and adenocarcinoma cells. Oncotarget. 2015 Nov 03;6(34):35652–35666. PubMed PMID: 26452257; PubMed Central PMCID: PMC4742132.
- Freitas AA, De Magalhaes JP. A review and appraisal of the DNA damage theory of ageing. Mutat Res. 2011 Jul-Oct;728(1–2):12–22. PubMed PMID: 21600302.
- Noguchi M, Yu D, Hirayama R, et al. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem Biophys Res Commun. 2006 Dec 22;351(3):658–663. PubMed PMID: 17083915.
- Quanz M, Herbette A, Sayarath M, et al. Heat shock protein 90alpha (Hsp90alpha) is phosphorylated in response to DNA damage and accumulates in repair foci. J Biol Chem. 2012 Mar 16;287(12):8803–8815. PubMed PMID: 22270370; PubMed Central PMCID: PMC3308794.
- Tilstra JS, Robinson AR, Wang J, et al. NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest. 2012 Jul;122(7):2601–2612. PubMed PMID: 22706308; PubMed Central PMCID: PMC3386805.
- Tsutsumi S, Scroggins B, Koga F, et al. A small molecule cell-impermeant Hsp90 antagonist inhibits tumor cell motility and invasion. Oncogene. 2008 Apr 10;27(17):2478–2487. PubMed PMID: 17968312; PubMed Central PMCID: PMC2754825.
- Li W, Sahu D, Tsen F. Secreted heat shock protein-90 (Hsp90) in wound healing and cancer. Biochim Biophys Acta. 2012 Mar;1823(3):730–741. PubMed PMID: 21982864; PubMed Central PMCID: PMC3266443.
- Becker B, Multhoff G, Farkas B, et al. Induction of Hsp90 protein expression in malignant melanomas and melanoma metastases. Exp Dermatol. 2004 Jan;13(1):27–32. PubMed PMID: 15009113.
- Eustace BK, Jay DG. Extracellular roles for the molecular chaperone, hsp90. Cell Cycle. 2004 Sep;3(9):1098–1100. PubMed PMID: 15326368.
- Tchkonia T, Zhu Y, van Deursen J, et al. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013 Mar;123(3):966–972. PubMed PMID: 23454759; PubMed Central PMCID: PMC3582125.
- Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009 Feb;9(2):81–94. PubMed PMID: 19132009.
- Thuringer D, Hammann A, Benikhlef N, et al. Transactivation of the epidermal growth factor receptor by heat shock protein 90 via Toll-like receptor 4 contributes to the migration of glioblastoma cells. J Biol Chem. 2011 Feb 4;286(5):3418–3428. PubMed PMID: 21127066; PubMed Central PMCID: PMC3030348.
- Sidera K, Patsavoudi E. Extracellular HSP90: conquering the cell surface. Cell Cycle. 2008 Jun 1;7(11):1564–1568. doi:10.4161/cc.7.11.6054. PubMed PMID: 18469526.
- McCready J, Wong DS, Burlison JA, et al. An Impermeant Ganetespib Analog Inhibits Extracellular Hsp90-Mediated Cancer Cell Migration that Involves Lysyl Oxidase 2-like Protein. Cancers (Basel). 2014 Apr 30;6(2):1031–1046. doi:10.3390/cancers6021031. PubMed PMID: 24785146; PubMed Central PMCID: PMC4074815.
- Stellas D, El Hamidieh A, Patsavoudi E. Monoclonal antibody 4C5 prevents activation of MMP2 and MMP9 by disrupting their interaction with extracellular HSP90 and inhibits formation of metastatic breast cancer cell deposits. BMC Cell Biol. 2010 Jul 5;11:51. doi:10.1186/1471-2121-11-51. PubMed PMID: 20602761; PubMed Central PMCID: PMC2914660.
- Song HY, Dunbar JD, Zhang YX, et al. Identification of a protein with homology to hsp90 that binds the type 1 tumor necrosis factor receptor. J Biol Chem. 1995 Feb 24;270(8):3574–3581. PubMed PMID: 7876093.
- Felts SJ, Owen BA, Nguyen P, et al. The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J Biol Chem. 2000 Feb 4;275(5):3305–3312. PubMed PMID: 10652318.
- Chen B, Piel WH, Gui L, et al. The HSP90 family of genes in the human genome: insights into their divergence and evolution. Genomics. 2005 Dec;86(6):627–637. PubMed PMID: 16269234.
- Leskovar A, Wegele H, Werbeck ND, et al. The ATPase cycle of the mitochondrial Hsp90 analog Trap1. J Biol Chem. 2008 Apr 25;283(17):11677–11688. PubMed PMID: 18287101.
- Altieri DC, Stein GS, Lian JB, et al. TRAP-1, the mitochondrial Hsp90. Biochim Biophys Acta. 2012 Mar;1823(3):767–773. PubMed PMID: 21878357; PubMed Central PMCID: PMC3263322.
- Masuda Y, Shima G, Aiuchi T, et al. Involvement of tumor necrosis factor receptor-associated protein 1 (TRAP1) in apoptosis induced by beta-hydroxyisovalerylshikonin. J Biol Chem. 2004 Oct 8;279(41):42503–42515. PubMed PMID: 15292218.
- Pridgeon JW, Olzmann JA, Chin LS, et al. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007 Jul;5(7):e172. PubMed PMID: 17579517; PubMed Central PMCID: PMC1892574.
- Masgras I, Sanchez-Martin C, Colombo G, et al. The chaperone TRAP1 as a modulator of the mitochondrial adaptations in cancer cells. Front Oncol. 2017;7:58. PubMed PMID: 28405578; PubMed Central PMCID: PMC5370238.
- Leav I, Plescia J, Goel HL, et al. Cytoprotective mitochondrial chaperone TRAP-1 as a novel molecular target in localized and metastatic prostate cancer. Am J Pathol. 2010 Jan;176(1):393–401. PubMed PMID: 19948822; PubMed Central PMCID: PMC2797899.
- Gao JY, Song BR, Peng JJ, et al. Correlation between mitochondrial TRAP-1 expression and lymph node metastasis in colorectal cancer. World J Gastroenterol. 2012 Nov 7;18(41):5965–5971. PubMed PMID: 23139614; PubMed Central PMCID: PMC3491605.
- Zhang B, Wang J, Huang Z, et al. Aberrantly upregulated TRAP1 is required for tumorigenesis of breast cancer. Oncotarget. 2015 Dec 29;6(42):44495–44508. PubMed PMID: 26517089; PubMed Central PMCID: PMC4792571.
- Amoroso MR, Matassa DS, Agliarulo I, et al. TRAP1 downregulation in human ovarian cancer enhances invasion and epithelial-mesenchymal transition. Cell Death Dis. 2016 Dec 15;7(12):e2522. PubMed PMID: 27977010; PubMed Central PMCID: PMC5260997.
- Baines CP, Kaiser RA, Purcell NH, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005 Mar 31;434(7033):658–662. PubMed PMID: 15800627.
- Basso E, Fante L, Fowlkes J, et al. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005 May 13;280(19):18558–18561. PubMed PMID: 15792954.
- Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005 Mar 31;434(7033):652–658. PubMed PMID: 15800626.
- Kang BH, Altieri DC. Compartmentalized cancer drug discovery targeting mitochondrial Hsp90 chaperones. Oncogene. 2009 Oct 22;28(42):3681–3688. PubMed PMID: 19648961; PubMed Central PMCID: PMC2766018.
- Kang BH, Plescia J, Song HY, et al. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J Clin Invest. 2009 Mar;119(3):454–464. PubMed PMID: 19229106; PubMed Central PMCID: PMC2648691.
- Landriscina M, Laudiero G, Maddalena F, et al. Mitochondrial chaperone Trap1 and the calcium binding protein Sorcin interact and protect cells against apoptosis induced by antiblastic agents. Cancer Res. 2010 Aug 15;70(16):6577–6586. PubMed PMID: 20647321.
- Condelli V, Piscazzi A, Sisinni L, et al. TRAP1 is involved in BRAF regulation and downstream attenuation of ERK phosphorylation and cell-cycle progression: a novel target for BRAF-mutated colorectal tumors. Cancer Res. 2014 Nov 15;74(22):6693–6704. PubMed PMID: 25239454.
- Lisanti S, Tavecchio M, Chae YC, et al. Deletion of the mitochondrial chaperone TRAP-1 uncovers global reprogramming of metabolic networks. Cell Rep. 2014 Aug 7;8(3):671–677. PubMed PMID: 25088416; PubMed Central PMCID: PMC4127146.
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011 Nov 25;334(6059):1081–1086. PubMed PMID: 22116877.
- Rutkowski DT, Hegde RS. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J Cell Biol. 2010 May 31;189(5):783–794. PubMed PMID: 20513765; PubMed Central PMCID: PMC2878945.
- Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005 Jan 6;569(1–2):29–63. PubMed PMID: 15603751.
- Eletto D, Dersh D, Argon Y. GRP94 in ER quality control and stress responses. Semin Cell Dev Biol. 2010 Jul;21(5):479–485. PubMed PMID: 20223290; PubMed Central PMCID: PMC3676867.
- Marzec M, Eletto D, Argon Y. GRP94: an HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim Biophys Acta. 2012 Mar;1823(3):774–787. PubMed PMID: 22079671; PubMed Central PMCID: PMC3443595.
- Brown MK, Naidoo N. The endoplasmic reticulum stress response in aging and age-related diseases. Front Physiol. 2012;3:263. PubMed PMID: 22934019; PubMed Central PMCID: PMC3429039.
- Naidoo N, Ferber M, Master M, et al. Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J Neurosci. 2008 Jun 25;28(26):6539–6548. PubMed PMID: 18579727; PubMed Central PMCID: PMC2925257.
- Paz Gavilan M, Vela J, Castano A, et al. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol Aging. 2006 Jul;27(7):973–982. PubMed PMID: 15964666.
- Nuss JE, Choksi KB, DeFord JH, et al. Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem Biophys Res Commun. 2008 Jan 11;365(2):355–361. PubMed PMID: 17996725; PubMed Central PMCID: PMC2238339.
- Minakshi R, Rahman S, Jan AT, et al. Implications of aging and the endoplasmic reticulum unfolded protein response on the molecular modality of breast cancer. Exp Mol Med. 2017 Nov 10;49(11):e389. PubMed PMID: 29123254.
- Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006 Dec;26(24):9220–9231. PubMed PMID: 17030611; PubMed Central PMCID: PMC1698520.
- Selkoe DJ. Folding proteins in fatal ways. Nature. 2003 Dec 18;426(6968):900–904. PubMed PMID: 14685251.
- Zhu G, Lee AS. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J Cell Physiol. 2015 Jul;230(7):1413–1420. PubMed PMID: 25546813; PubMed Central PMCID: PMC4725317.
- Ryu EJ, Harding HP, Angelastro JM, et al. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J Neurosci. 2002 Dec 15;22(24):10690–10698. PubMed PMID: 12486162.
- Forman MS, Lee VM, Trojanowski JQ. ‘Unfolding’ pathways in neurodegenerative disease. Trends Neurosci. 2003 Aug;26(8):407–410. PubMed PMID: 12900170..
- Reddy PH, Williams M, Tagle DA. Recent advances in understanding the pathogenesis of Huntington’s disease. Trends Neurosci. 1999 Jun;22(6):248–255. PubMed PMID: 10354600.
- Jaronen M, Goldsteins G, Koistinaho J. ER stress and unfolded protein response in amyotrophic lateral sclerosis-a controversial role of protein disulphide isomerase. Front Cell Neurosci. 2014;8:402. PubMed PMID: 25520620; PubMed Central PMCID: PMC4251436.
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006 Dec 14;444(7121):860–867. PubMed PMID: 17167474.
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008 Jul 24;454(7203):455–462. PubMed PMID: 18650916; PubMed Central PMCID: PMC2727659.
- Staron M, Yang Y, Liu B, et al. gp96, an endoplasmic reticulum master chaperone for integrins and Toll-like receptors, selectively regulates early T and B lymphopoiesis. Blood. 2010 Mar 25;115(12):2380–2390. doi:10.1182/blood-2009-07-233031. PubMed PMID: 19965672; PubMed Central PMCID: PMC2845896.
- Schaiff WT, Hruska KA, Jr., McCourt DW, et al. HLA-DR associates with specific stress proteins and is retained in the endoplasmic reticulum in invariant chain negative cells. J Exp Med. 1992 Sep 1;176(3):657–666. PubMed PMID: 1512535; PubMed Central PMCID: PMC2119345.
- Mao C, Wang M, Luo B, et al. Targeted mutation of the mouse Grp94 gene disrupts development and perturbs endoplasmic reticulum stress signaling. PLoS One. 2010 May 26;5(5):e10852. PubMed PMID: 20520781; PubMed Central PMCID: PMC2877114.
- Chhabra S, Jain S, Wallace C, et al. High expression of endoplasmic reticulum chaperone grp94 is a novel molecular hallmark of malignant plasma cells in multiple myeloma. J Hematol Oncol. 2015 Jun 25;8:77. PubMed PMID: 26108343; PubMed Central PMCID: PMC4483199.
- Huang CY, Batzorig U, Cheng WL, et al. Glucose-regulated protein 94 mediates cancer progression via AKT and eNOS in hepatocellular carcinoma. Tumour Biol. 2016 Apr;37(4):4295–4304. PubMed PMID: 26493996.
- Crowley VM, Huard DJE, Lieberman RL, et al. Second generation Grp94-Selective inhibitors provide opportunities for the inhibition of metastatic cancer. Chemistry. 2017 Nov 7;23(62):15775–15782. PubMed PMID: 28857290.
- Laberge RM, Sun Y, Orjalo AV, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015 Aug;17(8):1049–1061. PubMed PMID: 26147250; PubMed Central PMCID: PMCPMC4691706.
- Herranz N, Gallage S, Mellone M, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015 Sep;17(9):1205–1217. PubMed PMID: 26280535; PubMed Central PMCID: PMCPMC4589897.
- Ovadya Y, Krizhanovsky V. Strategies targeting cellular senescence. J Clin Invest. 2018 Apr 2;128(4):1247–1254. PubMed PMID: 29608140; PubMed Central PMCID: PMC5873866.
- Schafer MJ, White TA, Iijima K, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017 Feb 23;8:14532. PubMed PMID: 28230051; PubMed Central PMCID: PMC5331226.
- Jeon OH, Kim C, Laberge RM, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017 Apr 24 PubMed PMID: 28436958. DOI:10.1038/nm.4324.
- Samaraweera L, Adomako A, Rodriguez-Gabin A, et al. A novel indication for panobinostat as a senolytic drug in NSCLC and HNSCC. Sci Rep. 2017 May 15;7(1):1900. PubMed PMID: 28507307; PubMed Central PMCID: PMC5432488.