
ABSTRACT
The tumor suppressor protein p53 is central to the cellular stress response and may be a predictive biomarker for cancer treatments. Upon stress, wildtype p53 accumulates in the nucleus where it enforces cellular responses, including cell cycle arrest and cell death. p53 is so dominant in its effects, that p53 enforcement – or – restoration therapy is being studied for anti-cancer therapy. Two mechanistically distinct small molecules that act via p53 are the selective inhibitor of nuclear export, selinexor, and MDM2 inhibitor, nutlin-3a. Here, individual cells are studied to define cell cycle response signatures, which captures the variability of responses and includes the impact of loss of p53 expression on cell fates. The individual responses are then used to build the population level response. Matched cell lines with and without p53 expression indicate that while loss-of-function results in altered cell cycle signatures to selinexor treatment, it does not diminish overall cell loss. On the contrary, response to single-agent nutlin-3a shows a strong p53-dependence. Upon treatment with both selinexor and nutlin-3a there are combination effects in at least some cell lines – even when p53 is absent. Collectively, the findings indicate that p53 does act downstream of selinexor and nutlin-3a, and that p53 expression is dispensable for selinexor to cause cell death, but nutlin-3a response is more p53-dependent. Thus, TP53 disruption and lack of expression may not predict poor cell response to selinexor, and selinexor’s mechanism of action potentially provides for strong efficacy regardless of p53 function.
Introduction
The karyopherin-β protein, exportin-1 (XPO-1) is a key regulator of the nuclear export of proteins that contain the leucine-based nuclear export signal motif LxxLxL [Citation1]. Cargo proteins include cell survival and death regulators, for example NFκb/RelA and survivin, and key tumor suppressor proteins such as p53, p73, FOXO3a, pRB, p21CIP1, and p27KIP [Citation2–Citation4]. p53 is a central regulator of cellular responses to various stress stimuli. When intact, p53 signaling exerts dominant effects on cell cycle arrest, apoptosis, and senescence from within the nucleus where it acts to transactivate gene targets that enforce these cell fates. The p53 gene (TP53) is the most frequently mutated tumor suppressor across all cancers, and when its normal function is lost, it enables tumorigenesis and cancer progression and affects the response of cells to different anti-cancer therapeutics [Citation5–Citation7]. Because of the common importance of TP53 in cancers, it is vital to understand how the response and fate of cancer cells to different therapies are linked to TP53 status.
p53 restoration or functional enforcement therapy is an emerging anti-cancer approach. Multiple methods are being explored to test its utility, including the nuclear entrapment of p53 using small molecules that prevent its export via binding and blocking the function of XPO-1, termed selective inhibitors of nuclear export (SINE) [Citation8–Citation11]. Additionally, there are small molecules that block the ubiquitination of p53 via inhibition of the E3-ubiquitin ligase MDM2 (HDM2), namely nutlin-3a [Citation12]. Nuclear accumulation of p53, cell cycle effects, apoptosis, and senescent-like growth arrest are noted after treatment of cells with selinexor [Citation13–Citation15] or nutlin-3a [Citation12,Citation16], consistent with the involvement of p53. Some studies indicate cancer cells with mutated TP53 show response to treatment with SINE, and others report that the response shows some p53-dependence [Citation14,Citation15,Citation17]. The cell models vary among these studies and inherent differences in stress signaling, cell cycle checkpoints, and apoptotic pathways may account for overall responses. Treatment of cells with nutlin-3a shows strong p53-dependent responses that include cell cycle arrest and apoptosis [Citation12,Citation18,Citation19] and there is a growing body of literature showing p53-independent effects [Citation20–Citation22].
Here, we investigate cell cycle responses and cell fates after treatment with selinexor and nutlin-3a. Multiple p53 expression-matched cell lines are used, including two that express fluorescent ubiquitin-based cell cycle indicators, FUCCI [Citation23]. This systematic approach is used to define how loss of p53 expression impacts the cell cycle response and cell fates after treatment and to establish the collective cellular responses, or phenomic signature. The SINE, selinexor (KPT-330) is progressing through several clinical trials for blood and solid cancers [Citation10,Citation24–Citation28] and is used here, along with nutlin-3a. When p53 is expressed, both selinexor and nutlin-3a show strong cell cycle effects and at least some cell death. When p53 expression is lost, the cell cycle effects shift toward more proliferation initially, but selinexor remains potently effective at killing cells, while nutlin-3a appears less effective. The data support that selinexor may remain effective in the treatment of cancers regardless of TP53 function.
Results
Selinexor results in potent anti-cancer effects regardless of p53 expression
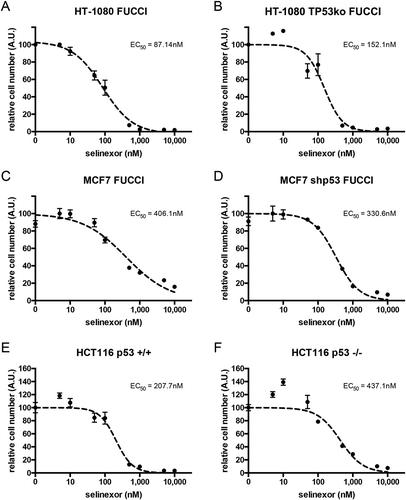
Three sets of paired cell lines with or without TP53 expression were treated with increasing concentrations of selinexor and ATP was measured after 72 hours as a surrogate for cell viability (Figure 1). HT-1080, MCF7, and HCT116 cells are TP53 wildtype [Citation29–Citation33]. For TP53 disrupted HT-1080, CRISPR/Cas9 was used and a clone was established (see Materials and Methods); HT-1080 TP53ko (Figs. S1 versus S2 and Fig. S3). For MCF7 an established clone with stable, high-level knock-down of p53 via small hairpin RNA is used; MCF7 shp53 [Citation34,Citation35] (Figs. S4 versus S5 and Fig. S3). For HT-1080 and MCF7, the absence of p53 expression is substantiated further by the lack of strong p21CIP1 induction after treatments (Fig. S3). For HCT116, p53 -/- cells are via targeted homologous recombination; HCT116 p53-/- [Citation36]. All cell lines show a dose-dependent response to selinexor. The effective concentration for 50% cell viability (EC50) for selinexor in wildtype HT-1080 is calculated as 87.1nM and is 152.1nM in TP53-disrupted cells (). For MCF7 and MCF7 shp53, the EC50 values are 406.1nM and 330.6nM, respectively (), and for HCT116 +/+ and HCT116 -/-, the values are 207.7nM and 437.1nM ().
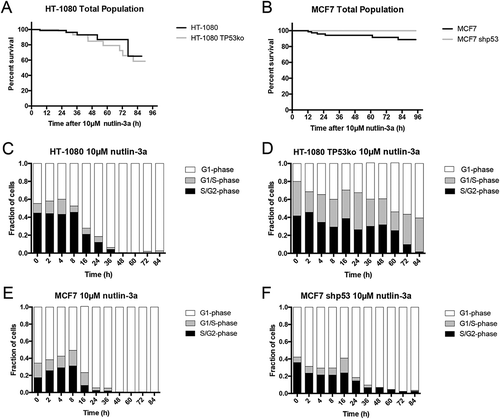
Figure 1. Loss of p53 expression does not prevent cell loss after treatment with selinexor. Three p53 expression-matched cell lines were treated with a titration of selinexor from 1nM to 10μM for 72 hours and the relative remaining cell population was subjected to quantification of ATP. (A, B) Wildtype HT-1080 cells have a calculated EC50 value of 87.1nM, matched HT-1080 cells without p53 expression have an EC50 of 152.1nM. (C, D) Wildtype MCF7 cells have a calculated EC50 value of 406.1nM, matched MCF7 cells without p53 expression have an EC50 of 330.6nM. (E, F) Wildtype HCT116 cells have a calculated EC50 value of 207.7nM, matched HCT116 cells without p53 expression have an EC50 of 437.1nM. Note, all cell lines show a strong response and are mostly lost at 1μM, except wildtype MCF7, which show some survival (C).
Cells without p53 die faster after treatment with selinexor
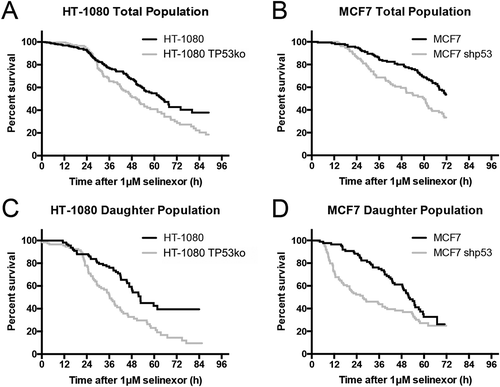
To follow cell loss directly over time we used long-term time-lapse microscopy and longitudinal tracking after treatment with 1 μM selinexor; 1 μM provides nearly maximal effects (), has been used extensively in vitro [Citation8,Citation13,Citation17], and is relevant in patients [Citation24,Citation37]. TP53-matched HT-1080 and MCF7 cell lines expressing FUCCI are used; HCT116 FUCCI cells lines could not be obtained due to poor degradation of the G1-phase indicator peptide, mKO2-hCdt1(30/120). The FUCCI system was validated previously in HT-1080 and MCF7 cells by time-lapse microscopy showing that both the G1- and S/G2/M-phase (mAG-hGem(1/110) probes accumulate and degrade properly throughout the cell cycle [Citation13,Citation38]. For both HT-1080 cell lines there is little change in survival until approximately 24 hours, followed by a precipitous decrease, with HT-1080 TP53ko (grey line) reaching 18% survival at 90 hours compared to 37% for HT-1080 (black line) (Figure 2(a)). HT-1080 wildtype cell loss is less rapid than cells without p53, particularly between 24–48 hours, and again at later times after 70 hours. Matched MCF7 cell lines are like HT-1080 in that there is initially a delay, followed by a decrease in survival where more cells lacking p53 (grey line) are lost faster than wildtype cells (black line); approximately 33% remaining at 72 hours versus 53% (). Direct observation demonstrates that, as published previously, some treated HT-1080 wildtype cells remain in interphase after treatment and die, while others first progress through cell division, and then die or arrest in the next cell cycle [Citation13]. To understand the population response further, the daughter cell population from some initial cell divisions was analyzed. Survival curves normalized to the time of cell division (time 0) show that after an initial delay period, more HT-1080 without p53 are lost faster than wildtype cells; approximately 10% survival versus 38% (). MCF7 matched cell lines show a similar result, that more cells lacking p53 are lost at earlier time-points, but at 72 hours both MCF7 cell lines show approximately 25% survival (). Cell cycle-associated cell fates occur after selinexor treatment in wildtype HT-1080 cells [Citation13]. Because p53 is a central regulator of cell cycle arrest and cell death and accumulates in the nucleus after selinexor treatment, we next asked how response is altered when p53 is removed.
Figure 2. Single cell longitudinal tracking of selinexor response indicates faster and greater cell loss without p53 expression. (A, B) Individual matched HT-1080 and MCF7 cells were tracked and population survival curves were plotted. After an initial delay, cells without p53 expression (black lines) are lost faster than wildtype cells (grey lines). Overall cell loss is greater in cells without p53; HT-1080 40%, and HT-1080 TP53ko 20% survival – and – MCF7 50%, and MCF7 shp53 30% survival. (C, D) Daughter cell populations were parsed out to document any sensitivity. For both HT-1080 and MCF7, daughter cells with p53 expression (grey lines) are lost somewhat faster than the respective total population (A, B) and daughters lacking p53 (black lines) show faster and more extensive cell loss. (A, B) >150 cells tracked for each cell line. (C, D) >70 daughter cells tracked for each cell line.
Loss of p53 expression results in changes in cell cycle distribution after selinexor treatment
Matched HT-1080 and MCF7 cell lines expressing FUCCI are used to monitor cell cycle state over time after treatment with selinexor. Mock-treated wildtype and HT-1080 TP53ko cells show no trend and little change in FUCCI distribution until the cultures become dense after 24 hours of growth (Figure 3(a,b)), Videos S1 and S2, Fig. S6). Analyses of single selinexor-treated cells show that as cells are lost, wildtype HT-1080 accumulate strongly in G1-phase (, Video S3, Fig. S7A), while HT-1080 without TP53 expression instead show an expansion of the G1/S-phase (grey) between 2–16 hours after treatment, that is lost concomitant with an increase in the S/G2-phase (black) population after 24 hours (, Video S4, Fig. S7B). At 84 hours, wildtype cells are >80% G1-phase, compared to approximately 25% for HT-1080 without p53 ( versus D). This indicates that in contrast to selinexor treated HT-1080 wildtype cells, which most often arrest or die in G1-phase [Citation13], HT-1080 cells without p53 either progress slowly or arrest in G1/S- or S/G2-phase and eventually die, accounting for the predominance of yellow and green FUCCI cells over time as the total population is lost ( ).
Figure 3. Cell cycle signatures after treatment with selinexor indicate p53-dependent G1-phase arrest. Cell cycle distribution was monitored over time in the p53 matched cell lines using the FUCCI system. (A, B) Mock (0.1% DMSO) treated HT-1080 and HT-1080 TP53ko cell lines show no obvious change in cell cycle distribution over time until the cultures become highly dense (Videos S1 and S2), then the populations become somewhat enriched in G1-phase cells (white). (C, D) After treatment with selinexor, wildtype HT-1080 cells persistently accumulate to 80% G1-phase (white) and lose G1/S- (grey) and S/G2-phase (black) cells as the overall cell population is lost (Video S3), but HT-1080 TP53ko cells are only 20–25% G1-phase and instead show an accumulation of G1/S- and S/G2-phase cells as the overall cell population is lost (Video S4). (E, F) Mock (0.1% DMSO) treated MCF7 and MCF7 shp53 cell lines show no obvious change in cell cycle distribution over time until the cultures become highly dense after 24 hours, at which point the populations are enriched for G1-phase cells. (G, H) After treatment with selinexor, wildtype MCF7 cells persistently accumulate to >95% G1-phase and lose G1/S- and S/G2-phase cells as the overall cell population is lost (Video S7). MCF7 shp53 cells also accumulate to >95% G1-phase G1-phase as the overall population is lost, and this is after an initial increase in G1/S- and S/G2-phase up to 16 hours (Video S8). >120 cells tracked for each condition and time point.
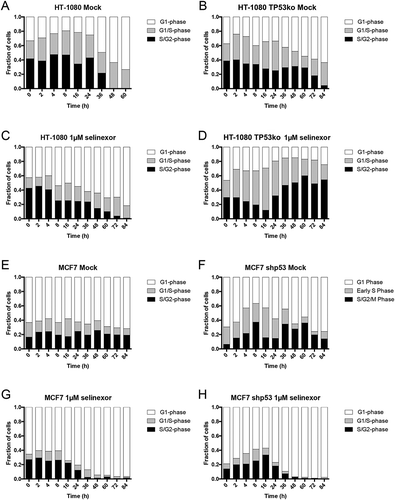
Mock-treated MCF7 cell lines show no trend in FUCCI distribution (, Videos S5 and S6, Fig. S8). After selinexor treatment, wildtype MCF7 cells accumulate to > 90% G1-phase after 36 hours, and they show a steady decrease in G1/S- and S/G2-phase cells during this time, which are nearly absent after 48 hours (, Video S7, Fig. S9A). MCF7 cells without p53 expression also accumulate to >90% G1-phase after 36 hours (, Video S8, Fig. S9B). In MCF7 without p53 the G1/S- and S/G2-phase populations increase for the first 16 hours, before decreasing and becoming nearly absent after 36 hours (). MCF7 and HT-1080 matched lines show similar FUCCI trends in response to selinexor treatment. Wildtype cells tend to accumulate in G1-phase arrest, while cells with disrupted p53 seem to maintain some proliferation, even while cells are lost (). Taken together with the cell survival data (), MCF7 wildtype cells treated in G1-phase may show a strong and immediate arrest response, and cells treated in S/G2-phase may first progress through cell division, like HT-1080 cells [Citation13], before they die predominantly in G1-phase (). MCF7 cells lacking p53 may also respond like wildtype MCF7, but at least some cells treated in G1-phase progress to cell division, explaining the initial increase in the G1/S- and S/G2-phase population, but unlike HT-1080 TP53ko, these cells either arrest or die in the next G1-phase ( versus D, Videos S3, S4 and S7, S8).
Loss of TP53 expression shifts selinexor cell fate signatures toward cell death
The fates of individual cells were monitored longitudinally in each matched cell line population. For both HT-1080 and MCF7, loss of p53 results in some increase of cells progressing to cell death (Figures 4(a) and ) and fewer cells that survive to the end of the time-lapse, which we term “arrest” (). Each matched pair shows a similar fraction of the total population that divides after treatment, termed “division”. Cell fates are scored from the time of treatment with selinexor and are classified as parental (single cells at the time of treatment) or daughter (cells born from cell divisions after treatment). Loss of p53 expression in parental MCF7 cells results in increased cell divisions and decreased death (). Nearly 30% of parental MCF7 cells arrest after treatment, but less than 1.0% arrest when p53 is absent (); we note in parental HT-1080 that effects of p53 loss on cell responses are less significant than in MCF7 cells. When tracking the fate of daughter cells, loss of p53 in both HT-1080 and MCF7 results in increased cell death and less arrest at the end of the time-lapse. No daughter cell in any of the cell lines was observed to continue to a second cell division (). Related, we immunoblotted and performed immunofluorescence staining for the apoptosis indicator cleaved PARP in the HT-1080 and MCF7 cell line pairs and immunoblotted for the anti-apoptotic protein MCL1. Corroborating the single cell tracking data, cleaved PARP is more increased after 48 hours of treatment in cell lines without p53 while MCL1 levels are less before treatment and are more degraded after treatment (Fig. S10).
Figure 4. Cell fate distribution after treatment with selinexor changes when p53 expression is lost, but cells remain death sensitive. (A-C) Cell fates for the total, parental, and daughter cell populations for HT-1080 and MCF7 p53-matched cell lines. Cell fates were classified into death (black), arrest (grey, cell remains at end of time-lapse), or division (white). For the total population, cells without p53 expression show increased cell death, with a corresponding decrease in arrest. When the populations are parsed into parental and daughter cells, there is a propensity for the daughters to die, with 60% or more being lost, and the remained in an arrested state; no daughter cells progress to a second cell division. (D-F) Cell cycle-associated fates of parental cells were tracked. (D) TP53 wildtype cells show predominantly death and arrest if treated in G1-phase, that upon loss of p53 results in increased cell divisions. (E) When parental HT-1080 cells are treated in G1/S-phase the percent of cells that progress to cell division increases for wildtype cells, and remains unchanged for HT-1080 TP53ko. Essentially all MCF7 cells treated in G1/S-phase progress to cell division. (F) For S/G2-phase cells, more than 90% progress to cell division, regardless of p53 expression status. (G-I) Cell cycle-associated cell fates of the daughter cells were tracked. (G) TP53 wildtype daughter cells resulting from a parent cell treated in G1-phase show 80% or greater arrest, whereas daughter cells without p53 expression show at least 70% cell death. (H) When parental TP53 wildtype HT-1080 or MCF7 cells are treated in G1/S-phase, any resulting daughter cells show increased cell death compared to treatment in G1-phase, where arrest is most common in the daughter cells, whereas daughter cells without p53 expression continue to show strong cell death. (I) All daughter cells resulting from a parent treated in S/G2-phase show 60% or greater cell death and 40% or more arrest. >150 cells tracked for each cell line.
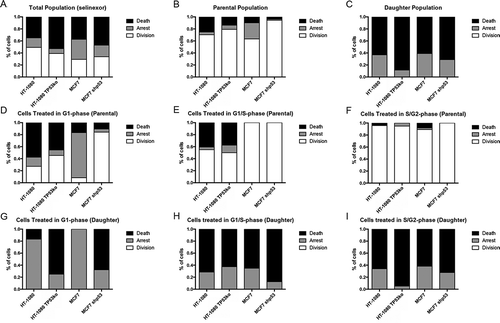
Cell fates upon selinexor treatment were further parsed by cell-cycle stage at the moment of treatment. Parental wildtype HT-1080 cells treated in G1-phase (white) show approximately 60% death, 15% arrest in G1-phase, and 25% progress to cell division. When p53 is lost in HT-1080, fewer parental cells treated in G1-phase show death and arrest, and instead nearly 50% progress to cell division (). For the parental MCF7 cell population treated in G1-phase () the shift in cell fate distribution is dramatic upon p53 loss. Wildtype parental cells show approximately 75% arrest when treated in G1-phase, but only 5% arrest when p53 is absent (). Instead of arresting, greater than 80% of MCF7 without p53 expression initially progress to cell division after treatment with selinexor. Parental HT-1080 and HT-1080 without p53 that are treated in G1/S-phase (grey) show a similar cell fate distribution to each other, with approximately 50% of these cells progressing to cell division; the MCF7 cell lines show 100% progression to cell division (). When cells are treated with selinexor in S/G2-phase (black), nearly all progress to cell division regardless of p53 expression status (), consistent with that observed previously for wildtype HT-1080 [Citation13].
The fate of all daughter cells as a function of cell cycle stage when treated can be tracked and correlated with p53 expression status similarly to the parental cells. Cell death of daughter cells from a parent was treated in G1-phase is > 4-fold higher in cells lacking TP53 expression, whereas daughter cells with p53 expression more often arrest; no cells progress to another division (). We published previously that daughter cells of HT-1080 treated with selinexor in G1-S- or S/G2-phase either die or arrest [Citation13]. We find here that daughter cells from a parent treated in G1/S- and S/G2-phase progress to cell death or arrest regardless of p53 expression ().
Inhibition of MDM2 by nutlin-3a results in strong cell cycle arrest but less death compared to inhibition of XPO-1
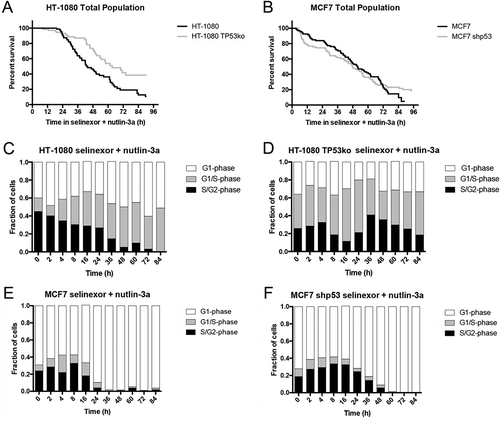
Cell fate signatures upon inhibition of XPO-1 indicate that when functional p53 is expressed it strongly influences cell cycle arrest, however, it is none-the-less dispensable for cell death and for achieving a potent population response (, , and Videos S3, S4 and S7, S8). Another small molecule whose molecular action is proposed to work through enforcing p53 is nutlin-3a. Nutlin-3a binds to MDM2 and prevents it from ubiquitylating substrate proteins, particularly p53, effectively resulting in its stabilization, nuclear accumulation, and subsequent transactivation of p53 gene targets [Citation18,Citation39]. When wildtype HT-1080 and MCF7 cells are treated with 10μM nutlin-3a, cell loss is marginal compared to selinexor treatment (Figure 5(a,b) versus ), and G1-phase arrest is favored, particularly in MCF7 cells (, Fig. S5A, Videos S9 and S10). When p53 expression is lost in HT-1080, overall survival appears similar to wildtype (, Fig. S11A) and in MCF7 shp53 there is little if any cell loss ( and Fig. S11A). For HT-1080 TP53ko, rather than the strong G1-phase arrest found in wildtype HT-1080, there is continued proliferation as observed via longitudinal tracking of FUCCI expressing cells ( versus D, Video S11). Interestingly, MCF7 shp53 cells remain sensitive to arrest in G1-phase after treatment with nutlin-3a, like wildtype MCF7 ( versus F; Video S12), although they do not die (, Fig. S11A).
Figure 5. Treatment with nutlin-3a favors G1-phase accumulation regardless of p53 expression. (A, B) Cell survival curves indicate treatment with nutlin-3a results in 35–40% cell loss in HT-1080 cells regardless of p53 expression, and approximately 10% loss in wildtype MCF7 cells, and no loss in MCF7 shp53 cells (black line = wildtype, grey line = p53 absent). (C-F) FUCCI status over time after nutlin-3a treatment indicates a trend toward G1-phase (white) arrest for all cell lines. (C) Wildtype HT-1080 cells accumulate to approximately 80% G1-phase 24 hours after treatment, and are nearly entirely G1-phase after that; this is sooner and stronger than after treatment with selinexor (, Video S9). (D) HT-1080 TP53ko show slower accumulation in G1-phase than wildtype cells, and reach approximately 60% (Video S10). (E, F) MCF7 and MCF7 shp53 cells both show approximately 70% G1-phase 24 hours after treatment, and are nearly entirely G1-phase after that; this is like treatment with selinexor but with different kinetics at early time points post-treatment (, Videos S11 and S12). ≥100 cells tracked for each condition and time point.
Longitudinal tracking was used to parse out cell populations to quantify cell fates and observe whether there are cell cycle-associated responses after treatment with nutlin-3a. For HT-1080 cells, p53 expression does not significantly affect the fate of the parent cell population, no strong cell cycle-associated fates are observed, and >90% of cells in the total population and parent cells progress to cell division after treatment with nutlin-3a before arresting (Fig. S11A, B, D-F). Death of HT-1080 daughter cells remained approximately 20% regardless of p53 expression status (Fig. S11C). For MCF7 cells, p53 expression more strongly affects cell fates than for HT-1080. Wildtype parental MCF7 arrest strongly regardless of what cell cycle phase they are treated in and upon loss of p53 expression the treated cells more frequently progression to division (Fig. S11D-F). For wildtype MCF7 there are no daughter cells from parents treated in G1- and G1/S-phase (Fig. S11G, H) and daughters arising from parents treated in S/G2-phase show approximately 70% death and 30% arrest (Fig. S11G-I). In MCF7 cells without p53, daughter cells tend to arrest regardless of the cell cycle phase upon treatment of their parent with a small population that progresses to another cell division (Fig. S11G-I). Nearly all HT-1080 daughter cells resulting from a parent cell treated in G1-phase progress to cell death, compared to less than 5% for daughter cells resulting from a parent treated in G1/S- or S/G2phase (Fig. S11G-I). HT-1080 TO53ko daughter cells from a parent treated in G1-phase show approximately 25% cell death, greatly decreased than this population in wildtype HT-1080, indicating a role for p53 that is consistent with nutlin-3a treatment. Interestingly, unlike wildtype HT-1080 cells, survival remains unchanged for HT-1080 TP53ko daughter cells regardless of cell cycle phase of the parent upon treatment (Fig. S11G-I), suggesting this population dies independently of p53 and regardless of cell cycle stage when the parent cell is treated.
Co-inhibition of XPO1 and MDM2 alters cell fate signatures and increases cell death
Selinexor and nutlin-3a treatment each result in nuclear accumulation of p53 in wildtype cells and can work through p53-dependent effects, yet the cell fate and cell cycle signatures are distinct ( versus ; versus FUCCI distributions; versus Fig. S11). Chemotherapy agents that are mechanistically distinct but that each can work via p53 induction may effectively combine to reduce cell survival (e.g. ref [Citation40]), and both selinexor and nutlin-3a show some effects on cells without p53 expression (e.g. , , and ). HT-1080 and MCF7 wildtype, and matched cell lines lacking p53 expression, were treated with a combination of selinexor and nutlin-3a. Longitudinal tracking reveals strong cell death occurs in all cell lines (, Fig. S12A, Videos S13-16). We note that for p53 wildtype cell lines, cell loss is accelerated with the drug combination compared to either drug alone ( and versus ). As for the single drug treatments, the entire cell populations were parsed into parental and daughter cell populations. HT-1080 and HT-1080 TP53ko parent cell survival decreases slightly after combination treatment (, Figs. S11B, 6B), and all daughter cells show 80% or greater cell death, which is increased over single drug treatments for all but HT-1080 TP53ko daughters (Fig. S12C). MCF7 show a significant increase in the death of both parent and daughter cells ( versus Fig. S12B, C).
Figure 6. Selinexor combined with nutlin-3a alters cell survival and cell cycle signatures compared to treatment with single agents. (A, B) Cell survival curves indicate that selinexor and nutlin-3a combine to decrease cell survival except in HT-1080 TP53ko cells (A, grey line), which show a small decrease in cell loss compared to with selinexor treatment alone (). (C, D) HT-1080 and HT-1080 TP53ko show approximately 50% and 30% G1-phase (white), considerably less than after treatment with single agents, and a correspondingly increased G1/S- (grey) and S/G2-phase (black) populations (compare to and , Videos S13 and S14). (E, F) Different than HT-1080 and HT-1080 TP53ko cells, MCF7 and MCF7 shp53 accumulate to nearly 100% G1-phase as they die. The G1-phase arrest appears slightly greater for wildtype MCF7 cells after treatment for 24 hours compared to single agents and to MCF7 shp53 cells (see and for comparison, Videos S15 and S16). 100 cells tracked for HT-1080, HT-1080 TP53ko, and MCF7 shp53, 80 cells tracked for MCF7.
The cell cycle status of combination treated cell lines was measured over time. HT-1080 wildtype cells, which die rapidly after combination treatment, display a unique FUCCI signature compared to the same cells treated singly with selinexor or nutlin-3a ( versus ). After single drug treatments, the HT-1080 cell population accumulates in a G1-phase state, and while selinexor treatment results in cell loss, nutlin-3a does not (, and and ). The HT-1080 cell population at later time-points after combination treatment shows comparatively enriched G1/S- and S/G2-phase cells, indicating that rather than progressing to cell division and arresting/dying in the subsequent G1-phase as occurs after single drug treatments ( and Fig. S11), these cells are arresting or slowly progressing and more frequently dying, which is supported by time-lapse microscopy (, Video S13). Interestingly, HT-1080 TP53ko cells show high cell loss in the drug combination, with a FUCCI distribution over time similar to single agent selinexor, in agreement with the live-cell microscopy showing cell death occurs from different cell cycle stages as the cells proliferate (, Video S14). Wildtype MCF7 cells show strong G1-phase accumulation by 24 hours, like after treatment with selinexor or nutlin-3a alone and >90% cell loss (, and , Video S15). MCF7 shp53 cells also accumulate in a G1-phase state after combination treatment, like after treatment with selinexor or nutlin-3a, but cell survival decreases to approximately 20% , Video S16).
Cell cycle-associated cell fate signatures indicate that 80% of parental wildtype HT-1080 and MCF7 cells treated with the selinexor-nutlin-3a combination in G1-phase eventually die (Fig. S12D) while those treated in G1/S- or S/G2-phase most often first progress to cell division (Fig. S12E, F). Essentially all p53 wildtype cells born into the selinexor-nutlin-3a combination die regardless of which cell cycle stage they were treated in (Fig. S12E-I). When the selinexor-nutlin-3a combination is used on cell lines without p53 expression there is decreased cell death of parental cells compared to selinexor alone (Fig S12D-F versus ), but 80% or more of all daughter cells die (Fig. S12C). For all cell lines, daughter cells predominantly die regardless of which cell cycle stage their parent is treated in (Fig. S12G-I), suggesting there are compound stresses that can occur in the selinexor-nutlin-3a combination independent of p53 expression which can lead to a stronger overall response compared to single drug treatments.
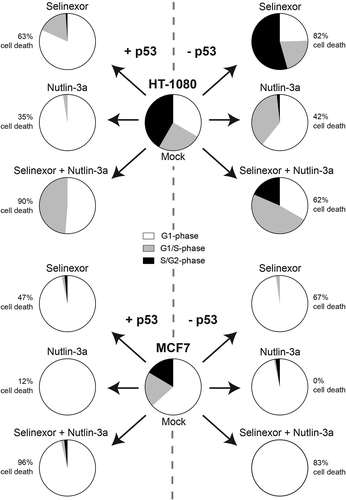
ATP concentration of cells treated with selinexor, nutlin-3a, or selinexor-nutlin-3a was measured over time. This surrogate assay for metabolically active cells shows that in wildtype HT-1080 and MCF7, each drug alone has strong activity (Fig. S13A, C). Selinexor also decreases ATP on the matched p53 deficient cell lines, but nutlin-3a shows a reduced effect. In agreement with cell survival via single cell longitudinal tracking, when selinexor and nutlin-3a are combined ATP levels notably decrease, indicating that p53 activity is dispensable for these two drug mechanisms to act together on cells to decrease growth and/or kill cells (, Fig. S13). Staurosporine is used as a potent cell death inducer in these experiments and the CDK4/6 inhibitor, PD-0332991 is used to block cell growth; some level of cell loss occurs at late time-points after PD-0332991 treatment. The cell viability measure supports the longitudinal tracking that in contrast to nutlin-3a, selinexor strongly reduces cell survival regardless of p53 expression, and that the selinexor-nutlin-3a combination show effects in the absence of p53. summarizes the findings here for selinexor and nutlin-3a in wildtype and p53-deficient HT-1080 and MCF7 cell models. Selinexor and nutlin-3a treatment engage p53 function if present, causing strong cell cycle effects and/or cell death. For selinexor treatment, when p53 is removed, longitudinal tracking and cell cycle signatures indicate greater continued proliferation and enhanced cell death response. For Nutlin-3a, p53 loss shows some effects on cell cycle signatures but little impact on cell death. The selinexor-nutlin-3a combination further decreases cell survival in all cell lines.
Discussion
XPO-1 inhibition by selinexor and MDM2 inhibition by nutlin-3a can each result in cell cycle arrest and cell death in cancer-derived cell lines. These cellular responses are consistent with the function of p53, which accumulates in the nucleus after treatment with these drugs (refs [Citation2, Citation3] and Figs. S1, 4 and S3). For selinexor, the requirement of wildtype p53 expression for anti-cancer activity, and if cell responses are altered when p53 is lost, is an important area to study as strong responses in p53-deficient cells could yield some cancer selectivity over normal cells. For nutlin-3a, studies indicate that functional p53 is important for cell cycle arrest and apoptosis, including in sarcomas [Citation12,Citation19,Citation41]. Using FUCCI with longitudinal tracking and ATP measurements, we show that cell fate signatures for either drug change when wildtype p53 expression is removed, and that with selinexor, cell death is increased in both rate and extent (, , ). The observation that each p53 matched cell line dies after treatment with selinexor indicates that SINE molecules may be effective as a single agent regardless of p53 expression or functionality. Alternatively, the efficacy of nutlin-3a in the cell lines tested here appears generally more dependent on wildtype p53, suggesting that for nutlin-3a to work maximally on cells as a single agent, normally functional p53 is important (, and Fig. S13).
Figure 7. Summary of the effects of loss of p53 expression on treatment with selinexor, nutlin-3a, and selinexor with nutlin-3a. For selinexor treatment alone, overall cell death is increased 20% in HT-1080 (top) and MCF7 (bottom) cells when p53 expression is absent. For HT-1080, p53 plays an important role in cell-cycle distribution after selinexor treatment; G1-phase (white), G1/S-phase (grey), S/G2-phase (black). For MCF7, cells accumulate in G1-phase regardless of treatment and p53 expression. Cell death is least in all cell lines for nutlin-3a treatment, and for MCF7 cell death is dependent on p53 expression. For selinexor combined with nutlin-3a treatment, cell death is increased in all cell lines compared to singe drug treatments, except for HT-1080 TP53ko.
TP53 mutational/functional status is frequently sought after as a predictive biomarker for response to anti-cancer therapy. We find here that selinexor treatment results in altered cell cycle responses and death kinetics in the absence of p53; these data extend off other studies showing that cell lines and a xenograft model with mutated p53 do respond to selinexor [Citation15,Citation17]. Together, the data challenge the idea that loss of p53 function, especially for treatments known to induce p53 stabilization, translates into poorer response, thereby limiting therapeutic potential. Nutlin-3a does show decreased cell cycle arrest and cell death in the absence of p53 (, and Fig. S13), suggesting that wildtype p53 status may have some predictive value for response as is suggested [Citation42,Citation43], although p53-independent effects are noted [Citation20,Citation21]. The established MCF7 shp53 cell line used is a high-level knockdown (Fig. S3). We note a low level of p21CIP1 is detectable in MCF7 shp53 cells that may be very modestly increased after treatment with the drugs. This is interesting as MCF7 cell fates strongly associate with G1-phase where p21CIP1 functions to arrest cells, whereas HT-1080 TP53ko cells show no detectable p21CIP1 and often progress to S/G2-phase and die rather than in G1-phase like HT-1080 wildtype cells. Further, it is possible that any cell cycle arrest and death that occurs in MCF7 shp53 cells after the treatments, including nutlin-3a alone, is via very low level induction of p53 or the p53-related protein p73, whose association with the E3 ubiquitin ligase is also blocked by nutlin-3a.
Over the duration of the treatments here there are surviving cells after selinexor or nutlin-3a, suggesting there could be a cell population in each case that is inherently death resistant or that executes pro-survival signaling responses differently from dying cells. This surviving population may be important in the context of long-term response. When selinexor and nutlin-3a are combined there are fewer survivors, especially in p53 wildtype cells where less than 10% remain (). In the case of wildtype cells, the two drugs could act independently to enforce p53 function, resulting in stronger cellular responses; combination treatment does show high level p53 induction (Fig. S3). Also, some evidence shows MDM2 (HDM2) is a cargo protein of XPO-1 [Citation44,Citation45], this may provide the potential for some effects when XPO-1 and MDM2 inhibitors are combined. Combination effects are less intuitive in cells without p53 expression, but may be possible. Without p53-dependent cell cycle arrest or senescence, which may provide a protective effect against drug action, continued proliferation in both drugs may provide combined effects from p53-independent stress mechanisms exerted by each drug ().
We note that loss of p53 expression increases the extent of overall cell death after treatment with selinexor similarly in HT-1080 and MCF7 cells, with approximately 20% greater population loss (). While cell death signaling is not the focus of this study, selinexor results in greater PARP cleavage in cells without TP53 expression (Fig. S10), consistent with the single cell longitudinal tracking and indicative of apoptotic cell death mechanisms. We also note the loss of MCL1, an anti-apoptotic Bcl2 family member after treatment with selinexor (Fig. S10). Overall, cell death in HT-1080 cells is greater than MCF7 cells after treatment with selinexor or nutlin-3a. This is likely due to inherent differences in cell death signaling in MCF7 cells due to loss of caspase-3 and strong function of XIAP [Citation46,Citation47]. For cells treated with selinexor, we posit that wildtype p53 exerts a dominant effect, contributing to cell cycle arrest in G1-phase and presumably cell death (). When p53 expression is removed, the cells show a decreased tendency to arrest, leading to lowered protection against cell death. Instead, the cells continue to proliferate in the presence of selinexor and accumulate stress, which may include extensive DNA damage [Citation48] that could combine with the action of other XPO-1 cargo proteins [Citation2–Citation4] and effect cell survival. TP53 wildtype cells present a mixture of potent growth arrest, mostly in G1-phase, and cell death in response to combined selinexor and nutlin-3a. Here, cell death is generally enhanced compared to treatments with single drugs, even in the absence of p53 expression, revealing that nutlin-3a can exert some effects in these cells. Thus, while nutlin-3a is best known to increase p53 levels to enforce cell response, it may still combine well with selinexor in some cancer lines without TP53 expression.
There are numerous TP53 mutations in cancers falling into different classes, some which destroy p53 transactivation of target genes, and others that appear to alter it. It will be interesting in future work to evaluate if cancer cells and tumors harboring different classes of TP53 mutations show unique cell response signatures and altered cell survival after treatment with selinexor, which ultimately could help identify those cancers that may respond the strongest to treatment with selinexor.
Materials and methods
Cell lines, plasmids, and TP53 disruption
HT-1080 (ATCC CCL-121) cell lines are grown in MEM with Earle’s salts (Corning; 10–010-CV), sodium pyruvate (Sigma), non-essential amino acids (Sigma), 1% penicillin/streptomycin (Sigma; P/S), and 10% FBS (Sigma). MCF7 (ATCC HTB-22) and MCF7 shp53 are grown in RPMI (Corning; 10–040-CV), 10% FBS, and 1% P/S. HCT116 p53 +/+ and HCT116 p53 -/- are grown in McCoys5a (Corning; 10–050-CV), 1% P/S, and 10% FBS.
HT-1080 FUCCI are as described previously [Citation13]. TP53 was disrupted in HT-1080 FUCCI using CRISPR/Cas9 to obtain the knockout, HT-1080 TP53ko FUCCI line (see below). MCF7 FUCCI and MCF7 shp53 FUCCI were created by infection of cells with lentiviral particles to express the mKO2-hCdt1(30/120) and mAG-hGem(1/110) fluorescent peptides. Single expressing clones were obtained by dilution cloning. The parental MCF7 shp53 cell line is from R. Agami (Netherlands Cancer Institute).
For disruption of p53 expression in HT-1080 FUCCI, a Cas9 expression plasmid, lentiCas9-Blast, and a validated targeting vector, lentiGUIDE-puro-P53 sgRNA (nucleotides ACTTCCTGAAAACAACGTTC) for the end of exon 2 of human TP53 [Citation49,Citation50] were purchased from the Functional Genomic Shared Resource (University of Colorado Cancer Center). Plasmid DNAs were purified (Qiagen), and Cas9 expression and targeting vectors were co-transfected into HT-1080 (FuGene VI, Promega). 1μg/ml puromycin (Gold Biotechnology) and 10μg/ml blasticidin (Gold Biotechnology) were added 24 hours after transfection and cells were maintained for 7 days. Single clones were obtained by successive rounds of dilution cloning. Loss of TP53 expression was determined initially by immunofluorescent screening for p53 signal after 16 hours treatment with 10μM etoposide (Selleckchem), a known inducer of p53. Clones were subjected to multiple functional tests for p53 induction: 1) 8 hours treatment with 1μM selinexor, 2) 8 hours treatment with 10μM nutlin-3a, and 3) 8 hours combined selinexor and nutlin-3a. Disrupted clones show no p53 by immunostaining and immunoblotting (HT1080 TP53ko); absence of p53 expression was confirmed throughout the duration of the project.
Microscopy and cell tracking analyses
Fixed cell, immunofluorescence microscopy and live cell microscopy was performed using an inverted Olympus IX81 epifluoresence microscope with Prior Lumen200 Pro metal halide lamp, Hamamatsu ORCA R2 CCD camera, motorized Prior ProScan III stage, and 20 × 0.70NA air, 40 × 0.75NA air, or 100 × 1.40NA oil objectives with optical filters for DAPI (Chroma), Alexa488/EGFP (Chroma), Alexa568/mCherry (Chroma) and Alexa647/Cy5 (Semrock) and SlideBook 6.0 software (3i). For live cells, a stage-top incubation chamber (InVivo Scientific) was used described previously (see refs [Citation13, Citation49]). Images were acquired every 10 minutes binned 2 × 2 to limit exposure times. Autofocusing was performed using a phase-contrast algorithm. Control conditions are included in each experiment to confirm normal growth. A Yokogawa CV1000 spinning disk confocal microscope with 488 and 561 nm lasers, Olympus 20 × 0.75NA objective, and Hamamatsu ImageEM X2 C9100-14 1024 × 1024 EM-CCD was also used.
For FUCCI status, two investigators scored the cells blindly at distinct time-points after treatment. Fluorescence intensity values in the red and green channels were measured and based on signal over background cells were scored as G1- (red), G1/S- (yellow), or S/G2-phase (green). Two investigators independently tracked all live cells. Each cell was tracked longitudinally and fates were defined as; 1) death, cell rounding accompanied with blebbing and cell fragmentation, 2) arrest, cells remain in interphase, and, 3) cell division, cell enters mitosis and completes division. All cells and conditions were imaged for at least 90 hours to capture the full response. Survival curves were plotted for each condition until the timepoint after which no additional cell loss was observed. All plots were generated using GraphPad Prism software. Cell numbers analyzed in each case are provided in the figure legends.
Antibodies, small molecules, immunofluorescence and immunoblotting
For immunofluorescence, mouse monoclonal p53 antibody (D01, Santa Cruz Biotechnology, https://www.scbt.com/scbt/product/p53-antibody-do-1) was used at 1:200 dilution and mouse monoclonal cleaved PARP (Asp214) (BD Pharmingen, 552,597) was used at 1:200 dilution. Donkey anti-mouse secondary antibodies conjugated with AlexaFluor 647 (LifeTech) were used at 1:500 dilution. Cells were grown on 22 × 22 millimeter #1.5 glass coverslips (VWR 48,366–227) and fixed in 3.7% formaldehyde (Mallincrokdt) in PBS (pH 7.4) for 20 minutes at room temperature, washed at least 3 times in PBS, permeablized in 0.5% Triton X100 (Sigma) in PBS for 5 minutes, washed at least 3 times in PBS, blocked in 4% BSA (Fisher, fraction V) in PBS for 60 minutes at room temperature, incubated in primary antibody diluted in 4% BSA/PBS for 60 minutes at room temperature, washed at least 3 times in PBS, incubated in secondary antibody diluted in 4% BSA/PBS for 60 minutes at room temperature, washed at least 3 times in PBS, and counterstained with 1μM DAPI (Sigma) in PBS for 5 minutes at room temperature, washed in distilled water, and mounted in ProLong Gold or ProLong Diamond antifade reagent (Invitrogen) on glass microscope slides (VWR, 16,004–422). The XPO-1 specific inhibitor selinexor (KPT-330) is from Karyopharm Therapeutics, Inc. (Newton, MA), dissolved at 10mM into anhydrous dimethyl sulfoxide (Hybrimax DMSO, Sigma) and used at 1μM. Nutlin-3a (Selleckchem) is dissolved at 10mM into DMSO and used at 10μM. PD-0332991 (Selleckchem) is dissolved at 10mM into DMSO and used at 10μM. Staurosporine (Sigma) is dissolved at 1mM in DMSO and used at 1μM.
For immunoblotting, cell lysate was obtained using RIPA lysis buffer from cells grown in 35mm dishes treated as indicated. Total protein was quantified using Bradford assay (ThermoFisher) and 10μg was subjected to SDS-PAGE using Invitrogen pre-cast Bis-Tris 4–12% gels and MES buffer. Lysate with LDS sample buffer (Invitrogen, NP0007) were separated at 150 volts and the proteins transferred using 100 milliamps via wet transfer to nitrocellulose membranes. Membranes were rinsed in TBS with 0.1% Tween-20 (TBST), blocked for 1 hour at room temperature in 5% non-fat dry milk in TBST. The same p53 antibody as for immunofluorescence was used at 1:1000 in 5% non-fat dry milk/TBST. CDKN1A/p21CIP1 mouse monoclonal antibody was from BD Pharmingen (#556,430) and used at 1:1000. PARP rabbit polyclonal antibody was from Cell Signaling (#9542) and used at 1:1000. MCL1 rabbit polyclonal antibody was from Santa Cruz Biotechnology (sc-819) and used at 1:1000. Cytoplasmic dynein mouse monoclonal antibody was from MilliporeSigma (MAB1618) and used at 1:1000. Primary antibodies were incubated overnight at 4 Celsius and washed off via three five minute incubations in TBST. Secondary sheep anti-mouse and donkey anti-rabbit HRP (GE Healthcare) were used at 1:2000 in milk/TBST for 1 hour at room temperature, washed in TBST, and protein was detected using ECL Western Lightening Plus (Perkin Elmer) exposed to x-ray film (Genemate).
ATP measurements and plotting
Cells (1000) were plated into 96 well white-walled plates (ThermoScientific) with glass or optical plastic bottoms, grown overnight, and treated as indicated. ATP luminescence (CellTiter-Glo 2.0, Promega) was read using a Biotek plate reader within 10 minutes of sample preparation. The luminescence signal at each time-point was normalized to control (0.1% DMSO) cells, which is defined as 100 to facilitate interpretation of effects on cell viability. All values were plotted using GraphPad Prism. EC50 concentrations were calculated by dose-response analysis using the Hill equation [Citation51].
Supplemental Material
Download Zip (26 MB)Acknowledgments
We acknowledge Sakaue-Sawano et al. for the FUCCI reporters via MTA. We thank Karyopharm Therapeutics, Inc. (Newton, MA) for selinexor and for their generous gift to the University of Colorado Boulder for the study of cancer biology. We also acknowledge Dongjoo Park in the Orth laboratory for discussions, Xinyi Fu for help with immunoblotting, and the Light Microscopy Facility at the University of Colorado Boulder, Porter B047A, B, and B049.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- la Cour T, Kiemer L, Molgaard A, et al. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng Des Sel. 2004;17:527–536.
- Senapedis WT, Baloglu E, Landesman Y. Clinical translation of nuclear export inhibitors in cancer. Semin Cancer Biol. 2014;27:74–86.
- Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2012;83:1021–1032.
- Cheng Y, Holloway MP, Nguyen K, et al. XPO1 (CRM1) inhibition represses STAT3 activation to drive a survivin-dependent oncogenic switch in triple-negative breast cancer. Mol Cancer Ther. 2014;13:675–686.
- Hill R, Rabb M, Madureira PA, et al. Gemcitabine-mediated tumour regression and p53-dependent gene expression: implications for colon and pancreatic cancer therapy. Cell Death Dis. 2013;4:e791.
- Gizatullin F, Yao Y, Kung V, et al. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res. 2006;66:7668–7677.
- Lukin DJ, Carvajal LA, Liu WJ, et al. p53 Promotes cell survival due to the reversibility of its cell-cycle checkpoints. Mol Cancer Res. 2015;13:16–28.
- Neggers JE, Vercruysse T, Jacquemyn M, et al. Identifying drug-target selectivity of small-molecule CRM1/XPO1 inhibitors by CRISPR/Cas9 genome editing. Chem Biol. 2015;22:107–116.
- Nakayama R, Zhang YX, Czaplinski JT, et al. Preclinical activity of selinexor, an inhibitor of XPO1, in sarcoma. Oncotarget. 2016;7:16581–16592.
- Razak ARA, Soerensen MM, Mahipal A, et al. First-in-class, first-in-human phase I trial of KPT-330, a selective inhibitor of nuclear export (SINE) in patients (pts) with advanced solid tumors. J Clin Oncol 2013;31:15_suppl: abstr 2505-2505.
- Lassen UN, Soerensen MM, Kung AL, et al. A phase 2 study on efficacy, safety and intratumoral pharmacokinetics of oral selinexor (KPT-330) in patients with recurrent glioblastoma (GBM). J Clin Oncol 2015;33:15_suppl: abstr 2044-2044.
- Vassilev LT. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle. 2004;3:419–421.
- Marcus JM, Burke RT, DeSisto JA, et al. Longitudinal tracking of single live cancer cells to understand cell cycle effects of the nuclear export inhibitor, selinexor. Scientific Reports. 2015;5:14391.
- Mendonca J, Sharma A, Kim HS, et al. Selective inhibitors of nuclear export (SINE) as novel therapeutics for prostate cancer. Oncotarget. 2014;5:6102–6112.
- Yoshimura M, Ishizawa J, Ruvolo V, et al. Induction of p53-mediated transcription and apoptosis by exportin-1 (XPO1) inhibition in mantle cell lymphoma. Cancer Sci. 2014;105:795–801.
- Purvis JE, Karhohs KW, Mock C, et al. p53 dynamics control cell fate. Science. 2012;336:1440–1444.
- Azmi AS, Aboukameel A, Bao B, et al. Selective inhibitors of nuclear export block pancreatic cancer cell proliferation and reduce tumor growth in mice. Gastroenterology. 2013;144:447–456.
- Allen MA, Andrysik Z, Dengler VL, et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. eLife. 2014;3:e02200.
- Feng FY, Zhang Y, Kothari V, et al. MDM2 Inhibition sensitizes prostate cancer cells to androgen ablation and radiotherapy in a p53-dependent manner. Neoplasia. 2016;18:213–222.
- Lau LM, Nugent JK, Zhao X, et al. HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73 function. Oncogene. 2008;27:997–1003.
- Carrillo AM, Hicks M, Khabele D, et al. Pharmacologically increasing Mdm2 inhibits DNA repair and cooperates with genotoxic agents to kill p53-Inactivated ovarian cancer cells. Mol Cancer Res. 2015;13:1197–1205.
- Conradt L, Henrich A, Wirth M, et al. Mdm2 inhibitors synergize with topoisomerase II inhibitors to induce p53-independent pancreatic cancer cell death. Int J Cancer. 2013;132:2248–2257.
- Sakaue-Sawano A, Kurokawa H, Morimura T, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–498.
- Garzon R, Savona M, Baz R, et al. A phase 1 clinical trial of single-agent selinexor in acute myeloid leukemia. Blood. 2017;129:3165–3174.
- Walker CJ, Oaks JJ, Santhanam R, et al. Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph+ leukemias. Blood. 2013;122:3034–3044.
- Chen C, Siegel D, Gutierrez M, et al. Safety and efficacy of selinexor in relapsed or refractory multiple myeloma and Waldenstrom’s macroglobulinemia. Blood. 2018;131:855–863.
- Gounder MM, Zer A, Tap WD, et al. Phase IB study of selinexor, a first-in-class inhibitor of nuclear export, in patients with advanced refractory bone or soft tissue sarcoma. J Clin Oncol. 2016;34:3166–3174.
- Abdul Razak AR, Mau-Soerensen M, Gabrail NY, et al. First-in-Class, First-in-Human phase I study of selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J Clin Oncol. 2016;34:4142–4150.
- Moran DM, Maki CG. Nutlin-3a induces cytoskeletal rearrangement and inhibits the migration and invasion capacity of p53 wild-type cancer cells. Mol Cancer Ther. 2010;9:895–905.
- Wasielewski M, Elstrodt F, Klijn JG, et al. Thirteen new p53 gene mutants identified among 41 human breast cancer cell lines. Breast Cancer Res Treat. 2006;99:97–101.
- Liu Y, Bodmer WF. Analysis of P53 mutations and their expression in 56 colorectal cancer cell lines. Proc Natl Acad Sci U S A. 2006;103:976–981.
- Chang BD, Xuan Y, Broude EV, et al. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999;18:4808–4818.
- Clewell RA, Sun B, Adeleye Y, et al. Profiling dose-dependent activation of p53-mediated signaling pathways by chemicals with distinct mechanisms of DNA damage. Toxicol Sci. 2014;142:56–73.
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553.
- Loewer A, Batchelor E, Gaglia G, et al. Basal dynamics of p53 reveal transcriptionally attenuated pulses in cycling cells. Cell. 2010;142:89–100.
- Bunz F, Dutriaux A, Lengauer C, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501.
- Lapalombella R, Sun Q, Williams K, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012;120:4621–4634.
- Yano S, Li S, Han Q, et al. Selective methioninase-induced trap of cancer cells in S/G2 phase visualized by FUCCI imaging confers chemosensitivity. Oncotarget. 2014;5:8729–8736.
- Bozzi F, Conca E, Laurini E, et al. In vitro and in silico studies of MDM2/MDMX isoforms predict Nutlin-3A sensitivity in well/de-differentiated liposarcomas. Lab Invest. 2013;93:1232–1240.
- Paek AL, Liu JC, Loewer A, et al. Cell-to-cell variation in p53 dynamics leads to fractional killing. Cell. 2016;165:631–642.
- Van Maerken T, Rihani A, Van Goethem A, et al. Pharmacologic activation of wild-type p53 by nutlin therapy in childhood cancer. Cancer Lett. 2014;344:157–165.
- Henze J, Muhlenberg T, Simon S, et al. p53 modulation as a therapeutic strategy in gastrointestinal stromal tumors. PLoS One. 2012;7:e37776.
- Barone G, Tweddle DA, Shohet JM, et al. MDM2-p53 interaction in paediatric solid tumours: preclinical rationale, biomarkers and resistance. Current Drug Targets. 2014;15:114–123.
- Henderson BR, Eleftheriou A. A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Exp Cell Res. 2000;256:213–224.
- Lain S, Midgley C, Sparks A, et al. An inhibitor of nuclear export activates the p53 response and induces the localization of HDM2 and p53 to U1A-positive nuclear bodies associated with the PODs. Exp Cell Res. 1999;248:457–472.
- Yang XH, Sladek TL, Liu X, et al. Reconstitution of caspase 3 sensitizes MCF-7 breast cancer cells to doxorubicin- and etoposide-induced apoptosis. Cancer Res. 2001;61:348–354.
- Lin YF, Lai TC, Chang CK, et al. Targeting the XIAP/caspase-7 complex selectively kills caspase-3-deficient malignancies. J Clin Invest. 2013;123:3861–3875.
- Burke RT, Marcus JM, Orth JD. Inhibition of exportin-1 function results in rapid cell cycle-associated DNA damage in cancer cells. Oncotarget. 2017;8:39460–39475.
- Procopio MG, Laszlo C, Al Labban D, et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol. 2015;17:1193–1204.
- Nikkila J, Kumar R, Campbell J, et al. Elevated APOBEC3B expression drives a kataegic-like mutation signature and replication stress-related therapeutic vulnerabilities in p53-defective cells. Br J Cancer. 2017;117:113–123.
- Gadagkar SR, Call GB. Computational tools for fitting the Hill equation to dose-response curves. J Pharmacol Toxicol Methods. 2015;71:68–76.