
ABSTRACT
Membrane lipid rafts are highly ordered microdomains and essential components of plasma membranes. In this work, we demonstrate that azurin uptake by cancer cells is, in part, mediated by caveolin-1 and GM-1, lipid rafts’ markers. This recognition is mediated by a surface exposed hydrophobic core displayed by azurin since the substitution of a phenylalanine residue in position 114 facing the hydrophobic cavity by alanine impacts such interactions, debilitating the uptake of azurin by cancer cells. Treating of cancer cells with azurin leads to a sequence of events: alters the lipid raft exposure at plasma membranes, causes a decrease in the plasma membrane order as examined by Laurdan two-photon imaging and leads to a decrease in the levels of caveolin-1. Caveolae, a subset of lipid rafts characterized by the presence of caveolin-1, are gaining increasing recognition as mediators in tumor progression and resistance to standard therapies. We show that azurin inhibits growth of cancer cells expressing caveolin-1, and this inhibition is only partially observed with mutant azurin. Finally, the simultaneous administration of azurin with anticancer therapeutic drugs (paclitaxel and doxorubicin) results in an enhancement in their activity, contrary to the mutated protein.
Introduction
Azurin is a protein from bacterial origin (Pseudomonas aeruginosa) which in the last years has been studied as an anticancer agent. We and others have identified different modes of action that account for the therapeutic effects of both the entire protein and one lead peptide, p28 [Citation1–Citation5]. The 28 amino acid sequence was first identified as the domain responsible for the penetration of azurin into cancer cells [Citation6] and further studied as an anticancer peptide. Indeed, p28 has already completed two phase I clinical trials in both adult patients with various tumors and in children with tumors of the central nervous system [Citation7,Citation8]. Both trials ended with positive indications in relation to the possible use of this peptide as an anticancer agent.
Mechanistically, the uptake of azurin or the peptide was suggested as being energy-dependent, with no observable loss of membrane integrity, independent of membrane-bound glycosaminoglycans, dependent on the cholesterol within the cell membrane and strongly associated with caveolae [Citation2]. The depletion of cholesterol from plasma membranes using methyl-β-cyclodextrin; the disruption of microtubules with nocodazole; or the inhibition of late endosomes/lysosomes activity with monensin, all led to a decrease in the penetration of p28 [Citation2,Citation4].
Caveolae are a subset of lipid rafts with unique physical and biological properties. These membrane microdomains are enriched in glycosphingolipids (including sphingomyelin, ceramide and gangliosides, like GM-1) and cholesterol that can act as lipid-ordered platforms within the plasma membrane [Citation9,Citation10]. In particular, caveolae are defined as wide pits in the plasma membrane that contain caveolin-1 (Cav1) in oligomers of 140–150 proteins. Caveolin-1 and caveolae in general are now recognized as important mediators for signal transduction, plasma membrane organization and composition. Additionally, they have been associated with the phenomenon of drug resistance in cancer either by contributing to altered membrane lipid and protein composition, higher membrane order and altered signaling pathways [Citation11,Citation12].
Research with azurin or the derived peptide in the past years have demonstrated that a number of signaling pathways associated with tumor progression and angiogenesis, such as FAK/Src or PI3K/Akt signaling are attenuated after treatment in several cancer models [Citation3,Citation5,Citation13]. Also, functional evidences associated to adhesion to extracellular matrices, invasion and migration of cells are also weakened [Citation5,Citation14], linking the cellular responses to azurin to its possible effects at lipid rafts, which may act as the main gate of azurin to cancer cells. Moreover, binding to both caveolin-1 and GM-1, structural components of caveolae/lipid rafts, is strongly associated to hydrophobic enriched regions in the binding partners [Citation15,Citation16], and azurin harbours in its tertiary structure two sheets arranged around an hydrophobic core region with the particular characteristic of being exposed and available for interactions [Citation17]. As such, in this work we evaluated the impact that azurin treatment has at the membrane level, particularly at the lipid raft organization level in terms of membrane order and lipid packaging, and identified a new region of azurin outside p28 that is also involved, at least in part, in the uptake of this protein by cancer cells.
Results
F114A substitution alters the uptake rate of azurin in cancer cells
In order to study the importance of the hydrophobic core in the process of cell uptake by azurin, we first analyzed this aromatic core by a computational analysis. In ) the residues that contribute most to hydrophobic core are highlighted in blue on the amino acid sequence representation. We analyzed three aromatic residues of the hydrophobic patch (Y108, F111, F114) ()), and the computational results showed that F114 is facing the central cavity of the hydrophobic core on a loop with the hydrophobic ring exposed in the azurin hydrophobic cavity (,)). This positioning of F114 makes it an ideal place to introduce a mutation in order to study the importance of possible hydrophobic interactions mediated by this region. On the other hand, Y108 and F111 are located on a β-sheet and a mutation there might affect azurin structure and functionality. Furthermore, while previous studies regarding azurin but not related to its anticancer activity had shown that the mutation F114A did not alter significantly the protein structure, the in silico prediction of the mutation F114A reveals changes in the hydrophobic core of azurin that reduces the hydrophobicity in the surface of the protein ( [Citation18]. For all these reasons, we choose to mutate azurin in the F114 residue, replacing it by alanine. The exposure of cells (MCF-7 and HeLa) to 50 and 100µM of both proteins for 30 minutes and 2 hours clearly demonstrated that the mutant protein is much less efficient in entering the cells when compared to the WT protein ()). In parallel, both WT and mutated protein were labeled with the fluorescent molecule Atto 390 NHS ester by coupling it with the amines of proteins. Labeled proteins (10µM) were mixed with unlabeled protein (to a final concentration of 100µM) and added to MCF-7 cells for 2 hours, after which cells were imaged under the confocal microscope. It is possible to see that while the WT protein is detected both at the plasma membrane and inside the cells, lower intensity is observed for the mutated protein. This suggests that the F114A mutant protein is less efficient in the processes of recognition and uptake by the cancer cells than the WT protein ()).
Figure 1. Point mutation of phenylalanine 114 delays azurin entry in cancer cells. (a) Amino acids sequence of azurin from Pseudomonas aeruginosa. Hydrophobic patches are highlighted in blue. (b) 3D structural view of azurin depicting surface hydrophobicity (top view) and ribbon 3D structure (lateral view) of both WT azurin (left panel) and F114A mutant azurin (right panel), identifying the structural positioning of phenylalanine amino acid residue side chain. The amino acid change was generated using SPDB Viewer (v4.01). Surface hydrophobicity was generated using PyMol. (c) Entry of azurin WT and F114A mutant in MCF-7 and HeLa cells. Cells were exposed to 50–100µM of both proteins for 30 minutes (left) and 2 hours (right), after which cells were lysed and protein entry inside cells was determined by Western blot. A cropped representative image of Western blot is depicted. Full-length Western blot images are displayed as supplementary information. (d) Confocal scanning microscopy of Atto 390-labelled WT or F114A azurin proteins. Labeled proteins (10µM) were mixed to non-labeled proteins (to a final concentration of proteins of 100µM), and MCF-7 cells were exposed for 2 hours before fixing and imaging. Cell nuclei are displayed labelled with NucRed and presented in red, labeled WT and mutated azurin proteins are displayed in green
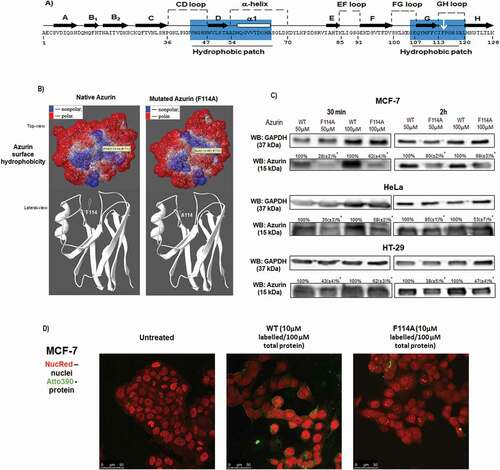
Blocking GM-1 ganglioside reduces the penetration of azurin in cancer cells
CTxB, a component of a heat-labile enterotoxin produced by Vibrio cholerae, is a probe commonly used to label and/or detect GM1 ganglioside which can be used as marker for lipid rafts, since the GM-1 ganglioside has an abundant localization in these membrane microdomains [Citation19,Citation20]. GM-1 is important for recognition and trafficking of a great number of protein and viruses. To evaluate the involvement of GM-1 in the recognition and cell entry of azurin in cancer cells (due to the apparent hydrophobic nature involved in this process), we hypothesized that pre-treating the cells with CTxB might decrease the availability of GM-1 to mediate the entry of azurin in cancer cells, if this route is used by the protein. MCF-7 and HT-29 cancer cells were exposed to CTxB during 10 minutes before the addition of 50μM of WT azurin or the mutated protein ()). The controls were the cells exposed to bacterial proteins in the absence of CTxB. In both models, we observed that when cells were firstly subjected to CTxB a decrease in the total levels of WT and F114A azurin inside cancer cells was observed suggesting that availability of GM-1 is important for the entry of azurin in the cells. For the WT protein, a decrease of about 40% was observed, whereas for the mutated protein the decrease was more accentuated (~50%). Despite it is not possible to rule-out that the effect is due to a change in membrane raft morphology due to lipid-raft crosslinking by CTxB, our results indicate that the intact GM-1/raft morphology is needed for an entirely efficient azurin entry and that the hydrophobicity provided by this phenylalanine aromatic residue and its exposure to the surface of the protein may be important to mediate the entry of the protein by means of its interaction with GM-1. Having determined that the presence of GM-1 is important for the recognition and entry of azurin, we went on to study how both proteins impacted the distribution of GM-1 at the plasma membrane. Using a fluorescence tagged (Alexa488) form of CTxB, cells were treated for both a short (30 minutes) as well as a longer (48 hours) period of time with the different azurin proteins after which cells were labeled with Alexa488-CTxB (10 minutes), before fixing and observed under the scanning confocal microscope. Overall, we observed that untreated cells have a more selective plasma membrane staining (), upper panel) which upon treatment with the WT protein becomes much more undefined at the membrane level (), middle panel). Our results seem to indicate that the involvement of GM-1 in the entry of azurin dislodges, at least in part, this ganglioside from the plasma membrane. As for the F114A azurin mutant, the results seem to be intermediate between the untreated cells and cells treated with the WT protein (), lower panel). The increase in the intracellular staining appears to be maintained but at a more moderate level, nevertheless there is also a more evident membrane staining than in the cells treated with the WT protein. The signal intensity in the cells was quantified in the plasma membrane and in the cytoplasm in the interior of the cells (excluding the nucleus region), and represented as a ratio of the plasma membrane signal intensity over the cytoplasm intensity signal for the different proteins ()). As it is seen, the treatment with the WT protein decreases the ratio of signal in the plasma membrane, contrary to the F114A protein, particularly right after the addition of the proteins (30 minutes). Being a regularly marker used to assess lipid rafts localization, the displacement here observed suggests that WT azurin may alters the membrane profile of these ordered domains.
Figure 2. Phenylalanine 114 is important for interaction with GM-1. (a) Azurin interacts with GM-1. Blocking GM-1 with CTxB impairs azurin entry for both WT and F114A azurin mutant. GM-1 was blocked by adding CTxB for 10 minutes, before being treated with 50μM of WT or the mutated azurin proteins for 30 minutes, in MCF-7 (left) and HT-29 (right) cells. A cropped representative image of Western blot is depicted and results are presented as the ratio of band intensity of target protein between azurin treated samples and control samples, both normalized to their respective GAPDH or actin band intensity (* p < 0.05; two-tailed t-student test). Full-length Western blot images are displayed as supplementary information. (b) The effects of WT and F114A azurin in the cells’ lipid raft organization. Cells were grown and treated with both azurin proteins at 100µΜ, 30 minutes or 48h. The glycosphingolipid GM-1 of lipid rafts is marked with CTxB-Alexa488 (green) and the nuclei are stained with DAPI (blue). CTxB-Alexa488 was added for 10 minutes at a final concentration of 1μg/mL right before cells were fixed in formaldehyde and prepared for visualization under the confocal fluorescence microscope. (c) Fluorescence signal intensity was quantified in Image J software for at least 20 cells from experiments performed in different days, as explained in Material and Methods Section. Results are represented as an average of the ratio between fluorescence intensity signal in the plasma membrane and the intracellular signal, excluding the nucleus, ± SD. Results were compared by analysis of variance ANOVA using GraphPad Prism (ver 6)
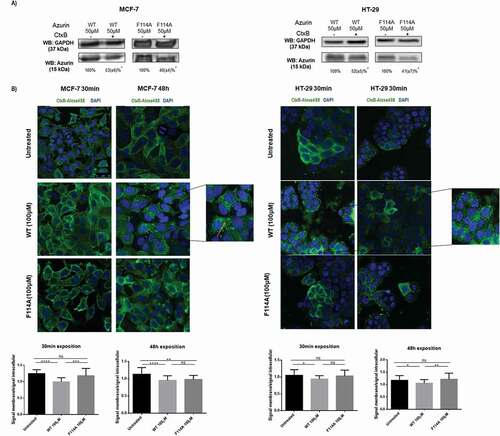
Silencing of caveolin-1 inhibits cell entry of WT azurin
Previous reports had described that azurin co-localizes with caveolin-1 almost immediately after penetration into cells. Caveolin-1, together with GM-1, is a component of caveolae, a particular type of lipid rafts, in particular. Being an important mediator of several signaling pathways, numerous studies already indicated that part of the mechanism by which caveolin-1 does so is through the binding to several other proteins, therefore controlling part of their signaling activity [Citation16,Citation21,Citation22]. Furthermore, not only caveolin-1 is recognized as an important mediator of endocytosis, it has been strongly associated to the entry of azurin in cancer cells [Citation3,Citation4]. Therefore, to understand how caveolin-1 protein levels in the membrane could also control the velocity of the entry process for azurin, we silenced the CAV1 gene with siRNA to test if under those conditions azurin was less capable to enter in cancer cells. After silencing of CAV1 expression, cells were exposed to 50μM of WT azurin or the mutated F114A, for 30 minutes, as for the previous assay. As seen in ), we observed a decrease in the levels of WT azurin entry for both MCF-7 and HeLa cells but for F114A mutated azurin, no decrease was observed. The protein levels were normalized by the respective actin level. It is interesting to note that for F114A azurin, the same levels of azurin are detected in the cells non-silenced for caveolin-1 (Control siRNA) when compared to the cells subjected to the treatment with WT azurin after silencing caveolin-1. In addition, when compared with its own control, no decrease in the entry of the mutant protein was observed, which suggests that the mediation of Cav1 for azurin entry is more important to the WT protein than for that mutated in the F114 amino acid residue. Nevertheless, when CAV1 gene expression is down-regulated, the entire membrane organization may be altered, for instance caveolae that are caveolin-1-enriched smooth invaginations of the plasma membrane that form a subset of lipid rafts, can no longer be formed in the absence of caveolin-1. Therefore, such inhibition may cause alterations that can explain the differences observed for the F114A mutant protein.
Figure 3. Phenylalanine 114 is important for interaction with caveolin-1. (a) Entry of WT azurin and mutated F114A azurin upon silencing of caveolin-1 by siRNA. Cells were treated with 50μM of WT or the mutated azurin proteins for 30 minutes. Control siRNAs with proteins are the control conditions. A cropped representative image of Western blot is depicted and results are presented as the ratio of band intensity of target protein between azurin treated samples and control samples, both normalized to their respective GAPDH or actin band intensity (* p < 0.05). Full-length Western blot images are displayed as supplementary information. (b) Energy transfer efficiencies of WT and F114A mutant azurin proteins labeled with Att0 390 in the presence of FITC-labed CSD (Caveolin Scaffolding Domain) peptide. Values were calculated as described in the Methods section. (c) Co-immunoprecipitation of caveolin-1 and azurin in MCF-7 and HeLa cells treated with azurin 100 µM for 30 minutes. An antibody to caveolin-1 was incubated with total cell lysates and used to precipitate it from both control and azurin treated total cell lysates. Proteins were separated in SDS-Page gels transferred to membranes which were probed with both anti-caveolin-1 and anti-azurin antibodies. Full-length Western blot images are displayed as supplementary information. (d) Azurin causes a decrease in the protein levels of caveolin-1. A single dose of azurin (50-100 µM) for 48 hours leads to a dose-dependent decrease in two different cancer cell types (MCF-7 and HeLa). A cropped representative image of Western blot is depicted and results are presented as the ratio of band intensity of target protein between azurin treated samples and control samples, both normalized to their respective GAPDH band intensity (* p < 0.05)
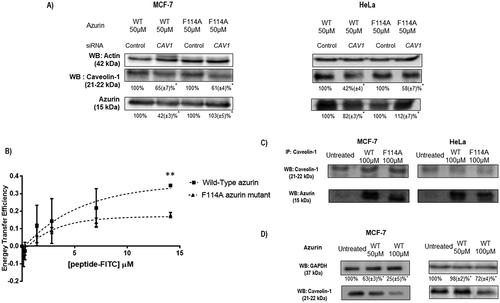
Azurin binds to caveolin-1 scaffolding domain in vitro
One particular domain of caveolin-1 that is involved in multiple interactions with other proteins is its caveolin-scaffolding domain (CSD). In order to study possible interactions between the CSD and WT azurin or F114A azurin, we carried out FRET measurements between WT and F114A mutant azurin labeled with Atto390 NHS and CSD labeled with FITC. We collected fluorescence spectra for WT and mutant azurin labeled proteins in the absence and in the presence of increasing concentrations of FITC-CSD labeled peptide (Supplementary Information). The occurrence of energy transfer was readily visualized as a decrease in the donor fluorescence of about 35% for the WT for the highest concentration of the FITC-peptide (14μM). For the mutated protein, we observed a decrease in the intensity of the protein of only about 17%, indicating that in the same experimental conditions, a higher energy transfer occurs for the WT protein than for the F114A mutant, suggesting a higher interaction with the WT protein. The FRET efficiency was, at this concentration, 35% for the WT and only 17% for the F114A mutant protein ()).
Furthermore, we also observed that the WT azurin is immunoprecipitated with caveolin-1 in much higher levels that the mutated protein after 30 minutes of exposure to each protein ()) in both MCF-7 and HeLa cells, reflecting both the decreased capacity to recognize and enter in cancer cells and a diminished ability of the mutant protein of azurin to bind to caveolin-1.
Azurin leads to a decrease in caveolin-1 total protein levels
The mechanism of azurin/p28 cell entry has been associated to caveolin-1 and lipid rafts. However, those indications were related mainly to the interactions between the protein and the peptide almost immediately after the cells were exposed to any of them, but no information is available regarding the impact of azurin on the total protein levels of caveolin-1. We have demonstrated that azurin causes an increase in endocytosis in breast cancer cells [Citation14], while in lung cancer cells the exposure to azurin led to a decrease in the membrane stiffness probed by Atomic Force Microscopy (AFM) to which alterations at the caveolin-1 levels may be related [Citation5]. In this work, azurin treatment of MCF-7 and HeLa cancer cells for 48h led to a decrease in the levels of caveolin-1, in a dose-dependent manner ()). Shorter treatment times produced similar results, while onset times for depletion of caveolin-1 levels were observed to be cell line dependent (data not shown).
Azurin decreases plasma membrane order
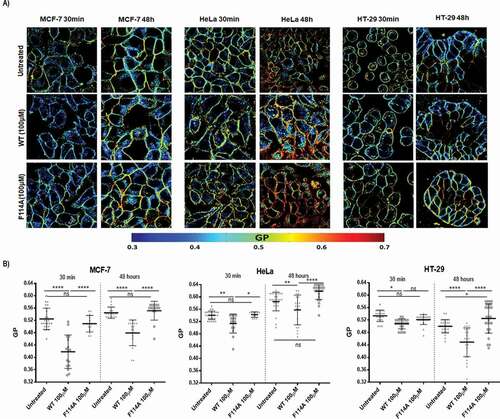
Having determined that exposition to WT azurin leads to a decreased content of lipid raft components GM-1 and Cav1, we next sought to investigate if the interactions of azurin with caveolar lipid raft components could be associated to changes in the organization of the plasma membrane of the cancer cell lines under study. It has been demonstrated that variations in the order of the plasma membranes in living cells can be detected using the environment-sensitive fluorescent probe Laurdan and two-photon excitation microscopy [Citation19,Citation20]. Laurdan fluorescence emission spectra is sensitive to changes in membrane order by exhibiting a 50nm red shift as the order changes from the liquid-ordered to a liquid-disordered state (see references [Citation23,Citation24]). The changes in emission spectra are the result of alterations in the penetration of water molecules within the lipid bilayer and it can be quantified by calculating the generalized polarization (GP) value as demonstrated in e.g [Citation23]. The value of GP can vary between 1 and −1 (complete exposure to bulk water). Therefore, higher GP values are associated to higher membrane ordering and a less fluid plasma membrane.
To determine Laurdan GP values several images were acquired ()) and Laurdan fluorescence spectral shifts in the plasma membrane were quantified through the GP function as stated in the Methods section. Laurdan GP measurements were made for exposure times of 30 minutes and 48 hours to both WT and mutant F114A azurin proteins. In both time points, exposure to azurin WT caused a decrease in the GP values measured in the plasma membranes, indicating that exposition to azurin causes a decrease in the plasma membrane order ()). On the other hand, the F114A azurin mutant did not altered the GP values when compared to untreated cells, in accordance to what was observed in previous sections. The GP values found in control conditions are in line with published values for these cell lines [Citation20].
Figure 4. Impact of WT or F114A mutant azurin protein on the membrane fluidity of cancer cell lines MCF-7, HeLa and HT-29. (a) Cells were loaded with 5μM of Laurdan after incubation with azurin proteins at 100μM for 30 minutes or 48h. Laurdan GP values were determined as described in Material and Methods. Representative Laurdan GP images are shown. (b) Average GP values after incubation with azurin proteins are shown for the plasma membranes for the three cell lines. WT azurin causes a decrease in the average GP value in all three cell lines, either from 30 minutes or 48 hours of exposure. Average GP values are expressed as mean ± SD from at least 15 individual cells in each condition. Results were compared by analysis of variance ANOVA using GraphPad Prism (ver 6)
Azurin enhances the activity of chemotherapeutic drugs
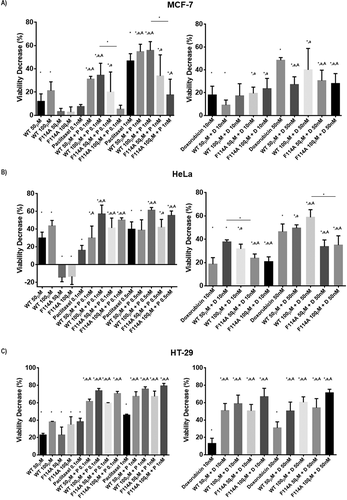
With the above demonstration of the interaction of azurin with caveolin-1 and GM-1 and taking in consideration that caveolin-1/lipid rafts are important mediators of numerous cellular processes, including the resistance to anticancer drugs, we determined the degree of cytotoxicity for two chemotherapeutic drugs (paclitaxel and doxorubicin), alone or in the presence of both WT or mutated azurin. Interestingly, in MCF-7 and HeLa cells the proteins alone had different behaviors in terms of cytotoxicity. One single delivery of the WT protein results in a viability loss of about 20% for MCF-7 and 40% for HeLa cells, whereas the F114A mutant did not produce such an effect, causing a lower reduction in cell viability (,)). However, for the HT-29 colon cancer cell line, these differences were not observed with both proteins showing a similar effect in terms of cytotoxicity. Such a result might be explained by the low expression of caveolin-1 in that cell line (supplementary Figure 1). Overall, for the combined treatments, the results point to the increase in the cytotoxicity observed when the proteins are present, using drug concentrations that inhibit cell proliferation in values close to 20–50% (). In MCF-7 and HeLa cells, an increase is seen particularly for paclitaxel at the lower doses tested (0.1 and 1nM), and the effects are more evident for the WT protein. Indeed, in the case of F114A mutated azurin the values seem to reflect the less pronounced effect demonstrated by the protein in terms of perturbing the organization of lipid rafts. On the other hand, for doxorubicin the effects of the combinations did not produce a significant increment in the action of the drug in these cell lines. However, in both cell lines the WT protein had a more pronounced effect that the F114A mutant. In the HT-29 cell line, the two proteins significantly enhanced the action of both tested drugs, possibly reflecting the higher cytotoxicity that the proteins alone present in these cells.
Figure 5. The effect of the combination of WT or F114A mutant azurin proteins with chemotherapeutic agents (paclitaxel and doxorubicin) was determined by MTT assay. Paclitaxel or Doxorubicin, WT azurin or F114A mutant azurin were added alone or in combination for 72hours in MCF-7 (a), HeLa (b) and HT-29 (c) cells. Concentrations are shown above each data point. MTT reagent was added to each well and percentage change in absorbance at 570nm in treated cells relative to untreated controls. Results are represented as percentage of viability decrease (VD), determined as 100% (control) – % of proliferation for each treatment condition. Values represent the mean ± SD. * p < 0.05 each condition vs untreated cells; a p < 0.05 combination vs protein alone; A p < 0.05 combination vs drug alone
Discussion
The microdomains present in the plasma membranes of cells, such as caveolae/lipid rafts, are important mediators in signal transduction pathways associated to several cellular phenomena such as membrane trafficking, cytoskeletal organization, motility, polarity and endocytosis [Citation10,Citation25,Citation26]. Many of these signaling pathways were in the last years identified as targets of azurin, which has been proposed as a therapeutic protein in anticancer therapies [Citation3,Citation5,Citation13]. The entire protein and one lead peptide, p28, target p53 and signaling pathways mediated by VEGF/FAK, Src, PI3K/Akt, and the EGFR signaling, in all cases contributing to their attenuation, therefore altering the capacity of several cancer cell models to progress. Over the years, the mechanisms by which azurin orderly enters in the cells without disrupting the membrane have been identified, pointing to an important interaction with caveolae/lipid rafts. Firstly, the p28 amino acid fragment was identified as a mediator of azurin entry [Citation6], but later other domains of azurin were also associated to its anticancer activity, namely the C-terminal region (aa 96–113). This domain has a structural similarity with ephrinB2 at the GH loop, and a peptide composed of these amino acids, demonstrated significant cytotoxicity against the prostate cancer cell line DU142 expressing a functional form of ephrinB2, contrary to the natural ligand [Citation27]. Azurin-derived peptides comprising this region were also later improved and used to radiosensitize cancer cells [Citation28]. In this work, we propose that the C-terminal region of azurin, which is rich in hydrophobic amino acid residues like phenylalanine and tyrosine, also contributes to the recognition of azurin in cancer cell lines, mainly due to its contribution to hydrophobic favored interactions. We computationally analyzed several point mutations in azurin, replacing three hydrophobic amino acids (Y108, F111 and F114) by alanine, and looked into the alterations that such mutations could cause to the hydrophobicity pattern exhibited at the topological surface of the protein ()). From these, the F114 residue demonstrated to be the one with the most impact, since its side chain is exposed to the surface of the protein and its replacement by alanine, led to an alteration in the hydrophobicity displayed at the surface of azurin ()). This mutated protein exhibited less effective penetration of cancer cells after exposure for 30 minutes and 2 hours ()).
An important component of lipid rafts is GM-1. GM-1 is a glycosphingolipid present in high abundance in lipid rafts [Citation15,Citation29]. Carbohydrate-protein interactions have been vastly studied, and it is known that in some sugars, the clustering of three or more adjacent C-H groups caused by the characteristic steric disposition of hydroxyl groups creates hydrophobic patches on the sugar surface that can establish apolar interactions with hydrophobic epitopes in proteins, most notably the aromatic rings of tryptophan, tyrosine and phenylalanine residues [Citation30]. The aromatic amino acids identified within the C-terminal amino acid sequence of azurin might interact with these sugar components of caveolae in the first recognition steps to enter cancer cells. In fact, pre-treatment of cells with tunicamycin which inhibits the N-linked glycosylation significantly reduced the entry of the peptide [Citation2]. The demonstration that there is a decrease in the levels of these proteins within cancer cells, when GM-1 is blocked with a powerful ligand such as CTxB reinforces those observations ()). It should be noted that there seems to be a greater decrease in the entry of the mutated protein in comparison to the WT protein in all cell lines, when these cells are previously exposed to CTxB. With these results one can infer that one of the mechanisms of azurin recognition by cancer cells acts at the level of GM-1 ganglioside and aromatic amino acids on the azurin structure, being F114 important for that recognition, which seems, according to our results, to occur both in caveolin-1 positive and negative cell lines. Indeed, in cell lines expressing low levels of caveolin-1, like HT-29, blocking GM-1 seems to be enough to delay azurin entry. GM-1 is already recognized as a mediator of protein accumulation in lipid membranes in other pathologies like in in Alzheimer´s disease where it facilitates the accumulation of amyloid β-protein and related peptides in neural plasma membrane cells [Citation31–Citation33].
CTxB is also a marker widely used to assess lipid rafts in cancer. An Alexa488-CTxB version was used to assess what happens to these membrane microdomains upon exposure to both proteins. Contrary to untreated cells, where the membrane staining of CTxB is clear, in cells treated with WT protein, GM-1 is dislocated from the plasma membrane within 30 minutes of exposure, an effect that is maintained for at least 48 hours ()). For the mutated protein, that observation is less clear than for the WT protein. The membrane staining is maintained to a higher extent than the WT protein, suggesting once again a stronger affinity of WT azurin to GM-1 than the mutated protein. This higher affinity then leads to a higher internalization of this lipid raft marker ()).
Interestingly, when we silenced caveolin-1 with siRNA, the WT protein was also affected in the entry process, reinforcing that caveolin-1 is another important mediator in that process ()). Regarding the F114A mutant, its entry was not delayed in the cells in which CAV1 expression was down-regulated; instead, it is close to the wt protein levels, which points to the different behavior of the two proteins in the absence of caveolin-1. Nevertheless, when caveolin-1 is silenced with siRNA, the molecular organization of the membrane is likely to be altered due to the structural role played by caveolin-1, which may lead to different observations when compared to an assay where both proteins are added to the cells for 30 minutes with no previous changes induced.
The interaction of each protein with the CSD was also analyzed using an FITC-labeled CSD peptide since the interactions of several proteins with caveolin-1 and other lipid rafts’ components are mainly determined by hydrophobic motifs in the proteins, being the CSD a mediator of such interactions with multiple signaling proteins. We used FRET to determine possible interactions of WT azurin with the FITC-CSD peptide, observing a higher energy transfer efficiency for the wt protein than for the mutant protein which suggests that azurin interacts directly with caveolin-1 through this domain. These results also suggest that the wildtype and mutated azurin show significant differences in affinity for the interaction with caveolin-1, offering an additional explanation for the less effective entry of the mutant protein relatively to the WT protein. In the cellular context, after exposition of both proteins to the cells for 30 min, when caveolin-1 is immunoprecipitated, azurin is detected in complex with it, being the WT protein detected in higher levels than the mutant, reflecting the lower levels of the protein that bind to and are up taken by the cells. Furthermore, when the total levels of caveolin-1 are analyzed by Western blot, after 48 hours of exposure to the WT protein, it is possible to see that caveolin-1 is less abundant in the cells. Combined with the displacement observed for GM-1, the decrease in the caveolin-1 protein suggests that additional changes in the profile of membrane protein composition, mainly within lipid rafts, are altered by azurin, which prompted us to analyze the membrane fluidity of cells treated with azurin.
Lipid rafts are clusters of specific lipids, cholesterol and sphingolipids, forming highly condensed relatively ordered nano-domains, distinctive from the rest of the membrane [Citation34], very important for the lateral organization of cellular membranes. The complexity of lipid and protein packing, rotation and lateral diffusion gives rise to what is called the cell membrane fluidity [Citation35]. Indeed, the biophysical features of membranes severely impact many of the phenomena they regulate, and may have a huge impact on drug resistance [Citation36]. After identifying the molecular changes above mentioned, we assessed the effects of azurin on the organization of plasma membranes, by assessing the membrane fluidity with the fluorescent probe Laurdan. Our results indicate clearly that the fluidity of the plasma membrane increases after exposure to WT azurin, as evaluated from changes in the Laurdan GP values. The entry of azurin preferentially through caveolae/lipid rafts, probably removing caveolin-1 from the membrane and perturbing the raft organization, seems to cause a structural change in the plasma membrane organization, possibly decreasing the fraction of liquid-ordered membrane/domains (). These results are in accordance with the previous results obtained by us and others, in which the exposure to azurin not only attenuates signaling pathways associated to motility, adhesion and invasiveness but also to biophysical changes at the plasma membrane level, effects attributed to the presence of lipid rafts. Indeed, plasma membranes define the boundaries of live cells, playing a major role in all cell functions and in the communications cell establish with the exterior. Being caveolin-1 a major integral protein of caveolae, the ability to target it can be of extreme importance to establish new therapeutic strategies. Its role in cancer is still controversial, with reports indicating that cancer development and progression can be either associated to an increase in caveolin-1 levels or its absence, varying across different models [Citation37–Citation42]. However, it is becoming clear that the development of the multidrug resistance phenomenon is strongly associated to its increase, as well as to the increase in the membrane stiffness, blocking the ability of several drugs to efficiently penetrate into the cells to reach their intracellular targets [Citation11,Citation12]. In this context, we tested if the alterations caused at the plasma membrane order here observed could be related to an increase in the sensitivity to two chemotherapeutic drugs, paclitaxel and doxorubicin (). Indeed, the p28 peptide was very recently associated to such an increase in the sensitivity to these drugs in different cell models [Citation43], mainly due to its effects of p53 stabilization. Therefore, we tested in parallel both the WT and the F114A mutant protein. Interestingly, from the three cell lines tested, in which all of them the delay in F114A entry was observed, in the caveolin-1 negative HT-29 cell lines, no significant differences were observed in the toxicity generated by the proteins alone. On the contrary, for caveolin-1 expressing MCF-7 and HeLa cells, significant differences were observed, with the mutant displaying much lower cytotoxicity than the WT protein, pointing to the identified decrease in the interaction with the CSD. In terms of potentiating the effects of both drugs, such different effects were also only detected in these cell lines, albeit in HT-29 the effects were more accentuated in all the combinations tested, indicating maybe that the higher toxicity levels created by the proteins were dominating. In MCF-7 and HeLa cells, the combinations with the WT protein led to a potentiation of the chemotherapeutic drugs, with a statistical significant difference in favor of the WT protein for the highest concentrations of proteins and drugs observed in most cases (,)). Therefore, in these cases, the molecular effects of WT azurin at the membrane level seem to contribute positively to the action of the drugs. Indeed, recently the interaction of doxorubicin with model cell membranes was evaluated to determine how the interaction of the drug alone could impact the organization of the plasma membranes in cancer cells and how it could affect its delivery efficacy and the contribution to chemoresistance. It was seen that this drug has a preference to locate in more ordered microdomains, where for example, P-glycoprotein is also preferentially located. For the authors, this could explain how the drug is available to be a substrate for efflux by this protein which is also located in these structures [Citation44]. It is then possible that, by decreasing the more ordered domains, one of the mechanisms by which azurin enhances the activity of this drug is by perturbing this preferential localization of the drug reducing its availability to be efflux pumps.
In sum, the combination of azurin with these drugs benefited their cytotoxic effect and statistical differences were observed between the WT and the mutant azurin tested in the lines where caveolin-1 is more expressed. A number of recent studies indicate that acting at the membrane level may be a new strategy to target cancer cells, enhancing the cytotoxicity of chemotherapeutic drugs, and by doing so, to also decrease the severe systemic toxic effects they cause, since lower concentrations are needed to achieve the same therapeutic response. Drugs that target the membrane lipid composition and/or organization are now receiving attention as adjuvants for cancer therapy [Citation45,Citation46] (reviewed in [Citation36,Citation47]). In this context, the action of azurin over different cancer cell models is a contribution to the broad anticancer action this protein demonstrates. In particular, it would be interesting in the future to evaluate the effects over drug resistant cells, where the biophysics of the plasma membranes seems to be a major contributor to that phenomenon.
Materials and methods
Human cancer cell lines and cell cultures
Three human cancer cell models have been used: the MCF-7 breast cancer cell line, the HeLa cervical cancer cell line and the HT-29 colon cancer cell line. These cell lines were purchased from European Collection of Authenticated Cell Cultures. All of them were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco® by Life Technologies), supplemented with 10% of heat-inactivated Fetal Bovine Serum (FBS; Gibco® by Life Technologies), 100IU/mL penicillin and 100mg/mL streptomycin (PenStrep, Invitrogen). These cell lines were passed between 2 to 3 times per week, by chemical detaching with 0.05% of trypsin. Cells were grown at 37°C in a humidified chamber containing 5% of CO2 (Binder CO2 incubator C150).
Hydrophobic surface analysis of azurin
Surface properties of azurin from P. aeruginosa PAO 1 (PDB entry 1jzg) were evaluated using the program PyMol. This program was used to identify and score clusters of hydrophobic atoms (named as hydrophobic patches). Swiss-Pdb Viewer (Deep View) was used to produce azurin 3D structure cartoons.
Construction of a site-directed mutation in the azurin gene of P. aeruginosa PAO1
The pWH844 vector with azurin-encoding gene from Pseudomonas aeruginosa PAO 1 [Citation13], was extracted using the ZR Plasmid MiniprepTM-Classic kit (ZymoResearch), according to manufacturer’s instructions. The Quick Change II site-directed mutagenesis kit (Agilent Technologies) was used for the site-directed mutagenisi. Forward and reverse primers used for the substitution of Phenyalalanine at position 114 by Alanine were respectively: 5´-GTA CAT GTT CTT CTG CAC CGC GCC GGG CCA CTC CGC GCT G-3´ and 5´-CAG CGC GGA GTC GCC CGG CGC GGT GCA GAA GAA CAT GTA C-3´. PCR was carried out in a 50μL mixture using 30 ng template plasmid DNA from the plasmid pWH844 with the WT azu gene, 5μL of 10x Reaction Buffer, 1.25μL of each primer (at 10μM), 1μL of dNTP mix and double-distilled water (ddH2O) to a final volume of 50μL. After that, 1μL of PfuTurbo DNA polymerase (2.5 U/μL) was added to the mixture.
Bacteria growth, over-expression, extraction and purification of WT azurin or mutated protein
The continuous production of azurin was performed as described in Bernardes et al., 2013. For the mutated protein F114A, a final concentration of 0.5mM of IPTG was added to induce protein overexpression.
Protein extraction and western blot analysis
For protein extraction, plates with incubated cells, after the desired incubation times with the azurin proteins, were placed on ice and wells were washed twice with PBS 1x. Then, cells lysed in 100μL of Catenin Lysis Buffer (CLB; 1% Triton X-100, 1% Nonidet-P40 in deionized PBS) supplemented with 1:7 proteases inhibitor (Roche Diagnostics GmbH) and 1:100 phosphatases inhibitor (Cocktail 3, Sigma Aldrich) for 10 minutes at 4°C. Then, the cells were scratched, collected and vortexed three times (10 seconds each), centrifuged (14.000 rpm, 4°C, 10 minutes; B. Braun Sigma-Aldrich 2K15) and the pellet was discarded, collecting the supernatant containing proteins. Total protein quantification was done using by a Quantification Protein Kit (Bradford, BioRad). 10-20µg of total protein per sample were prepared with Laemmli buffer. Proteins were transferred onto nitrocellulose membranes (BioRad) using Trans-Blot TurboTM system (BioRad). Membranes were blocked with 5% (w/v) non-fat dry milk in PBS containing 0.5% (v/v) Tween-20 (PBS-T) for 1 hour, incubated with different primary antibodies: β-actin (1:1000, sc-1616) or GAPDH (G-9) (1:1000, sc – 365062) were used as loading controsl; caveolin-1 (N-20) (1:1000, sc-894) In order to evaluate azurin expression, an anti-azurin antibody was produced through immunization of one goat with purified azurin, obtained as described above (1:1000 dilution). The resulting immunized serum was then purified by protein A affinity chromatography (SicGen, Portugal) and purity was checked by SDS-PAGE. All the membranes were incubated overnight at 4°C and then washed three times for 5 minutes with PBS-T. These were then incubated for 1 hour with secondary antibodies (anti-rabbit, anti-mouse or anti-goat, 1:2000, Santa cruz Biotechnology), conjugated with horseradish peroxidase Proteins were detected through the addition of ECL reagent (Pierce) as a substrate and exposed captured the chemiluminescense by Fusion Solo (Viber Lourmat) equipment. When loading controls were necessary, samples were run in the same gels, and after transfer to the membrane, the membranes were cut according to the protein MWs and probed with the antibodies in separate. Three experiments were independently performed and representative results are shown. Signal quantifications were performed using ImageJ and results are presented as the ratio between the signal intensities in azurin treated samples to untreated cells, both normalized to the respective GAPDH or actin band intensities.
For co-immunoprecipitation experiments, lysed cells were incubated with 10μL of primary antibody anti-caveolin-1 (Cell Signaling, 3238) in an agitator overnight at 4°C. The next day, 100μL of beads (Protein G Agarose, Thermo Scientific) were incubated with the mixture of lysate and antibodies, in an agitator during 2 hours at room temperature. After that time, 500μL of IP buffer (Thermo Scientific) were added, in order to precipitate the mixture, and then it was centrifuged (2500xg during 3 minutes), 10 times. At every time, the supernatant was discarded. To elute the proteins from the beads, the pellet was incubated twice with 50μL of Elution Buffer (Thermo Scientific), each time during 5 minutes, and then it was centrifuged (2500xg during 2 minutes) and the supernatant was recovered. To neutralize the supernatant, 10μL of Neutralization Buffer (Thermo Scientific) were added.
To the pellet, that contains the beads, 60μL of sample buffer were added and to the supernatant with the Neutralization Buffer it was added 30μL of sample buffer. 20μL per sample were denatured at 95°C during 5 minutes, and then separated by electrophoresis in a SDS-PAGE. Western Blot was performed as previously described.
GM-1 inhibition with Cholera Toxin Subunit B (CTxB)
Breast MCF-7 and colon HT-29 cell were plated in 6-well plates with 5 × 105 cells per well and left to adhere and grow overnight in a CO2 incubator (5%) at 37°C. The following day, medium was collected and cells were treated with 1μg/mL of CTxB (Invitrogen, Alexa Fluor® 488 conjugate) in DMEM during 10 minutes. After this time, medium was again collected and cells were treated with 50μM of WT azurin or mutated protein in DMEM. The plates were placed for 30 minutes at 37°C.
Confocal microscopy-Cholera Toxin Subunit B (CTxB)
MCF-7, and HT-29 cell lines were seeded on µ-Slide 8 well IBIDI treated chambers (Ibidi) with 5 × 104 cells/well. These cells were left to adhere and grow overnight in a CO2 incubator (5%) at 37°C. In the next day, medium was collected and cells were treated with 100μM of WT azurin or mutated protein in medium containing 10% FBS and 1% PenStrep, for 48h. After this time, medium was again collected and cells were treated with 1μg/mL of CTxB (Invitrogen, Alexa Fluor® 488 conjugate) in DMEM during 10 minutes. Afterwards, the chambers were rinsed three times with PBS 1x. For fixation, cells in coverslips were immersed in 3.7% formaldehyde for 20 minutes at room temperature. After seven washing steps in PBS 1x, Vectashield with DAPI was added and cells were observed in a Leica TCS SP5 inverted confocal microscope (Leica Microsystems CMS GmbH; model no. DMI6000) with a 63x water (1.2-numerical-aperture) apochromatic objective. For measurement of CTxB-Alexa488 fluorescence, the sample was excited at 488 nm, while emission was collected within the 500–600 nm range. For measurement of DAPI fluorescence, the sample was excited by two-photon excitation at 780 nm with a Ti:sapphire laser (Mai Tai, Spectra-Physics, Darmstadt, Germany), while emission was collected within the 400–450 nm range. Signal intensity was quantified for each cell using the Image J software. In each cell, a mask identifying the plasma membrane was defined and signal from within these pixels was quantified as plasma membrane associated intensity. Next, the signal from the within each cell was also quantified, excluding the nucleus, and defined as the intracellular signal. For each condition, quantifications are presented as ratios between the plasma membrane signal over the intracellular signal. Results were compared by analysis of variance ANOVA (Newman-Keuls Multiple Comparisons, using GraphPad Prism ver 6).
Sirna transfection of human cancer cells lines
MCF-7 and HeLa cell lines were plated in 6-well plates with 5 × 105 cells/well. These cells were left to adhere and grow overnight in a CO2 incubator (5%) at 37°C. Prior to transfection, 100nM of Control siRNA (sc-37007, Santa Cruz Biotechnology) and Caveolin-1 siRNA (sc-29241, Santa Cruz Biotechnology) were mixed with Lipofectamine® 2000 (Thermo Fisher Scientific). For this, 25µL of each siRNA were added to 225µL of DMEM and 10µL of Lipofectamine® 2000 were added to 240µL of the same medium, according to the manufacturer’s instructions. After 5 minutes, the prepared solutions were mixed gently to form siRNA-lipofectamine complex. This mixture was incubated for 20 minutes at room temperature and added to 2mL of DMEM in the respective well. After 6–8 hours in a CO2 incubator (5%) at 37°C, the medium was removed and fresh medium containing 10% FBS and 1% PenStrep was added to each well. The appropriate time for observing the decrease in the Cav1 protein levels was determined by Western blot, determining that 24 hours post-transfection was adequate to perform the azurin entry assay. After this time, cells were treated with 50μM of WT azurin or mutated protein. The plates were placed for 30 minutes at 37°C. To determine the levels of azurin entry in Cav1-silenced cells, a Western blot was performed as described above (20µg of total protein per sample).
Interaction between Cav1-CSD and azurin: FRET measurements
To observe the interaction between the Caveolin-1 Scaffolding Domain (CSD; amino acids 82–101 of Cav 47) and WT or mutated azurin, we made used of Fluorescein-5-IsoThioCyanate (FITC)-labeled CSD peptide (Pepmic). WT and F114A azurin mutant proteins were labeled with Atto 390 NHS ester (Sigma), according to the manufacturer’s instruction. Briefly, 150μM of each protein was incubated with the dye (molar ratio 2:1), for 2h at room temperature. Reaction was stopped with by adding a solution of NH2OH 1.2M pH 8.5, for 1h at room temperature. After this, the mixture was centrifuged at 18000xg, 10 min, to remove any precipitated dye, before dialysis against 10mM Phosphate buffer pH 7.4, in slide-A-lyzer 3.5 kDa cut-off, overnight at 4°C. For FRET measurements, WT and F114A azurin concentration was 2μM. The donor (Atto390-WT or Atto390-F114A)-only fluorescence spectra were acquired with 390 nm excitation and measured over the emission wavelength range of 400 to 470 nm, since no acceptor (FITC-labeled CSD peptide) emits there. The FITC-labeled CSD peptide was titrated from 0 to 14μM. Fluorescence measurements were carried out with a SLMAminco 8100 Series 2 spectrofluorimeter (Rochester) with double excitation and emission monochromators (MC-400), in a right angle geometry. The light source was a 450-W Xe arc lamp and the reference a Rhodamine B quantum counter solution. Quartz cuvettes (1 × 1cm) from Hellma Analytics were used. The FRET efficiency, E, was calculated on the basis of the quenching of the donor fluorescence intensity in the FRET complex relative to the donor-only emission in the presence of the buffer. E was calculated using the following equation: E = 1-(FDA/FA) after all intensities were normalized to the intensities in the absence of FITC-peptide. All spectra were corrected for background and inner filter effects[Citation48].
Two-Photon excitation microscopy – GP determination
MCF-7, HeLa and HT-29 cells were cultured on µ-Slide 8 well IBIDI treated chambers (Ibidi) with 5 × 104 cells and treated with WT azurin or F114A mutated protein (100µM). After 48 hours, medium was collected and cells were washed twice with PBS. After that, DMEM with 5μM of Laurdan was added and the cells were incubated in a CO2 incubator at 37°C for 20 minutes. Samples were examined on a Leica TCS SP5 (Leica Microsystems CMS GmbH, Mannheim, Germany) inverted microscope (model no. DMI6000) with a 63× water (1.2-numerical-aperture) apochromatic objective. Two photon excitation microscopy data was obtained by using Leica TCS SP5 inverted microscope with a Ti:sapphire laser (Mai Tai, Spectra-Physics, Darmstadt, Germany) as the excitation light source. The excitation wavelength was set to 780 nm and the fluorescence emission was collected at 400–460nm and 470–550nm to calculate the GP images. Laurdan GP images were obtained through a homemade software based on a MATLAB environment, with the GP value defined as GP = (/400–460 – G. I470-530)/(/400–460 + G. I470-530), were G is the calibration factor for the experimental setup. G is obtained from imaging Laurdan in DMSO (with the predetermined GP = 0.01) using the same experimental conditions as those set for the measurements in living cells [Citation20]. Control conditions correspond to untreated cells. Dark counts were subtracted to all intensity values. In the analysis, only Regions of Interest (ROI) corresponding to the plasma membranes in each cell were selected, restricting therefore the analysis to this cellular component. At least 15 independent cells were analyzed per condition and all the experiments were done in independent days. Results were compared by analysis of variance ANOVA (Newman-Keuls Multiple Comparisons, using GraphPad Prism ver 6).
MTT cell viability assay
MTT [3-(4,5 dimethylthiazol-2-yl-2,5 tetrazolium bromide)] assays were used to determine the proliferation rate of MCF-7, HT-29 and HeLa human cancer cells lines after they were treated with WT azurin combined with drugs. The drugs used in these assays were paclitaxel, an antimitotic agent, and doxorubicin, a DNA-damaging drug (Sigma Life Science). All these cell lines were seeded in 96-well plates (3 replicates) with a density of 2 × 104 MCF-7 cells/well, 1 × 104 HeLa cells/well and 2 × 104 HT-29 cells/well. These cells were left to adhere and grow overnight in a CO2 incubator (5%) at 37°C. In the next day, medium was collected and cells were treated with 50 and 100μM of WT azurin together with 0.1, 0.5 or 1nM of paclitaxel or 10 or 50nM of doxorubicin in medium containing 10% FBS and 1% PenStrep. The plates were placed for 72 hours at 37°C. After this time, 20μL of MMT reagent (5mg/mL) were added to each well and incubated at 37°C for 3.5 hours. Reaction was stopped with the addition of 150μL of a solution 40mM HCL in isopropanol. MTT formazan formed was spectrophotometrically read at 590nm in a microplate reader (SpectroStarNano, BMG LABTECH). Untreated cells were used as control, in order to determine the relative cell viability of treated cells.
Statistical analysis
For Western blot experiments, three independent replicates were performed. All p-values were calculated using Student’s t-test (two-tailed distribution, two-sample equal variance). Values of p < 0.05 were considered statistically significant (*: p < 0.05). In MTT cell viablility experiments, results were compared by analysis of variance ANOVA (Newman-Keuls Multiple Comparisons, using GraphPad Prism ver 6 or Statistica ver 13).
Author contributions
This study was conceived by AMF and NB. The experiments were designed by AMF, NB and FF. NB, ARG, BC and CP performed the experiments. SNP and FF assisted in the design and execution of confocal and two-photon microscopy experiments and protein-peptide interactions. NB wrote the manuscript. All authors read and approved the final manuscript.
Supplemental Material
Download MS Word (2.5 MB)Acknowledgments
The work presented was supported by scientific projects (PTDC/EBBBIO/100326/2008, and FAPESP/20107/2014) financed by the Portuguese Science and Technology Foundation (FCT). FCT also provides a post-doctoral research grants for Nuno Bernardes (SFRH/BPD/98162/2013) and Sandra N Pinto (SFRH/BPD/92409/2013), and PhD grant for Ana Rita Garizo (SFRH/BD/122636/2016). Fabio Fernandes acknowledges financial support from FCT with grant IF/00386/2015. The authors acknowledge the support of the national infrastructure PPBI-Portuguese Platform of BioImaging (supported by POCI-01’0145-FEDER-022122). This work has received funding from European Structural & Investment Funds through the COMPETE Programme and from National Funds through FCT under the Programme grant SAICTPAC/0019/2015. Funding received by iBB-Institute for Bioengineering and Biosciences from FCT (UID/BIO/04565/2013) and from Programa Operacional Regional de Lisboa 2020 (Project N. 007317) is acknowledged.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Yamada T, Goto M, Punj V, et al. Bacterial redox protein azurin, tumor suppressor protein p53, and regression of cancer. PNAS. 2002;99:14098–14103.
- Taylor BN, Mehta RR, Yamada T, et al. Noncationic peptides obtained from azurin preferentially enter cancer cells. Cancer Res. 2009;69:537–546.
- Mehta RR, Yamada T, Taylor BN, et al. A cell penetrating peptide derived from azurin inhibits angiogenesis and tumor growth by inhibiting phosphorylation of VEGFR-2, FAK and Akt. Angiogenesis. 2011;14:355–369.
- Yamada T, Mehta RR, Lekmine F, et al. A peptide fragment of azurin induces a p53-mediated cell cycle arrest in human breast cancer cells. Mol Cancer Ther. 2009;8:2947–2958.
- Bernardes N, Abreu S, Carvalho FA, et al. Modulation of membrane properties of lung cancer cells by azurin enhances the sensitivity to EGFR-targeted therapy and decreased β1 integrin-mediated adhesion. Cell Cycle [Internet]. 2016;15:1415–1424.
- Yamada T, Fialho AM, Punj V, et al. Internalization of bacterial redox protein azurin in mammalian cells: entry domain and specificity. Cell Microbiol. 2005;7:1418–1431.
- Warso MA, Richards JM, Mehta D, et al. A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. Br J Cancer 2013 ;108:1061–1070.
- Lulla RR, Goldman S, Yamada T, et al. Phase i trial of p28 (NSC745104), a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in pediatric patients with recurrent or progressive central nervous system tumors: A pediatric brain tumor consortium study. Neuro Oncol. 2016;18:1319–1325.
- Mollinedo F, Gajate C. Lipid rafts as major platforms for signaling regulation in cancer. Adv Biol Regul. 2015;57:130–146.
- Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Caveolae and signalling in cancer. Nat Rev Cancer. 2015;15:225–237.
- Lavie Y, Fiucci G, Liscovitch M. Upregulation of caveolin in multidrug resistant cancer cells: functional implications. Adv Drug Deliv Rev. 2001;49:317–323.
- Quest FG, Lobos-González L, Nuñez S, et al. The caveolin-1 connection to cell death and survival. Curr Mol Med. 2013;13:266–281.
- Bernardes N, Ribeiro AS, Abreu S, et al. The bacterial protein azurin impairs invasion and FAK/Src signaling in P-cadherin-overexpressing breast cancer models. PLoS One. 2013;19:e69023. Available from:.
- Bernardes N, Ribeiro AS, Abreu S, et al. High-throughput molecular profiling of a P-cadherin overexpressing breast cancer model reveals new targets for the anti-cancer bacterial protein azurin. Int J Biochem Cell Biol [Internet]. 2014 [cited 2014 Mar 15];50:1–9.
- Pang H, Le PU, Nabi IR. Ganglioside GM1 levels are a determinant of the extent of caveolae/raft-dependent endocytosis of cholera toxin to the Golgi apparatus. J Cell Sci. 2004;117:1421–1430.
- Vihanto MM, Vindis C, Djonov V, et al. Caveolin-1 is required for signaling and membrane targeting of EphB1 receptor tyrosine kinase. J Cell Sci. 2006;119:2299–2309.
- Bernardes N, Chakrabarty AM, Fialho AM. Engineering of bacterial strains and their products for cancer therapy. Appl Microbiol Biotechnol. 2013 ;97:5189–5199. Available from
- Yanagisawa S, Banfield MJ, Dennison C. The role of hydrogen bonding at the active site of a cupredoxin : the Phe114Pro Azurin variant. Biochemistry. 2006;45:8812–8822.
- Gaus K, Gratton E, Kable EPW, et al. Visualizing lipid structure and raft domains in living cells with two-photon microscopy. Proc Natl Acad Sci U S A. 2003;100:15554–15559.
- Owen DM, Rentero C, Magenau A, et al. Quantitative imaging of membrane lipid order in cells and organisms. Nat Protoc. 2012;7:24–35.
- del Pozo MA, Balasubramanian N, Alderson NB, et al. Phospho-caveolin-1 mediates integrin-regulated membrane domain internalization. Nat Cell Biol. 2005 ;7:901–908.
- Norambuena A, Schwartz MA. Effects of integrin-mediated cell adhesion on plasma membrane lipid raft components and signaling. Mol Biol Cell. 2011 ;22:3456–3464.
- Parasassi T, De Stasio G, Ravagnan G, et al. Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of Laurdan fluorescence. Biophys J. 1991;60:179–189.
- Parasassi T, Krasnowska EK. Laurdan and Prodan as polarity-sensitive fluorescent membrane probes. J Fluoresc. 1998;8:365–373.
- Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50.
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39.
- Chaudhari A, Mahfouz M, Fialho AM, et al. Cupredoxin-cancer interrelationship: azurin binding with EphB2, interference in EphB2 tyrosine phosphorylation, and inhibition of cancer growth. Biochemistry. 2007;46:1799–1810.
- Micewicz E, Jung C, Schaue D, et al. Small Azurin derived peptide targets ephrin receptors for radiotherapy. Int J Pept Res Ther. 2011;17:247–257.
- Ichikawa N, Iwabuchi K, Kurihara H, et al. Binding of laminin-1 to monosialoganglioside GM1 in lipid rafts is crucial for neurite outgrowth. J Cell Sci. 2009;122:289–299.
- del Carmen Fernández-Alonso M, Díaz D, Berbis MÁ, et al. Protein-carbohydrate interactions studied by NMR: from molecular recognition to drug design. Curr Protein Pept Sci. 2012;13:816–830.
- Yamamoto N, Matsubara T, Sato T, et al. Age-dependent high-density clustering of GM1 ganglioside at presynaptic neuritic terminals promotes amyloid β-protein fibrillogenesis. Biochim Biophys Acta Biomembr. 2008;1778:2717–2726.
- Wakabayashi M, Matsuzaki K. Ganglioside-induced amyloid formation by human islet amyloid polypeptide in lipid rafts. FEBS Lett. 2009;583:2854–2858.
- Evangelisti E, Cascella R, Becatti M, et al. Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Sci Rep. 2016;6:32721.
- Sezgin E, Levental I, Mayor S, et al. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol. 2017;18:361–374.
- Sengupta P, Baird B, Holowka D. Lipid rafts, fluid/fluid phase separation, and their relevance to plasma membrane structure and function. Semin Cell Dev Biol. 2007 ;18:583–590.
- Peetla C, Vijayaraghavalu S, Labhasetwar V. Biophysics of cell membrane lipids in cancer drug resistance: implications for drug transport and drug delivery with nanoparticles. Adv Drug Deliv Rev. 2013 ;65:1686–1698.
- Lee SW, Reimer CL, Oh P, et al. Tumor cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene. 1998;16:1391–1397.
- Shi Y, Tan S-H, Ng S, et al. Critical role of CAV1/caveolin-1 in cell stress responses in human breast cancer cells via modulation of lysosomal function and autophagy. Autophagy. 2015;11:769–784.
- Logozzi M, De Milito A, Lugini L, et al. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients.PLoS One. 2009 ;4:e5219.
- Chanvorachote P, Pongrakhananon V, Halim H. Caveolin-1 regulates metastatic behaviors of anoikis resistant lung cancer cells. Mol Cell Biochem. 2014;399:291–302.
- Nam KH, Lee BL, Park JH, et al. Caveolin 1 expression correlates with poor prognosis and focal adhesion kinase expression in gastric cancer. Pathobiology. 2013;80:87–94.
- Bourseau-Guilmain E, Menard JA, Lindqvist E, et al. Hypoxia regulates global membrane protein endocytosis through caveolin-1 in cancer cells. Nat Commun . 2016;7:11371.
- Yamada T, Das Gupta TK, Beattie CW. P28-Mediated activation of p53 in G2-M phase of the cell cycle enhances the efficacy of DNA damaging and antimitotic chemotherapy. Cancer Res. 2016;76:2354–2365.
- Alves AC, Magarkar A, Horta M, et al. Influence of doxorubicin on model cell membrane properties: insights from in vitro and in silico studies.Sci Rep. 2017;7:6343.
- Colin D, Limagne E, Jeanningros S, et al. Endocytosis of resveratrol via lipid rafts and activation of downstream signaling pathways in cancer cells. Cancer Prev Res (Phila). 2011 ;4:1095–1106.
- Lee EJ, Yun UJ, Koo KH, et al. Down-regulation of lipid raft-associated onco-proteins via cholesterol-dependent lipid raft internalization in docosahexaenoic acid-induced apoptosis. Biochim Biophys Acta - Mol Cell Biol Lipids. 2014;1841:190–203.
- Escribá PV, Busquets X, Inokuchi J, et al. Membrane lipid therapy: modulation of the cell membrane composition and structure as a molecular base for drug discovery and new disease treatment. Prog Lipid Res. 2015;59:38–53.
- Lakowicz JR. Principles of fluorescence spectroscopy. Joseph R. Lakowicz, editor. 3rd ed.; 2006, Springer US.