
ABSTRACT
The anti-aging strategy is one of the main challenges of the modern biomedical science. The term “aging” covers organisms, cells, cellular organelles and their constituents. In general term, aging system admits the existence of nonfunctional structures which by some reasons have not been removed by a clearing system, e.g., through autophagy/mitophagy marking and destroying unwanted cells or mitochondria. This directly relates to the old kidney which normal functioning is critical for the viability of the organism. One of the main problems in biomedical studies is that in their majority, young organisms serve as a standard with further extrapolation on the aged system. However, some protective systems, which demonstrate their efficiency in young systems, lose their beneficial effect in aged organisms. It is true for ischemic preconditioning of the kidney, which is almost useless for an old kidney. The pharmacological intervention could correct the defects of the senile system provided that the complete understanding of all elements involved in aging will be achieved. We discuss critical elements which determine the difference between young and old phenotypes and give directions to prevent or cure lesions occurring in aged organs including kidney.
Abbreviations: AKI: acute kidney injury; I/R: ischemia/reperfusion; CR: caloric restriction; ROS: reactive oxygen species; RC: respiratory chain
KEYWORDS:
Introduction
Inevitable aging of the population, which is one of the natural consequences of increasing life expectancy in the 20th century, has become an important challenge for medicine in the 21st century. From 2000 to 2015, the number of people on our planet of age 60+ years increased by 48% from 607 to 901 million. By 2050, the total number of elderly is projected to double and reach approximately 2.1 billion people, which will account for over 20% of the population of the globe. The aging population is not the exclusive problem of the countries with developed economies. In general, the fractional increase of elderly in the general population can also be seen in countries with developing economies, albeit with lesser speed [Citation1]. Such a large fraction of older adults does not allow them to be categorically excluded from the workforce. Therefore, for medicine, the challenging problem is to maintain the health and longevity of older people so that they may continue to contribute to the economy. In turn, this poses a medical problem of offering effective treatment to cure old patients with age-related diseases.
One of the greatest problems in medicine is the treatment of acute kidney injury (AKI), which, according to the latest epidemiological data, occurs in every fifth hospitalized patient [Citation2]. As for many other diseases, the majority of patients with AKI are elderly. For example, an epidemiological study performed in a Calgary hospital demonstrated that the incidence of AKI in patients aged 18–49 years was 5 times lower than for the group aged 75+ years [Citation3]. In another large study examining 127,000 patients with AKI, the average age of ill people was 75.9 years, and only 10.7% of the patients were younger than 64 years [Citation4]. In contrast, more than 99% of experimental works on AKI have been utilizing animals of age just past puberty (6260 vs. 39 articles in Pubmed). This disparity between the age of real patients and the age of experimental animals may explain the fact that while numerous recent studies have demonstrated the effectiveness of particular drugs in rodent models of AKI, in a clinics, a renal replacement therapy introduced into wide practice in the 1950s remains the only practical and relatively effective treatment for AKI.
Interestingly, although studies examining the effect of aging on the kidney include more than 3000 articles, only 39 of these papers involved studies of AKI, and only 6 papers of these addressed the effectiveness (or ineffectiveness) of particular therapeutic approaches. Such a huge difference is likely related to the high cost and complexity of experiments in old animals. With great certainty, we can assume that research demonstrating an inability of therapeutic interventions to protect the kidneys of older animals has been underreported in the literature.
It is necessary to understand that the heterogeneous nature of AKI with regard to its effects on cells, subcellular structures and organisms, response to stress and the course of the disease complicates efforts to find an universal treatment, particularly for both young and old patients, and thus necessitates a personalized strategy to combat AKI pathologies. However, we assume that there is an universal propagation of the incidence of pathologies including aging. Sufficient data have been accumulated to prove that aging, and various pathologies are accompanied by accumulation of damaged and thereby detrimental biological structures (e.g., proteins, nucleic acids, lipids, organelles or cells) which begin to “coexist” with normal structures, thereby increasing the degree of heterogeneity of these structures. When the ratio of “bad” to “good” structures exceeds a certain threshold, a pathological phenotype characterizing a disease or aging appears. This aspect of heterogeneity is very important, and it will be addressed in some examples below. In general, we will focus our attention on the mechanism affording removal of damaged structures. Considering that preservation of mitochondrial structures and functions is essential to maintain a “young” phenotype and is fundamental for the protection of the entire organism, we will explore possible approaches to implement such protection. Firstly, we will discuss the importance of autophagy as the body’s primary mechanism for removing damaged macrostructures.
Autophagy
Autophagy is affected by AKI
Autophagy is an important adaptive mechanism to repair damage in various organs and tissues [Citation5] including the kidney. To date, the only available marker of autophagosomes in mammalian tissues is a microtubule-associated protein 1A/1B light chain 3B (commonly referred to as LC3). Its lipidated form, LC3 II, is bound to autophagosomal membrane [Citation6]. Increase of LC3 II level in renal tissue as well as the increased abundance of LC3 in tubular cell of LC3-GFP mice was shown in different models of AKI: cisplatin toxicity [Citation7,Citation8], kidney ischemia/reperfusion (I/R) [Citation9–Citation12], radio-contrast media-induced AKI [Citation13] and in septic AKI, caused either by the injection of LPS [Citation14,Citation15] or cecal ligation and perforation [Citation16].
However, augmented LC3 II level could reflect two distinct processes: enhanced autophagosome biogenesis on the one hand and retarded lysosomal digestion of autophagosome cargo on another hand. Two methods could be useful to address this question. First one is using tandem RFP-GFP-LC3 sensor capitalizing on pH difference between the acidic lysosome and the neutral autophagosome and the pH sensitivity differences exhibited by GFP (fluorescence is quenched in low pH environment) and RFP (stable in an acidic environment). The second monitors SQSTM1/p62 level. p62 serves as an adaptor between ubiquitin and LC3 II, therefore, playing a crucial role in delivering poly-ubiquitinated proteins to autophagosomes. However, its transcriptional regulation is independent of LC3, but its degradation occurs together with poly-ubiquitinated proteins as a result of autophagosomes fusion with lysosomes. As a result of lysosome dysfunction, both LC3 II and p62 destruction will be retarded yielding simultaneous increase in p62 and LC3 II levels [Citation17].
Several studies suggested that autophagosomal biogenesis rather than lysosomal insufficiency takes place during AKI. Indeed, after I/R of the kidney, the increase in LC3 II was not accompanied by an increase in p62 [Citation11]. An increase in lysosome abundance after kidney I/R was reported in another study [Citation18]. Ultimately, a comprehensive study utilizing RFP-GFP-LC3 mice and different lysosomal markers detected no signs of lysosomal dysfunction in renal I/R [Citation10].
Autophagy is a protective factor in AKI
Numerous data indicate that activation of autophagy is a protective mechanism. Most convincing is the data obtained from a variety of animal models, including post-ischemic, septic and cisplatin-induced AKI and hyperuremic AKI in knockout mice lacking autophagy-associated proteins, Atg5 or Atg7 in proximal tubules. Specifically, knockout of these proteins stops autophagosome formation [Citation19]. In these knockout animals, the severity of AKI was higher than in wild-type controls [Citation8,Citation9,Citation15,Citation20–Citation22]. Similar data were obtained after inhibition of autophagy. For example, chloroquine, an inhibitor of lysosomes, raised the severity of AKI [Citation15,Citation21,Citation23]. However, in doses of orders of magnitude smaller than used in other studies chloroquine was able to reduce the severity of AKI after kidney I/R [Citation24]. Physiological experiments with inhibition of autophagy by 3-methyladenine, which affects another step of the autophagy process by preventing the formation of autophagosomes via inhibition of phosphatidylinositol-3-kinase, also demonstrated the detrimental role of autophagy inhibition [Citation11,Citation13,Citation23,Citation25].
Although the impact of autophagy activation on acute consequences of AKI seems to be clear, the role of autophagy in AKI late-onset outcomes still to be determined. There are two conflicting articles studying the severity of fibrotic changes after kidney damage in Atg5-deficient mice. While one of them demonstrated augmented renal fibrosis after unilateral ureteral obstruction in KO mice [Citation26], in another study knockout of Atg5 in the S3 segment of the nephron resulted in reduced severity of fibrosis 30 days after I/R [Citation27].
Kidney inflammation and autophagy
Inflammation is a pivotal process in most of acute kidney insults like ischemia, nephrotoxicity of drugs and septic AKI [Citation28]. Chronic inflammation believed to be a common attribute of aged kidneys [Citation29], suggesting that cross-interference of autophagy and inflammation must be considered in analysis of age-associated kidney pathologies. By its nature, authophagy is closely associated with innate and adaptive immune responses as it is a key executive mechanism of elimination of a host cell invader [Citation30]. Also, in aseptic inflammation caused by kidney tissue injury, autophagy plays a protective role [Citation31], for instance by decreasing release of DAMPs and cytokines [Citation32], such as IL1b [Citation33]. Mitochondrial quality control (see below) through autophagy also provides immunomodulatory effects since as was recently suggested, it provides the linkage between mitochondrial metabolism and immune system, which was coined as immunometabolism [Citation34].
Although acute inflammation is a conventional response to AKI [Citation28], in senescent kidney, chronic inflammation accompanying by fibrosis is well-documented and it serves as an important element of chronic kidney disease progression [Citation35], which is highly prevalent in elderly population. Autophagy protects from aging-related kidney inflammation, and malfunctions in autophagic machinery leads to development of age-dependent kidney injury [Citation9,Citation36]. To note, inflammation-associated glomerular disorders are less dependent on autophagy [Citation37] than tubular injury. In most of the studies autophagic regulation of inflammation was shown to play a protective role for kidney damage, however in specific conditions, autophagy could induce inflammatory response via inflammasome activation [Citation38,Citation39] which may be deleterious.
Aging affects autophagy
Data on autophagy changes in the kidney during aging is scarce, nevertheless indicating age-associated insufficiency of the organ. Inverse correlation between cell senescence and autophagy activity was demonstrated in vitro. Potentially anti-aging drug rapamycin (see its effects below) downregulated expression of p16INK4a, p21, and α-SMA in primary proximal tubule cells while an inhibitor of lysosomes chloroquine increased their expression [Citation40]. p16INK4a and p21 were recognized as conventional markers of cell senescence, and accumulation of senescent cells is well documented in aged kidney [Citation41]. However, whether decreased autophagy directly drives renal senescence in vivo is not clear and requires further studies.
Robust accumulation of р62, ubiquitinated proteins [Citation42] and mitochondrial lessons [Citation36,Citation43] in aged rodent kidney were extensively reported, thus pointing to the autophagy insufficiency in aging. However, attempts intended to demonstrate a decrease of LC3 II levels, as well as downregulation of autophagy-associated gene expression, failed [Citation42,Citation43]. Taken together this data suggests that lysosomal dysfunction rather than a decrease in autophagosome biogenesis occurs within advanced age. Indeed, lysosomes occupy up to 60% more volume of proximal tubule cells cytoplasm in 25-month old rats vs. 2-month old rats [Citation44]. Apparently, the accumulation of enlarged lysosomes is the main morphological manifestation of lysosomal storage diseases [Citation45]. Note that in contrast to the situation with AKI where autophagy was activated mostly due to augmentation of the autophagosomal part, thus displaying the protective reaction of the organism to I/R, under aging instead, the autophagy insufficiency takes plays possibly due to some problems with lysosomal mechanism thus showing an inability to react to the aging-related challenge. Unfortunately, data on renal lysosome alterations in aging are even rarer than on autophagy. It is known that aging of the liver is accompanied by pronounced changes in lysosomes: a redistribution between isoforms of lysosomal protein LAMP2 occurs when LAMP2-A is playing a key role in chaperone-mediated autophagy, is substituted by the LAMP2-B and LAMP2-C isoforms. These changes are accompanied by an increase in the levels of p62 and oxidized proteins and, apparently, by a compensatory increase in the expression of TFEB, the main transcription factor regulating the biogenesis of lysosomes [Citation46].
Recently, another support of age-associated lysomal dysfunction was reported. In 24-month old mice, 1-day starvation did not increase autophagy flux, although such response takes place in 2-month old animals [Citation36]. Similarly, no increase in lysosome abundance was detected 24 hours after renal I/R in premature-aged OXYS rats, although it was typical for young wild-type rats [Citation18].
Would activation of autophagy protect the elderly?
Activation of autophagy could be achieved in different ways [Citation47]. The non-pharmacological way can bе accomplished by caloric restriction (CR) which results in robust upregulation of autophagy genes and accumulation of autophagosomes [Citation43,Citation48]. Moreover, the effect of CR was abolished under Becline-1 or Atg7 inhibition [Citation49]. Life-long CR increases lifespan in animals [Citation50] and, specifically, long-term (comparable with lifespan) CR can reduce the severity of age-related changes in the kidney associated with mitochondrial lessons [Citation43]. Shorter protocols, mainly involving the reduction of food consumption by 30–40% for several weeks, increase the tolerance of the organism to different damaging factors. In some studies, it has been shown that CR can reduce the severity of I/R-induced AKI [Citation51,Citation52] and in one work, using a long protocol (8 weeks), CR caused the increase of kidney resistance to cisplatin nephrotoxicity [Citation53]. This data indicates a promising clinical future for CR. However, in real clinical practice, there is no opportunity for such lengthy manipulations, except in the case of prevention of the planned operations associated with an increased risk of AKI. No doubt, CR is important as a recommendation of a lifestyle. However, it seems reasonable to use pharmacological drug mimicking CR effects.
Rapamycin mimics the anti-aging effects of CR including the autophagy activation. A key component regulating the activity of autophagy is a proteinaceous complex called mammalian target of rapamycin complex 1 (mTORC1). Rapamycin releases inhibitory block of biogenesis of autophagosomes from mTORC1 [Citation54]. Rapamycin administered late in life also extended the lifespan as CR did [Citation55,Citation56]. It is currently used as an immunosuppressant and seems to be very successful in its use for clinical treatment of AKI since it is the most down-stream effector in autophagy signaling pathway. Although AKI leads to the activation of autophagy in young animals, the use of rapamycin further increases the formation of autophagosomes, accompanied by more rapid clearance of damaged structures, and this additional stimulation of autophagy reduces the severity of AKI [Citation11,Citation21,Citation25].
Although the protective effect of rapamycin has been shown in several studies using young animals, data on the protection of old animals are not available yet. There is some reason for careful optimism arousing from in vitro studies showing that rapamycin reduces the expression of cell senescence markers in renal cells [Citation40] and restores the protective effect of hypoxic postconditioning in artificially aged cardiac myocytes [Citation57].
The obvious problem related to the use of rapamycin in the elderly is that in addition to autophagy, mTOR controls several other processes (described in detail in [Citation54]). Indeed, knockout of mTORC1 in proximal tubules lead to impaired renal function and lower tolerance to I/R-induced damage [Citation58]. Administration of rapamycin did not cause serious consequences as did the knockout, however, rapamycin slowed down the proliferation of kidney cells after damage. Everolimus (a rapamycin analog) increased the severity of AKI after I/R or cisplatin exposure and in parallel with the activation of autophagy, a decrease in proliferation in the kidney during the recovery period takes place [Citation59]. On the other hand, rapamycin did not increase the severity of AKI but delayed the time of full recovery after I/R [Citation60–Citation62]. Such unwanted results can greatly weaken the positive effect of rapamycin, increasing the risk of AKI-associated complications due to increased duration of the disease. It is important to note that in studies where rapamycin was able to protect kidneys from AKI, the animals received a single dose of rapamycin before ischemia while in studies showing a retarded recovery from AKI, the drug was repeatedly administered, both before and after ischemia. It seems attractive to develop a protocol that reduces its negative impact on proliferation. We cannot exclude that this side effect may be much stronger in the elderly, where proliferative and regenerative processes are retarded [Citation63]. For example, it has been shown that in telomerase-knockout mice, the time necessary to recover renal function after I/R was longer than in age-matched wild-type controls [Citation64]. Amplification of this effect by rapamycin could be disastrous. It is also remarkable that the effect of rapamycin was deleterious when it was administered to people [Citation65] and animals [Citation66] with preexisting glomerular pathologies which are quite common in elderly subjects [Citation67].
There are other ways to activate autophagy, such as administering drugs that act on different up-stream components of the mTORC1 signaling pathway. Among drugs already used in the clinic, such targeted action has been ascribed to metformin and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), both of which activate AMP kinase (AMPK). Pretreatment of animals with these compounds increased autophagy flux in the kidney [Citation68]. However, the efficiency of these drugs in old animals requires evaluation.
Mitochondria and kidney aging
What is the role of mitochondria in AKI?
AKI includes damage to the entire mitochondrial apparatus as it has been proven in animal studies. Among the morphological changes observed in the tubular epithelium after I/R, the mitochondrial swelling has been detected, and this was one of the earliest ultrastructural changes in renal cells caused by AKI [Citation69,Citation70]. Structural changes were accompanied by functional changes. The main functional feature of this organelle is its transmembrane potential. It is generated across the inner mitochondrial membrane due to the activity of the respiratory chain (RC) and coupling of oxygen reduction with proton transport from the mitochondrial matrix to intermembrane space. This proton gradient is used by the ATP synthase complex to produce ATP from ADP and inorganic phosphate. The transmembrane potential is the main marker for the mitochondrial quality control system, configured to utilize low potential mitochondria [Citation71].
A drop of the transmembrane potential in renal cortex cells is observed already 10 min after reperfusion following kidney ischemia [Citation72]. One hour after the start of reperfusion, ATP levels in the kidney tissue do not reach 50% of initial values [Citation73], and one day after induction of ischemic AKI, respiratory control, an index of mitochondrial functional state, is significantly lower than in intact animals [Citation74]. Decreased activity of respiratory chain complex I and a decline in respiratory control was also detected in mitochondria isolated from kidneys of rats receiving injections of gentamicin, a drug known for its nephrotoxicity [Citation75]. Mitochondrial changes were observed already three days after antibiotic administration, while renal failure developed only on the 6th day of the course, which suggests that mitochondrial dysfunction is the cause of kidney failure rather than its consequence. Changes in RC activity and a drop of the ATP concentration in the kidney were also observed in two different models of septic shock-induced AKI [Citation76,Citation77].
Mitochondrial dysfunction is deleterious for renal cells. A drop in the mitochondrial membrane potential and consequent fall in ATP levels may cause the cells of the proximal tubules to be unable to maintain their functionality and the structure of their cytoskeleton [Citation78]. This, in turn, leads to the disruption of tight junctions between cells in the tubules and causes a reverse leak of ultrafiltrate, ultimately resulting in uremia. Moreover, loss of tight junctions leads to a loss of polarity in tubular cells, specifically to Na/K-ATPase translocation from the basal to the apical membrane. The subsequent decrease in the sodium reabsorption causes polymerization of Tamm-Horsfall protein and activation of the tubular-glomerular feedback system [Citation79]. The latter effect leads to vasoconstriction of afferent arterioles thus reducing oxygen supply to the nephron, possibly leading to hypoxia/ischemia. In addition, when it becomes impossible to generate a proton gradient by the RC, the mitochondria build the membrane potential through the reverse activity of ATP-synthase, which begins to pump protons into the intramembrane space, using the energy of ATP hydrolysis [Citation80]. Thus, dysfunctional mitochondria not only cease their ATP production but also become an additional consumer of ATP, exacerbating the energy deficit of the cell.
Oxidative stress is another important consequence of mitochondrial dysfunction. An inability to accomplish electron transport along the RC causes some its components to mediate one-electron reduction of oxygen to form superoxide anion radical, ultimately leading to the accumulation of various highly reactive oxygen and nitrogen species (ROS and RNS; the mechanism and consequences of their formation by mitochondria have been reviewed in detail [Citation81]). Concomitantly, RC components are easily damaged by ROS and RNS. Thus, oxidative stress of a non-mitochondrial origin (for example, that caused by inflammatory cells through activation of NADPH oxidase) could disrupt RC function and evoke additional mitochondrial ROS production. A set of data indicates that mitochondrial ROS can activate extramitochondrial ROS-producing systems; a positive feedback relation between the different ROS-generating systems of the cell is discussed in detail elsewhere [Citation81,Citation82]. Each ROS-producing component (mitochondrial and non-mitochondrial), as well as cross-talks between these ROS sources, can cause the amplification of ROS levels and convert signaling function of ROS into pathological.
Oxidative stress is an essential feature of AKI. In experiments in vivo, it has been shown that 40-min ischemia caused a manifold increase in ROS production in the kidney [Citation72]. Similarly, an increase in ROS levels in the cortex of kidneys was observed 3 hours after a single injection of gentamicin [Citation83]. After 5–6 days of the introduction of the antibiotic, profound oxidative stress was indicated by increased protein carbonylation and oxidation of lipid membranes [Citation75,Citation83,Citation84]. Likewise, after induction of rhabdomyolysis, an increase in renal tissue of malondialdehyde, a product of lipid peroxidation, was recorded [Citation85]. Similar signs of oxidative stress were observed in the kidney after administration of the nephrotoxic drug cisplatin [Citation86] or radiocontrast media [Citation87] and also during endotoxemic shock [Citation77].
Detailed study of mitochondrial involvement in ROS generation showed that mitochondria isolated from the kidneys of rats treated with gentamicin had elevated ROS levels [Citation75]. When mitochondria isolated from a healthy animal were incubated with gentamicin, an increase in hydrogen peroxide production was observed [Citation88]. Combined staining of kidney slices exposed to 40-min ischemia with probes for energized mitochondria and ROS revealed that mitochondria were the major source of ROS [Citation72]. Another fluorescent dye sensitive to superoxide anion revealed the incidence of mitochondrial oxidative stress in the kidneys 36 hours after induction of sepsis [Citation76].
ROS generation in mitochondria or their vicinity carries great risk for the cell because ROS act as a special trigger, activating a pore complex in the inner membrane of mitochondria called mPTP, the mitochondrial permeability transition pore [Citation89]. Opening the pore makes the inner mitochondrial membrane permeable to molecules up to 1.5 kDa, which results in equilibration of the ions between the matrix and extramitochondrial space following by swelling of the mitochondrial matrix (which is a characteristic feature of ultrastructural changes induced by ischemic AKI in cells of the nephron) and dissipation of transmembrane potential. In addition to adverse effects on bioenergetics, the opening of the mPTP leads to the release from mitochondria of cytochrome C, Аpaf-1, apoptosis-inducing factor (AIF) and endopeptidase G what promotes the final steps of apoptosis [Citation90]. When the opening of the mPTP occurs in most of the mitochondria in the cell, acute depletion of the ATP pool will lead to necrotic cell death [Citation91].
Finally, mitochondrial dysfunction and specifically mitochondrial ROS can have a negative impact on renal hemodynamics [Citation92]. Endothelial dysfunction and impaired hemodynamics play an important role in the pathogenesis of AKI. Superoxide radical interacts with NO with a high affinity, thus lowering its stimulatory effect on guanylate cyclase, which regulates the tonus of the blood vessels. It has been shown that the decrease in expression of mitochondrial superoxide dismutase leads to more severe angiotensin II-induced hypertension [Citation93]. Another mechanism of vasoconstriction can be associated with the mPTP, which causes the release of mitochondrial calcium ions to the cytosol [Citation81]. Partially, due to this, mitochondria of smooth muscle cells of blood vessels in the kidneys are highly susceptible to reperfusion damage [Citation94]. Nephrotoxins that primarily target the proximal tubules, increase hydrogen peroxide production, which can leave the cell and to reach neighboring cells, spreading the damage from tubules to endothelium [Citation95].
Can we afford protection of the kidney by treating its mitochondria?
Mitochondrial antioxidants
Oxidative stress can damage various cellular structures, however, as mentioned above, the greatest danger is posed by excessive ROS formed in or reaching the mitochondria. Perhaps, a low concentration of conventional antioxidants in the mitochondria can explain their low efficacy in the clinic [Citation96]. Recently, mitochondria-targeted antioxidants, which carry both antioxidative properties and the ability to be accumulated in the mitochondria of cells, have become available. A comprehensive investigation of the effect of mitochondrial antioxidants from the SkQ family on renal pathologies demonstrated that SkQ1 introduced one day before I/R prevented the animals’ death [Citation97], and the administration of SkQ1 3 hour before and 1 hour after ischemia and every 12 hours during the post-ischemic period for 2 days improved kidney function [Citation98]. An even more pronounced nephroprotective potential was reported for SkQR1. This drug protected the kidney from damage in experimental rhabdomyolysis, I/R and gentamicin toxicity [Citation99,Citation100]. It prevented the animals’ death, diminished morphological lesions of the kidney, decreased the blood levels of creatinine and urea and maintained the normal production of erythropoietin impaired by AKI. It deserves the special attention that due to targeted delivery of antioxidants to the mitochondria, the protective effect of the drug was observed at very low doses – as little as 20–100 nmol/kg in the intraperitoneal injection.
SkQR1 increased the recovery rate of renal blood flow after I/R and reduced the resistivity of the renal vessels. The protective effect of the drug on both renal hemodynamics and renal function vanished after inhibition of NO synthesis indicating a possible key role of mitochondrial oxidative stress in renal vasculature damage [Citation92]. In addition, SkQR1 prevented the inflammatory changes observed in the kidney in experimental acute pyelonephritis [Citation101]. The antioxidant MitoQ, having a similar structure, also afforded nephroprotection. The drug was shown to slow down the biochemical processes associated with damage to the graft caused by prolonged cold ischemia [Citation102] as well as mitigate AKI after cisplatin treatment [Citation103] and endotoxemic shock [Citation77,Citation104].
In a septic model of AKI the nephroprotective potential of another mitochondria-targeted antioxidant, mitoTEMPO, has been revealed [Citation76]. Finally, the efficiency of the mitochondria-targeted antioxidant SS-31 (Bendavia) was shown in an ischemic model of AKI [Citation105,Citation106] (through reduction of severity of pathological changes and improvement of renal hemodynamics) and after ureteral obstruction (causing reduction of oxidative stress, infiltration of macrophages and apoptosis) [Citation107].
Mild uncoupling of oxidative phosphorylation
One more nephroprotective approach, which affords the protection of renal mitochondria from excessive ROS production, is a mild uncoupling of respiration and oxidative phosphorylation. This approach is based on the fact that mitochondrial ROS production is directly dependent on transmembrane potential [Citation108]. The higher the potential, the more hydrogen peroxide will be formed by mitochondria and vice versa. Uncoupling may be caused by a proton leak bypassing ATP synthase, so the transport of protons into the matrix becomes not associated with ATP synthesis. In this process, the transmembrane potential decreases, and generation of ROS decreases as well. The main problem in the use of uncouplers for this purpose is the selection of a dose that would result in a decrease in potential sufficient to drop ROS production, but not so great as to inhibit ATP synthesis to the levels insufficient to provide normal cell functioning. Among the existing drugs, this balance was found for 2,4-dinitrophenol, which showed the ability to prevent AKI caused by renal I/R [Citation98]. To increase the safety of this class of substances, mitochondria-targeted uncouplers were recently developed, which concentration in mitochondria depends on the transmembrane potential. Thus, drug-induced uncoupling will decrease the drug accumulation, thereby forming a negative reverse link preventing energy collapse of mitochondria. Currently, the nephroprotective potential of two such new compounds, dodecylrhodamine (С12R1) [Citation98] and mitoFluo [Citation109], has been proven. In the nearest future, we can expect the emergence of new uncouplers with an increasingly wide therapeutic window. The uncouplers were shown to prevent proinflammatory changes in endothelial cells in vitro and animal death induced inflammation, indicating an important role for mitochondria in the regulation of inflammatory response [Citation110,Citation111]. Translation these results from the bench to the bed implies the possibility of treatment with uncouplers of a late diagnosed AKI through inhibition of inflammatory processes in the organism.
Ischemic preconditioning
Another mitochondria-targeted strategy for nephroprotection originates from the phenomenon of ischemic preconditioning (IPC) when short episodes of ischemia precede a prolonged ischemic stroke, resulting in higher resistance to damage [Citation112]. To prevent I/R-induced renal failure, various protocols of IPC are used generally containing 1–6 episodes of I/R, each lasting 4–11 min [Citation113,Citation114]. The efficiency of the much shorter protocol (4 cycles at 15 s of ischemia and 15 hours reperfusion) [Citation18], and much longer protocol (30 min ischemia) [Citation115] have also been demonstrated. Several studies opened an incredible prospect for the use of IPC in the clinic. First, it was found that the protective effect was afforded not only after ischemic “training” before long-term ischemia but also immediately after an ischemic insult to the kidney [Citation116]. This allows the use of IPC not only for the prevention of AKI in situations associated with high risk of disease but also to treat an already existing AKI. Secondly, the phenomenon of remote preconditioning was discovered, in which ischemic “training” of one organ increased the tolerance of another organ to prolonged ischemic insult [Citation117,Citation118]. Short-term ischemia of the limbs was observed to be no less protective than IPC of the targeted organ [Citation119], which opened the possibility of using in clinical practice of highly simple and safe protocols based on minutes-long ischemia of a shoulder or hip induced by an inflatable cuff. Thirdly, it became practical to reproduce the protective effect of IPC with pharmacological agents (pharmacological preconditioning). In the heart, IPC desensitizes the mPTP, making its opening less likely [Citation120]. Further, it has been shown that the status of the mPTP plays a key role in determining whether cells will die or survive [Citation121,Citation122]. Conventional mPTP inhibitor, cyclosporine A prevented an I/R-induced drop of the transmembrane potential in isolated renal mitochondria [Citation72], and it also prevented the development of AKI in some in vivo models, demonstrating that kidney dysfunction is closely associated with mitochondrial dysfunction [Citation123,Citation124]. Further evidence for the important role of the mPTP in the pathogenesis of AKI came from the studies done on cyclophilin D (CyD)-knockout mice. CyD is one of the most important modulators of the mPTP complex. It binds to the mitochondrial component organized by a dimer of ATP synthase and makes it insensitive to mPTP triggers. Knockout of the CyD gene prevented mitochondrial dysfunction and energy deficiency under oxidative stress and protected from the consequences of I/R [Citation121].
In kidney mitochondria, CyD colocalizes with the β isoform of glycogen synthase kinase 3 (GSK-3β). Inhibition of GSK-3β activity through phosphorylation of serine-9 reduced phosphorylation of mitochondrial CyD, mitochondrial swelling and nephrotoxic effects of NSAID, 2-[(2,6-dichlorophenyl)amino]phenyl-acetate [Citation125]. Since GSK3β is phosphorylated by a large set of kinases (Akt/PkB, PkA, PkC, p70S6, etc.), it became possible to arrange a search for substances preventing the opening of the mPTP while targeting named kinases. Now, the protective effects of various GSK-3β inhibitors, operating at different levels of the signaling pathway, have been shown in various models of AKI, including I/R [Citation72,Citation126–Citation128], kidney transplantation [Citation129], gentamicin toxicity [Citation83,Citation130], cisplatin toxicity [Citation126], NSAID toxicity [Citation125], septic AKI [Citation131] and rhabdomyolysis [Citation132].
One proven endogenous mediator of IPC signaling pathways is erythropoietin (EPO). EPO receptor activation leads to phosphorylation of GSK-3β, presumably through the PI3K-Akt pathway. Increased production of EPO by the kidney is one of the ways to implement protective mechanisms in remote preconditioning [Citation117,Citation118]. Currently, substantial evidence has been accumulated proving the nephroprotective effect of human recombinant EPO in various models of AKI, including those caused by nephrotoxic drugs [Citation133–Citation135], I/R [Citation136,Citation137], rhabdomyolysis [Citation138] and experimental sepsis [Citation139,Citation140].
How do mitochondria change during aging?
One of the leading theories of aging postulates that the accumulation of oxidative damage in mitochondria due to uncompensated ROS production by the respiratory chain is the cause of aging [Citation141,Citation142]. In the kidney of 24-month old mice, the levels of 8-OHdG in mtDNA was greater, and the number of point mutations and deletions in mtDNA was much greater than in 3-month old animals. Life-long CR resulted in simultaneous normalization of the mitochondrial parameters and vanishing of physiological hallmarks of aging in the kidney [Citation43]. In addition, mice with a point mutation in the gene of mtDNA polymerase, which leads to a 3–5 fold increase in the number of point mutations and deletions in mtDNA vs. wild-type, demonstrate not only shorter lifespan but also a senescent specific phenotype [Citation143]. Additional powerful support for the mitochondrial theory of aging comes from a study in which two lines of mice with identical nuclear but different mitochondrial genomes were compared. It occurred that only differences in the mitochondrial genome could significantly affect the lifespan of animals and the severity of the signs of aging [Citation144].
In mitochondria isolated from kidneys of 23 to 27-month old mice, protein synthesis was two times slower than in mitochondria of young mice [Citation145]. This data correlates with the results of another study, where lower expression of TFAM, a mtDNA transcriptional factor, was detected in the kidneys of 21-month old and young Fischer 344 rats [Citation146]. That study demonstrated a decrease in cytoplasmic and nuclear levels of transcriptional factor Nrf2, which plays an important role in the expression of several antioxidant genes.
Aging has been found to be accompanied not only by the accumulation of damaged mtDNA copies but also by functional changes in the mitochondria of the kidneys. Particularly, a decline in the activity of mitochondrial enzymes, including those of the Krebs cycle [Citation147] and of the respiratory chain has been demonstrated [Citation148]. Aging was associated with lower respiration rates [Citation146,Citation149], a greater fraction of mitochondria with low potential [Citation18] and a lower rate of ATP production [Citation146].
Opponents of the mitochondrial theory of aging emphasize that, despite the successful reproduction of the aging phenotype in mtDNA mutator mice, a remarkable number of studies have failed to detect significant functional changes in mitochondria with age, and even when these have been detected, such changes did not exceed 10–20%. In response to this criticism, it has been suggested that perhaps the aging process is not characterized by a gross change in all cells, but rather by changes occurring in local cell communities bearing multiple copies of defective mtDNA [Citation150]. This argument emphasizes the importance of the heterogeneity of healthy vs. unhealthy biological structures including cells, mitochondria, and macromolecules. The key role of cellular heterogeneity in aging is consistent with the fact that accumulation of a quite small number of senescent cells in an organ can significantly affect healthy cells not touched by the aging process [Citation41]. An important discovery became the fact that maintenance of stemness is associated with the ability of the cell to separate old from new mitochondria through asymmetric mitochondrial fission with further placing of newly formed healthy mitochondria in a resting stem cell [Citation151,Citation152]. Thus, accumulation of mtDNA mutations may determine the aging which is mediated by senescence of stem cells.
A very challenging question remains: why dysfunctional mitochondria are accumulated in aging despite the presence of a system of autophagy, which detects and destroys “bad” mitochondria [Citation71] and thus prevents unnecessary heterogeneity in the mitochondrial population. It seems that increasing mitochondrial heterogeneity not only in stem but also in all cells is a sign of pathological phenotype including aging phenotype [Citation153]. We can propose that such heterogeneity is a result of either impaired asymmetric mitochondrial division (in other words, fragmentation or fission) or an impaired utilization of damaged mitochondrial fragments through autophagy (mitophagy) [Citation71].
Will mitochondria-targeted approaches be protective in the elderly?
As aging evidently affects mitochondria, a reasonable question arises as to whether mitochondria-targeted approaches can protect the aging kidney. Two works attempting to extend the protective effects of IPC and ischemic postconditioning to older animals yielded a disappointing result: these interventions were not protective in the kidney of old rats [Citation18,Citation154]. In one of these studies, aging was accompanied by an increase in the level of acetylation of the nuclei in the epithelial cells of the proximal tubules and a lack of growth in the number of lysosomes one day after I/R. Under basal conditions, there was a smaller amount of LC3 II in the kidney homogenates and a greater abundance of PINK1 and ubiquitinated proteins in the mitochondria of old versus young rats. The accumulation of mitophagic markers with age was accompanied by a growing number of low-potential mitochondria, suggesting that in older animals, the system of mitophagy retains the ability to detect dysfunctional mitochondria and mark them for further utilization, but loses the ability to destroy mitochondria [Citation18].
In the same study, IPC did not affect the membrane potential of mitochondria from kidneys of young rats. However, it led to an increase in the number of low potential mitochondria in old animals [Citation18]. This data emphasizes the importance of studying not only the average of each parameter over the population of mitochondria but also the distribution of parameter values within that population [Citation153]. The need for such analysis may be augmented by the existence of the phenomenon of ROS-induced ROS-release, in which oxidative stress in an individual mitochondrion induces mPTP opening in adjacent mitochondria, resulting in an even greater increase in ROS production and mPTP induction in mitochondria far away from the source of the original production of ROS [Citation155]. It can be assumed that this phenomenon makes the accumulation of even a small percentage of dysfunctional mitochondria highly sensitive to oxidative stress extremely dangerous for the cell. A small increase in ROS production that in young animals triggers a cascade of protective signaling, in older animals may provoke long-lasting and permanent opening of the mPTP in the population of “bad” mitochondria. This, in turn, will lead to increased oxidative stress in remote mitochondria, so that a low level of oxidative stress, such as that associated with IPC, can be amplified to a damaging level.
summarizes the concept of mitochondrial changes occurring on aging. The main point is the increasing heterogeneity with age when “bad” mitochondria progressively persist in the entire mitochondrial population as a result of inefficient mitophagy. Heterogeneity of the membrane potential in aging mitochondria can abolish the protective effect of mitochondria-targeted antioxidants. Most of these antioxidants use mitochondrial potential as the driving force for accumulation in the mitochondria. As a result, in the low potential mitochondria, their concentrations will be less, although these are precisely the mitochondria most in need of antioxidants to ameliorate oxidative stress. The opening of the mPTP only in this population of mitochondria may be sufficient to initiate cell death. Therefore, the efficacy of mitochondria-targeted antioxidants should definitely be tested in old animals.
Figure 1. Appearance and progressive increase of mitochondrial heterogeneity in the aged kidney. In the old kidney, the level of active GSK-3 is increased, which leads to higher percentage of the mitochondria with open pores and higher ROS generation. This results in higher number of dysfunctional mitochondria that are poorly eliminated because of ineffective autophagy. Mechanisms for effective mitochondria clearance induced by IPC or by CR are weak in the old kidney, i.g., because of active Akt. As a result, aged kidney contains more “bad” mitochondria, but they cannot be removed as it occurs in young tissue, leading to enrichment of a population with dysfunctional organelles and the formation of the senile phenotype.
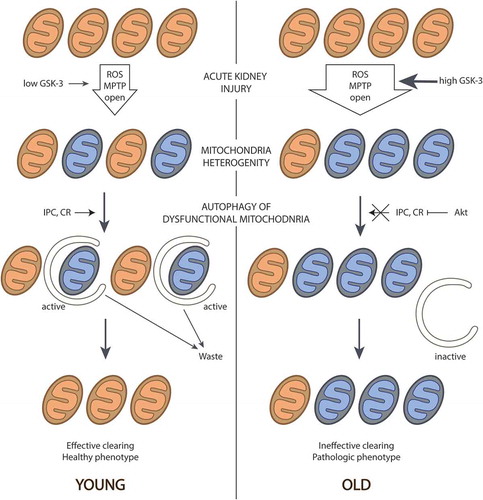
Undoubtedly, a more detailed study of heterogeneity in the mitochondrial population of different kidney cells and the effects of aging on this heterogeneity are required. Such study might lead to the development of mitochondrial antioxidants whose accumulation does not depend on mitochondrial membrane potential, or conversely, of ways to induce mitophagy of “bad”, low-potential mitochondria, for example, by increasing the expression of Parkin. The exacerbation of mitochondrial dysfunction by aging may cause nephroprotective approaches proven to be effective in young animals to be ineffective in older subjects. Thus, the growth of production of mitochondrial ROS may determine the essential for the aged kidney increase in resistance of renal vasculature [Citation156] and diminished vasodilatation response [Citation157]. The most widely used vasodilators mediate their activity through NO, thus necessitating the testing of their effectiveness in old animals. It is possible that the efficacy of NO-dependent dilatators could be resurrected by concomitant administration with mitochondrial antioxidants or uncouplers.
The role of Akt signaling in aging
The loss of the protective effect of IPC in old animals may be associated with changes in signaling pathways. Thus, it has been shown that in the aged kidney, the PI3K-Akt pathway is activated [Citation158]. One target of Akt is GSK-3β. Aging in the heart is accompanied by an increase in the content of both pAkt and pGSK-3β; however, for the kidney, there is no available data on this issue [Citation159]. Similarly, it has been shown that in the old heart, drugs that protect young animals from I/R by inhibiting GSK-3β were not able to increase the levels of the phosphorylated (inactive) form of the enzyme and were thus unable to reduce damage to the old heart. In one proposed mechanism for the protective effect of IPC, the mPTP opens for short periods of time, leading to a small regulatory increase of ROS production that triggers signaling that prevents the transition of the pore complex to the permanent open state [Citation160,Citation161]. In this context, excessive activation of the PI3K-Akt pathway and inhibition of GSK-3β activity in aging may play a negative role, preventing IPC from exerting its conventional protective effects. In addition, the increase in pGSK-3β during aging of the heart is associated with a proportional increase in CyD [Citation159], which after phosphorylation, inhibits mPTP. It is assumed that normally, the ratio of CyD to phosphoserine-9 on GSK-3β remains unchanged and inhibition of the pores does not occur.
Another target of the Akt signaling pathway is the mTORC1 complex [Citation54]. Via inhibition of TSC1/2 and/or the dissociation of PRAS40 from mTORC1, Akt increases the activity of mTORC1. It is important that the impact of AMPK, which inhibits mTORC1 in response to CR, is also implemented through the TSC1/2, however through activating phosphorylation. Thus, the inhibitory effect of Akt can compete with the activating effect of AMPK to reduce the activation of autophagy in response to starvation. Finally, it has been shown that Akt inhibits chaperone-mediated autophagy [Citation162]. In this process, unlike macroautophagy, special protein complexes on the surface of lysosomes recognize incorrectly folded proteins with attached chaperones with further their binding, unfolding and translocation into the lysosomal lumen for degradation [Citation163]. Inhibition of this pathway will increase the formation of protein aggregates, which will increase the burden on the autophagy system, increasing the probability that ubiquitin-labeled “bad” mitochondria will avoid destruction.
Concluding remarks
Currently, there is a huge gap between the two pools of publications. The first relates to the protection of young animals from AKI. The second, more limited, focuses on what happens to the kidney with aging. The evaluation of strategies for nephroprotection in old animals is virtually absent, although this is one of the most important issue limiting translation to the clinical situation. This is largely a logistical problem because even the design of conventional animal facilities does not support the maintenance of animals that have reached old age. Thus, old animals remain a unique object, and the diversity of pathologies that occur spontaneously in old animals of different strains makes their study extremely difficult. A radical revision of this approach is needed, particularly in the organization of animal facility design, to maintain different populations of old animals in sufficient quantities.
Another approach to solve this problem is to study animals that show signs of premature aging, such as OXYS rats or mtDNA mutator mice. However, such animal models will be unlikely to reproduce aging in its entirety. Some authors believe that the diabetic kidney exhibits a number of signs of aging, and therefore a strain of diabetic rodents can also offer some approximation to an aging model.
A third possible approach is to modify the protocols used to analyze clinical trial data. Within these trials, there is often a group of old patients, and by comparing the effect of any drug in young and old patients, it may become possible to understand what mechanisms are “turned off” in the elderly. Probably, this will be the most effective approach, as it will shift the focus of research from the simulation of aging in animals (which may be flawed, due to the fact that aging may be totally different in different species with different strategies of reproduction, as, for example, in primates or rodents) to the identification of the mechanisms that are disabled or attenuated in the elderly, followed by detailed examination of these specific signaling pathways in rodents.
A fourth approach is to pay more attention to the distribution of acquired data instead of averaging the effects (both positive and negative) of an intervention (physiological or pharmacologic) over the entire biological system including people, animals, their cells, organelles or macromolecules. The standard deviation of any measured value may reflect not only the skill of the investigator but also the diversity of biological responses in different biological systems. Presentation of the entire set of obtained data may help to specifically group the data thus revealing specific activation or inhibition of signaling pathways involved in many deleterious states such as aging of different organs including kidney.
Acknowledgments
The authors are very thankful to Dr. Kenneth W. Fishbein (LCI, NIA) for his valuable critique and help.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- United Nations. Department of Economic and Social Affairs, Population Division. World Population Ageing 2015. 2015; (ST/ESA/SER.A/390).
- Rewa O, Bagshaw SM. Acute kidney injury-epidemiology, outcomes and economics. Nat Rev Nephrol. 2014;10:193–207. PMID:24445744
- Bagshaw SM, Laupland KB, Doig CJ, et al. Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: a population-based study. Crit Care. 2005;9:R700–9. PMID:16280066
- Xue JL. Incidence and mortality of acute renal failure in medicare beneficiaries, 1992 to 2001. J Am Soc Nephrol. 2006;17:1135–1142. PMID:16495381
- Huang J, Klionsky DJ. Autophagy and human disease. Cell Cycle. 2007;6:1837–1849. PMID:17671424
- Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. PMID:11060023
- Periyasamy-Thandavan S, Jiang M, Wei Q, et al. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008;74:631–640. PMID:18509315
- Takahashi A, Kimura T, Takabatake Y, et al. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol. 2012;180:517–525. PMID:22265049
- Liu S, Hartleben B, Kretz O, et al. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy. 2012;8:826–837. PMID:22617445
- Li L, Wang ZV, Hill JA, et al. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J Am Soc Nephrol. 2014;25:305–315. PMID:24179166
- Zhang Y-L, Zhang J, Cui L-Y, et al. Autophagy activation attenuates renal ischemia-reperfusion injury in rats. Exp Biol Med. 2015;240:1590–1598. PMID:25898836
- Isaka Y, Suzuki C, Abe T, et al. Bcl-2 protects tubular epithelial cells from ischemia/reperfusion injury by dual mechanisms. Transplant Proc. 2009;41:52–54. PMID:19249473
- Ko GJ, Bae SY, Hong YA, et al. Radiocontrast-induced nephropathy is attenuated by autophagy through regulation of apoptosis and inflammation. Hum Exp Toxicol. 2016;35:724–736. PMID:26384705
- Li T, Liu Y, Zhao J, et al. Aggravation of acute kidney injury by mPGES-2 down regulation is associated with autophagy inhibition and enhanced apoptosis. Sci Rep. 2017;7. PMID:28860615. DOI:10.1038/s41598-017-10271-8
- Mei S, Livingston M, Hao J, et al. Autophagy is activated to protect against endotoxic acute kidney injury. Sci Rep. 2016;6. PMID:26916346. DOI:10.1038/srep22171
- Karagiannidis I, Kataki A, Glustianou G, et al. Extended cytoprotective effect of autophagy in the late stages of sepsis and fluctuations in signal transduction pathways in a rat experimental model of kidney injury. Shock. 2016;45:139–147.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. PMID:26799652
- Jankauskas SS, Pevzner IB, Andrianova NV, et al. The age-associated loss of ischemic preconditioning in the kidney is accompanied by mitochondrial dysfunction, increased protein acetylation and decreased autophagy. Sci Rep. 2017;7. PMID:28294175. DOI:10.1038/srep44430
- Wesselborg S, Stork B. Autophagy signal transduction by ATG proteins: from hierarchies to networks. Cell Mol Life Sci. 2015;72:4721–4757. PMID:26390974
- Kimura T, Takabatake Y, Takahashi A, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 2011;22:902–913. PMID:21493778
- Jiang M, Wei Q, Dong G, et al. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82:1271–1283. PMID:22854643
- Maejima I, Takahashi A, Omori H, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013;32:2336–2347. PMID:23921551
- Jiang M, Liu K, Luo J, et al. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol. 2010;176:1181–1192. PMID:20075199
- Todorovic Z, Medic B, Basta-Jovanovic G, et al. Acute pretreatment with chloroquine attenuates renal I/R injury in rats. PLoS ONE. 2014;9. PMID:24681567. DOI:10.1371/journal.pone.0092673
- Ling H, Chen H, Wei M, et al. The effect of autophagy on inflammation cytokines in renal ischemia/Reperfusion injury. Inflammation. 2016;39:347–356. PMID:26412257
- Li H, Peng X, Wang Y, et al. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G 2/M arrest and renal fibrosis. Autophagy. 2016;12:1472–1486.
- Baisantry A, Bhayana S, Rong S, et al. Autophagy induces prosenescent changes in proximal tubular S3 segments. J Am Soc Nephrol. 2016;27:1609–1616. PMID:26487561
- Rabb H, Griffin MD, McKay DB, et al. Inflammation in AKI: current understanding, key questions, and knowledge gaps. J Am Soc Nephrol. 2016;27:371–379. PMID:26561643
- Costello-White R, Ryff CD, Coe CL. Aging and low-grade inflammation reduce renal function in middle-aged and older adults in Japan and the USA. Age (Dordr). 2015;37. PMID:26187318. DOI:10.1007/s11357-015-9808-7.
- Lapaquette P, Guzzo J, Bretillon L, et al. Cellular and molecular connections between autophagy and inflammation. Mediators Inflamm. 2015;2015:398483. PMID:26221063
- Isaka Y, Takabatake Y, Takahashi A, et al. Hyperuricemia-induced inflammasome and kidney diseases. Nephrol Dial Transplant. 2016;31:890–896. PMID:25829326
- Deretic V, Kimura T, Timmins G, et al. Immunologic manifestations of autophagy. J Clin Invest. 2015;125:75–84. PMID:25654553
- Nakahira K, Haspel JA, Rathinam VAK, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. PMID:21151103
- O’Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16:553–565. PMID:27396447
- Bolignano D, Mattace-Raso F, Sijbrands EJG, et al. The aging kidney revisited: a systematic review. Ageing Res Rev. 2014;14:65–80. PMID:24548926
- Yamamoto T, Takabatake Y, Kimura T, et al. Time-dependent dysregulation of autophagy: implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy. 2016;12:801–813. PMID:26986194
- Hartleben B, Gödel M, Meyer-Schwesinger C, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084–1096. PMID:20200449. doi:10.1172/JCI39492.
- Kimura T, Jia J, Kumar S, et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017;36:42–60. PMID:27932448
- Dupont N, Jiang S, Pilli M, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011;30:4701–4711. PMID:22068051
- Baisantry A, Bhayana S, Wrede C, et al. The impact of autophagy on the development of senescence in primary tubular epithelial cells. Cell Cycle. 2016;15:2973–2979. PMID:27715411
- Sturmlechner I, Durik M, Sieben CJ, et al. Cellular senescence in renal ageing and disease. Nat Rev Nephrol. 2017;13:77–89. PMID:28029153
- Cui J, Bai X-Y, Shi S, et al. Age-related changes in the function of autophagy in rat kidneys. Age. 2012;34:329–339.
- Kume S, Uzu T, Horiike K, et al. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Investig. 2010;120:1043–1055.
- Sato T, Tauchi H, Kohtani K. Morphometric studies on the age changes of autofluorescent granules and lysosomes in the epithelial cells of rat kidneys. Mech Ageing Dev. 1984;25:237–242.
- Xu H, Ren D. Lysosomal Physiology. Annu Rev Physiol. 2015;77:57–80. PMID:25668017.
- Schneider JL, Villarroya J, Diaz-Carretero A, et al. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell. 2015;14:249–264. PMID:25620427
- Mariño G, Pietrocola F, Madeo F, et al. Caloric restriction mimetics: natural/physiological pharmacological autophagy inducers. Autophagy. 2014;10:1879–1882. PMID:25484097
- Wohlgemuth SE, Julian D, Akin DE, et al. Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res. 2007;10:281–292. PMID:17665967
- Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007;3:597–599.
- Lee SH, Min KJ. Caloric restriction and its mimetics. BMB Rep. 2013;46:181–187. PMID:23615258
- Mitchell JR, Verweij M, Brand K, et al. Short term dietary restriction and fasting precondition against ischemia reperfusion injury in mice. Aging Cell. 2010;9:40–53.
- Lempiäinen J, Finckenberg P, Mervaala EE, et al. Caloric restriction ameliorates kidney ischaemia/reperfusion injury through PGC-1α-eNOS pathway and enhanced autophagy. Acta Physiologica. 2013;208:410–421. PMID:23710679
- Ning Y-C, Cai G-Y, Zhuo L, et al. Beneficial effects of short-term calorie restriction against cisplatin-induced acute renal injury in aged rats. Nephron Exp Nephrology. 2013;124:19–27. PMID:24401898
- Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. PMID:19812304
- Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. PMID:19587680
- Speakman JR, Mitchell SE, Mazidi M. Calories or protein? The effect of dietary restriction on lifespan in rodents is explained by calories alone. Exp Gerontol. 2016;86:28–38. PMID:27006163
- Chen J, Gao J, Sun W, et al. Involvement of exogenous H2S in recovery of cardioprotection from ischemic post-conditioning via increase of autophagy in the aged hearts. Int J Cardiol. 2016;220:681–692. PMID:27393850
- Grahammer F, Haenisch N, Steinhardt F, et al. mTORC1 maintains renal tubular homeostasis and is essential in response to ischemic stress. Proc Natl Acad Sci. 2014;111:E2817–26. PMID:24958889
- Nakagawa S, Nishihara K, Inui KI, et al. Involvement of autophagy in the pharmacological effects of the mTOR inhibitor everolimus in acute kidney injury. Eur J Pharmacol. 2012;696:143–154. PMID:23022334
- Lieberthal W, Fuhro R, Andry CC, et al. Rapamycin impairs recovery from acute renal failure: role of cell-cycle arrest and apoptosis of tubular cells. Am J Physiol Renal Physiol. 2001;281:F693–706.
- Lui SL, Chan KW, Tsang R, et al. Effect of rapamycin on renal ischemia-reperfusion injury in mice. Transpl Int. 2006;19:834–839. PMID:16961776
- Goncalves GM, Cenedeze MA, Feitoza CQ, et al. The role of heme oxygenase 1 in rapamycin-induced renal dysfunction after ischemia and reperfusion injury. Kidney Int. 2006;70:1742–1749. PMID:17003813
- Demyanenko IA, Popova EN, Zakharova VV, et al. Mitochondria-targeted antioxidant SkQ1 improves impaired dermal wound healing in old mice. Aging. 2015;7:475–485. PMID:26187706
- Cheng H, Fan X, Lawson WE, et al. Telomerase deficiency delays renal recovery in mice after ischemia-reperfusion injury by impairing autophagy. Kidney Int. 2015;88:85–94. PMID:25760322
- Fervenza FC, Fitzpatrick PM, Mertz J, et al. Acute rapamycin nephrotoxicity in native kidneys of patients with chronic glomerulopathies. Nephrol Dial Transplant. 2004;19:1288–1292. PMID:15102967
- Coombes JD, Mreich E, Liddle C, et al. Rapamycin worsens renal function and intratubular cast formation in protein overload nephropathy. Kidney Int. 2005;68:2599–2607. PMID:16316336
- Hommos MS, Glassock RJ, Rule AD. Structural and functional changes in human kidneys with healthy aging. J Am Soc Nephrol. 2017;28:2838–2844. PMID:28790143
- Declèves AE, Sharma K, Satriano J. Beneficial effects of AMP-activated protein kinase agonists in kidney ischemia-reperfusion: autophagy and cellular stress markers. Nephron Exp Nephrol. 2014;128:98–110.
- Glaumann B, Glaumann H, Berezesky IK, et al. Studies on the pathogenesis of ischemic cell injury. II. Morphological changes of the pars convoluta (P1 and P2) of the proximal tubule of the rat kidney made ischemic in vivo. Virchows Arch B Cell Pathol. 1975;19:281–302.
- Glaumann B, Trump BF. Studies on the pathogenesis of ischemic cell injury. III. Morphological changes of the proximal pars recta tubules (P3) of the rat kidney made ischemic in vivo. Virchows Arch B Cell Pathol. 1975;19:303–323.
- Zorov DB, Popkov VA, Zorova LD, et al. Mitochondrial aging: is there a mitochondrial clock? J Gerontol A Biol Sci Med Sci. 2017;72:1171–1179. PMID:27927758
- Plotnikov EY, Kazachenko AV, Vyssokikh MY, et al. The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int. 2007;72:1493–1502.
- Vogt MT, Farber E. On the molecular pathology of ischemic renal cell death. Reversible and irreversible cellular and mitochondrial metabolic alterations. Am J Pathol. 1968;53:1–26.
- Wilson DR, Arnold PE, Burke TJ, et al. Mitochondrial calcium accumulation and respiration in ischemic acute renal failure in the rat. Kidney Int. 1984;25:519–526. PMID:6737843
- Morales AI, Detaille D, Prieto M, et al. Metformin prevents experimental gentamicin-induced nephropathyby a mitochondria-dependent pathway. Kidney Int. 2010;77:861–869. PMID:20164825
- Patil NK, Parajuli N, MacMillan-Crow LA, et al. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. AJP: Ren Physiol. 2014;306:F734–43. PMID:24500690
- Lowes DA, Webster NR, Murphy MP, et al. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br J Anaesth. 2013;110:472–480. PMID:23381720
- Molitoris BA. Actin cytoskeleton in ischemic acute renal failure. Kidney Int. 2004;66:871–883. PMID:15253754
- Schrier RW, Wang W, Poole B, et al. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Investig. 2004;114:5–14. PMID:15232604
- St-Pierre J, Brand MD, Boutilier RG. Mitochondria as ATP consumers: cellular treason in anoxia. Proc Natl Acad Sci. 2000;97:8670–8674. PMID:10890886
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94:909–950. PMID:24987008
- Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51:1289–1301. PMID:21777669
- Plotnikov EY, Grebenchikov OA, Babenko VA, et al. Nephroprotective effect of GSK-3β inhibition by lithium ions and δ-opioid receptor agonist dalargin on gentamicin-induced nephrotoxicity. Toxicol Lett. 2013;220:303–308. PMID:23651617
- Walker PD, Shah SV. Evidence suggesting a role for hydroxyl radical in gentamicin-induced acute renal failure in rats. J Clin Investig. 1988;81:334–341. PMID:3123518
- Plotnikov EY, Chupyrkina AA, Pevzner IB, et al. Myoglobin causes oxidative stress, increase of NO production and dysfunction of kidney’s mitochondria. Biochim Biophys Acta. 2009;1792:796–803. PMID:19545623
- Sahu BD, Kuncha M, Putcha UK, et al. Effect of metformin against cisplatin induced acute renal injury in rats: A biochemical and histoarchitectural evaluation. Exp Toxicologic Pathol. 2013;65:933–940. PMID:23395153
- Bin DS, Yang SK, Zhou QY, et al. Mitochondria-targeted peptides prevent on contrast-induced acute kidney injury in the rats with hypercholesterolemia. Ren Fail. 2013;35:1124–1129. PMID:23879473
- Walker PD, Shah SV. Gentamicin enhanced production of hydrogen peroxide by renal cortical mitochondria. Am J Physiol. 1987;253:C495–9.
- Bernardi P, Krauskopf A, Basso E, et al. The mitochondrial permeability transition from in vitro artifact todisease target. FEBS J. 2006;273:2077–2099.
- Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol. 2002;192:131–137. PMID:12115719
- Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. PMID:15800626
- Jankauskas SS, Andrianova NV, Alieva IB, et al. Dysfunction of kidney endothelium after ischemia/reperfusion and its prevention by mitochondria-targeted antioxidant. Biochemistry (Moscow). 2016;81:1538–1548. PMID:28259131
- Dikalova AE, Itani HA, Nazarewicz RR, et al. Sirt3 impairment and SOD2 hyperacetylation in vascular oxidative stress and hypertension. Circ Res. 2017;121:564–574. PMID:28684630
- Kwon O, Phillips CL, Molitoris BA. Ischemia induces alterations in actin filaments in renal vascular smooth muscle cells. Am J Physiol Renal Physiol. 2002;282:F1012–9. PMID:11997317
- Bienert GP, Schjoerring JK, Jahn TP. Membrane transport of hydrogen peroxide. Biochim Biophys Acta. 2006;1758:994–1003. PMID:16566894
- Koyner JL, Sher Ali R, Murray PT. Antioxidants. Do they have a place in the prevention or therapy of acute kidney injury? Nephron Exp Nephrology. 2008;109:e109–17. PMID:18802373
- Zorov DB, Plotnikov EY, Jankauskas SS, et al. The phenoptosis problem: what is causing the death of an organism? Lessons from acute kidney injury. Biochemistry (Mosc). 2012;77:742–753. PMID:22817538
- Plotnikov EY, Silachev DN, Jankauskas SS, et al. Mild uncoupling of respiration and phosphorylation as a mechanism providing nephro- and neuroprotective effects of penetrating cations of the SkQ family. Biochemistry (Moscow). 2012;77:1029–1037. PMID:23157263
- Jankauskas SS, Plotnikov EY, Morosanova MA, et al. Mitochondria-targeted antioxidant SkQR1 ameliorates gentamycin-induced renal failure and hearing loss. Biochemistry (Moscow). 2012;77:666–670. PMID:22817467
- Plotnikov EY, Chupyrkina AA, Jankauskas SS, et al. Mechanisms of nephroprotective effect of mitochondria-targeted antioxidants under rhabdomyolysis and ischemia/reperfusion. Biochimica Biophysica Acta Molecular Basis Disease. 2011; 1812:77–86. PMID:20884348. DOI:10.1016/j.bbadis.2010.09.008.
- Plotnikov EY, Morosanova MA, Pevzner IB, et al. Protective effect of mitochondria-targeted antioxidants in an acute bacterial infection. Proc Natl Acad Sci U S A. 2013;110:E3100–8. PMID:23898194
- Mitchell T, Rotaru D, Saba H, et al. The mitochondria-targeted antioxidant mitoquinone protects against cold storage injury of renal tubular cells and rat kidneys. J Pharmacol Exp Ther. 2011;336:682–692. PMID:21159749
- Mukhopadhyay P, Horváth B, Zsengellér Z, et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic Biol Med. 2012;52:497–506. PMID:22120494
- Ramsey H, Wu MX. Mitochondrial anti-oxidant protects IEX-1 deficient mice from organ damage during endotoxemia. Int Immunopharmacol. 2014;23:658–663. PMID:25466275
- Birk AV, Liu S, Soong Y, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. 2013;24:1250–1261. PMID:23813215
- Szeto HH, Liu S, Soong Y, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. 2011;22:1041–1052. PMID:21546574
- Mizuguchi Y, Chen J, Seshan SV, et al. A novel cell-permeable antioxidant peptide decreases renal tubular apoptosis and damage in unilateral ureteral obstruction. Am J Physiology-Renal Physiol. 2008;295:F1545–53. PMID:18784263
- Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18.
- Antonenko YN, Denisov SS, Silachev DN, et al. A long-linker conjugate of fluorescein and triphenylphosphonium as mitochondria-targeted uncoupler and fluorescent neuro- and nephroprotector. Biochimica Et Biophysica Acta - General Subjects. 2016; 1860:2463–2473. PMID:27450891. DOI:10.1016/j.bbagen.2016.07.014.
- Zakharova VV, Pletjushkina OY, Galkin II, et al. Low concentration of uncouplers of oxidative phosphorylation decreases the TNF-induced endothelial permeability and lethality in mice. Biochimica Biophysica Acta Molecular Basis Disease. 2017;1863:968–977. PMID:28131916
- Romaschenko VP, Zinovkin RA, Galkin II, et al. Low concentrations of uncouplers of oxidative phosphorylation prevent inflammatory activation of endothelial cells by tumor necrosis factor. Biochemistry Biokhimii︠A︡. 2015;80:610–619. PMID:26071781
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. PMID:3769170
- Kapitsinou PP, Haase VH. Molecular mechanisms of ischemic preconditioning in the kidney. Am J Physiol Ren Physiol. 2015. ajprenal.00224.2015. PMID:26311114. DOI:10.1152/ajprenal.00224.2015
- Cochrane J, Williams BT, Banerjee A, et al. Ischemic preconditioning attenuates functional, metabolic, and morphologic injury from ischemic acute renal failure in the rat. Ren Fail. 1999;21:135–145.
- Park KM, Chen A, Bonventre JV. Prevention of kidney ischemia/reperfusion-induced functional injury and JNK, p38, and MAPK kinase activation by remote ischemic pretreatment. J Biol Chem. 2001;276:11870–11876. PMID:11150293
- Weng X, Shen H, Kuang Y, et al. Ischemic postconditioning inhibits the renal fibrosis induced by ischemia-reperfusion injury in rats. Urology. 2012;80:484 e1–7. PMID:22578919
- Diwan V, Jaggi AS, Singh M, et al. Possible involvement of erythropoietin in remote renal preconditioning- induced cardioprotection in rats. J Cardiovasc Pharmacol. 2008;51:126–130. PMID:18287879
- Silachev DN, Isaev NK, Pevzner IB, et al. The mitochondria-targeted antioxidants and remote kidney preconditioning ameliorate brain damage through kidney-to-brain cross-talk. PloS One. 2012;7:e51553. PMID:23272118
- Sedaghat Z, Kadkhodaee M, Seifi B, et al. Remote preconditioning reduces oxidative stress, downregulates cyclo-oxygenase-2 expression and attenuates ischaemia-reperfusion-induced acute kidney injury. Clin Exp Pharmacol Physiol. 2013;40:97–103. PMID:23240616
- Hausenloy DJ, Lim SY, Ong SG, et al. Mitochondrial cyclophilin-D as a critical mediator of ischaemic preconditioning. Cardiovasc Res. 2010;88:67–74. PMID:20400621
- Devalaraja-Narashimha K, Diener AM, Padanilam BJ. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am J Physiol Renal Physiol. 2009;297:F749–59. PMID:19553348
- Ying Y, Padanilam BJ. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis? Cell Mol Life Sci. 2016;73:2309–2324. PMID:27048819
- Lemoine S, Pillot B, Rognant N, et al. Postconditioning with cyclosporine a reduces early renal dysfunction by inhibiting mitochondrial permeability transition. Transplantation. 2015;99:717–723. PMID:25793558
- Singh D, Chander V, Chopra K. Cyclosporine protects against ischemia/reperfusion injury in rat kidneys. Toxicology. 2005;207:339–347. PMID:15664262
- Bao H, Ge Y, Zhuang S, et al. Inhibition of glycogen synthase kinase-3Β prevents NSAID-induced acute kidney injury. Kidney Int. 2012;81:662–673. PMID:22258319
- Bao H, Ge Y, Wang Z, et al. Delayed administration of a single dose of lithium promotes recovery from AKI. J Am Soc Nephrol. 2014;25:488–500. PMID:24408869
- Talab SS, Elmi A, Emami H, et al. Protective effects of acute lithium preconditioning against renal ischemia/reperfusion injury in rat: role of nitric oxide and cyclooxygenase systems. Eur J Pharmacol. 2012;681:94–99. PMID:22342279
- Ma D, Lim T, Xu J, et al. Xenon preconditioning protects against renal ischemic-reperfusion injury via HIF-1 activation. J Am Soc Nephrol. 2009;20:713–720. PMID:19144758
- Zhao H, Yoshida A, Xiao W, et al. Xenon treatment attenuates early renal allograft injury associated with prolonged hypothermic storage in rats. FASEB J. 2013;27:4076–4088. PMID:23759444
- Jia P, Teng J, Zou J, et al. Intermittent exposure to xenon protects against gentamicin-induced nephrotoxicity. PLoS ONE. 2013;8. PMID:23737979. DOI:10.1371/journal.pone.0064329
- Wang Y, Huang WC, Wang CY, et al. Inhibiting glycogen synthase kinase-3 reduces endotoxaemic acute renal failure by down-regulating inflammation and renal cell apoptosis. Br J Pharmacol. 2009;157:1004–1013. PMID:19508392
- Lochhead KM, Kharasch ED, Zager RA. Anesthetic effects on the glycerol model of rhabdomyolysis-induced acute renal failure in rats. J Am Soc Nephrol. 1998;9:305–309.
- Meerwein C, Korom S, Arni S, et al. The effect of low-dose continuous erythropoietin receptor activator in an experimental model of acute Cyclosporine A induced renal injury. Eur J Pharmacol. 2011;671:113–119. PMID:21968143
- Eshraghi-Jazi F, Nematbakhsh M, Pezeshki Z, et al. Sex differences in protective effect of recombinant human erythropoietin against cisplatin-induced nephrotoxicity in rats. Iran J Kidney Dis. 2013;7:383–389. PMID: 24072151
- Salahudeen AK, Haider N, Jenkins J, et al. Antiapoptotic properties of erythropoiesis-stimulating proteins in models of cisplatin-induced acute kidney injury. AJP: Ren Physiol. 2008;294:F1354–65. PMID:18385271
- Nemoto T, Yokota N, Keane WF, et al. Recombinant erythropoietin rapidly treats anemia in ischemic acute renal failure. Kidney Int. 2001;59:246–251. PMID:11135077
- Sharples EJ, Patel N, Brown P, et al. Erythropoietin protects the kidney against the injury and dysfunction caused by ischemia-reperfusion. J Am Soc Nephrol. 2004;15:2115–2124. PMID:15284297
- Yang FL, Subeq YM, Chiu YH, et al. Recombinant human erythropoietin reduces rhabdomyolysis-induced acute renal failure in rats. Injury. 2012;43:367–373. PMID:22209169
- Mitra A, Bansal S, Wang W, et al. Erythropoietin ameliorates renal dysfunction during endotoxaemia. Nephrol Dial Transplant. 2007;22:2349–2353. PMID:17438005
- Stoyanoff TR, Todaro JS, Aguirre MV, et al. Amelioration of lipopolysaccharide-induced acute kidney injury by erythropoietin: involvement of mitochondria-regulated apoptosis. Toxicology. 2014;318:13–21. PMID:24561306
- Harman D. The aging process. Proc Natl Acad Sci U S A. 1981.;78:7124–7128. PMID: 6947277
- Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Moscow). 2005;70:200–214. PMID:15807660
- Trifunovic A, Wredenberg A, Falkenberg M, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. PMID:15164064
- Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, et al. Corrigendum: mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature. 2016;542:124. PMID:27926738
- Bailey PJ, Webster GC. Lowered rates of protein synthesis by mitochondria isolated from organisms of increasing age. Mech Ageing Dev. 1984;24:233–241. PMID:6425575
- Pokkunuri I, Ali Q, Asghar M. Grape powder improves age-related decline in mitochondrial and kidney functions in fischer 344 rats. Oxid Med Cell Longev. 2016;2016. DOI:10.1155/2016/6135319
- Yarian CS, Toroser D, Sohal RS. Aconitase is the main functional target of aging in the citric acid cycle of kidney mitochondria from mice. Mech Ageing Dev. 2006;127:79–84. PMID:16289253
- Choksi KB, Nuss JE, DeFord JH, et al. Mitochondrial electron transport chain functions in long-lived Ames dwarf mice. Aging. 2011;3:754–767. PMID:21934186
- Rocha-Rodrigues S, Santos-Alves E, Coxito PM, et al. Combined effects of aging and in vitro non-steroid anti-inflammatory drugs on kidney and liver mitochondrial physiology. Life Sci. 2013;93:329–337.
- Larsson N-G. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem. 2010;79:683–706. PMID:20350166
- Ahlqvist KJ, Hämäläinen RH, Yatsuga S, et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in polg mutator mice. Cell Metab. 2012;15:100–109. PMID:22225879
- Katajisto P, Döhla J, Chaffer CL, et al. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science. 2015;348:340–343. PMID:25837514
- Popkov VA, Plotnikov EY, Lyamzaev KG, et al. Mitodiversity. Biochemistry Biokhimiia. 2015;80:532–541. PMID:26071770
- Chen H, Xing B, Wang L, et al. Aged kidneys are refractory to ischemic postconditioning in a rat model. Ren Fail. 2014;36:1575–1580. PMID:25156634
- Zorov DB, Filburn CR, Klotz L-O, et al. Reactive oxygen species (Ros-Induced) Ros Release. J Exp Med. 2000;192:1001–1014. PMID:11015441
- Boddi M, Sacchi S, Lammel RM, et al. Age-related and vasomotor stimuli-induced changes in renal vascular resistance detected by Doppler ultrasound. Am J Hypertens. 1996;9:461–466. PMID:8735177
- Fuiano G, Sund S, Mazza G, et al. Renal hemodynamic response to maximal vasodilating stimulus in healthy older subjects. Kidney Int. 2001;59:1052–1058. PMID:11231360
- Kim D. The activation of NF-kB through Akt-induced FOX01 phosphorylation during aging and its modulation by caloric restriction. Biogerontology. 2008;9:33–47.
- Zhu J, Rebecchi MJ, Glass PSA, et al. Cardioprotection of the aged rat heart by GSK-3beta inhibitor is attenuated: age-related changes in mitochondrial permeability transition pore modulation. Am J Physiol Lung Cell Mol Physiol. 2011;300:H922–30. PMID:21217064
- Saotome M, Katoh H, Yaguchi Y, et al. Transient opening of mitochondrial permeability transition pore by reactive oxygen species protects myocardium from ischemia-reperfusion injury. Am J Physiology-Heart Circulatory Physiol. 2009;296:H1125–32. PMID:19202002
- Hausenloy D, Wynne A, Duchen M, et al. Transient mitochondrial permeability transition pore opening mediates ischemic preconditioning-induced protection. Circulation. 2004;109:1714–1717.
- Arias E, Koga H, Diaz A, et al. Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol Cell. 2015;59:270–284. PMID:26118642
- Moreno-Blas D, Gorostieta-Salas E, Castro-Obregón S. Connecting chaperone-mediated autophagy dysfunction to cellular senescence. Ageing Res Rev. 2018;41:34–41. PMID:29113832