
ABSTRACT
The SWI/SNF ATP-dependent chromatin-remodeling complex is an important evolutionarily conserved regulator of cell cycle progression. It associates with the Retinoblastoma (pRb)/HDAC/E2F/DP transcription complex to modulate cell cycle-dependent gene expression. The key catalytic component of the SWI/SNF complex in mammals is the ATPase subunit, Brahma (BRM) or BRG1. BRG1 was previously shown to be phosphorylated by the G1-S phase cell cycle regulatory kinase Cyclin E/CDK2 in vitro, which was associated with the bypass of G1 arrest conferred by BRG1 expression. However, it is unknown whether direct Cyclin E/CDK2-mediated phosphorylation of BRM/BRG1 is important for G1-S phase cell cycle progression and proliferation in vivo. Herein, we demonstrate for the first time the importance of CDK-mediated phosphorylation of Brm in cell proliferation and differentiation in vivo using the Drosophila melanogaster model organism. Expression of a CDK-site phospho-mimic mutant of Brm, brm-ASP (all the potential CDK sites are mutated from Ser/Thr to Asp), which acts genetically as a brm loss-of-function allele, dominantly accelerates progression into the S phase, and bypasses a Retinoblastoma-induced developmental G1 phase arrest in the wing epithelium. Conversely, expression of a CDK-site phospho-blocking mutation of Brm, brm-ALA, acts genetically as a brm gain-of-function mutation, and in a Brm complex compromised background reduces S phase cells. Expression of the brm phospho-mutants also affected differentiation and Decapentaplegic (BMP/TGFβ) signaling in the wing epithelium. Altogether our results show that CDK-mediated phosphorylation of Brm is important in G1-S phase regulation and differentiation in vivo.
Abbreviations: A-P: Anterior-Posterior; BAF: BRG1-associated factor; BMP: Bone Morphogenetic Protein; Brg1: Brahma-Related Gene 1; Brm: Brahma; BSA: Bovine Serum Albumin; CDK: Cyclin dependent kinase dpp: decapentaplegic; EdU: 5-Ethynyl 2'-DeoxyUridine; EGFR: Epidermal Growth Factor Receptor; en: engrailed; GFP: Green Fluorescent Protein; GST: Glutathione-S-Transferase; HDAC: Histone DeACetylase; JNK: c-Jun N-terminal Kinase; Mad: Mothers Against Dpp; MAPK: Mitogen Activated Protein Kinase; MB:: Myelin Basic Protein; nub: nubbin; pH3: phosphorylated Histone H3; PBS: Phosphate Buffered Saline; PBT: PBS Triton; PFA: ParaFormAldehydep; Rb: Retinoblastoma protein; PCV: Posterior Cross-Vein; Snr1: Snf5-Related 1; SWI/SNF: SWitch/Sucrose Non-Fermentable; TGFβ: Transforming Growth Factor β; TUNEL: TdT-mediated dUTP Nick End Labelling; Wg: Wingless; ZNC: Zone of Non-Proliferating Cells
KEYWORDS:
Introduction
The SWI/SNF complex is a multi-subunit ATP-dependent chromatin remodeler, which acts to regulate gene expression by modulating nucleosome positioning, thereby “opening up” chromatin and facilitating the binding of the transcriptional machinery such as the TATA binding-protein [Citation1–Citation3]. In mammals, this 2MDa complex consists of either of the two ATPase catalytic subunits, Brahma (BRM) or Brahma-related gene-1 (BRG1), which share 74% identity [Citation4], together with 9–12 associated proteins known as BRG1-associated factors (BAFs) [Citation5]. The BRM/BRG1 subunits exhibit intrinsic ATP-dependent chromatin remodeling activity, independent of the other subunits, and are crucial for the complex’s primary function [Citation6]. In the genetically amenable vinegar fly, Drosophila melanogaster, the SWI/SNF complex has only one ATPase homologue, Brm, which shares 52% and 56% identity with mammalian BRG1 and BRM, respectively [Citation7].
This complex regulates several cellular processes; including signaling pathways, genomic stability, and the cell cycle (reviewed in [Citation8]). Although its main function is the modulation of gene transcription, SWI/SNF can exert its regulatory functions through mechanisms such as the compaction of mitotic chromatids [Citation9,Citation10], activation of kinases to mediate the DNA damage response [Citation11,Citation12] and acting as a scaffold for other proteins involved in DNA repair [Citation13]. Due to SWI/SNF’s role in a diverse range of cellular processes, deregulation of any of the subunits of this complex may lead to the development of diseases such as cancer. Indeed, several of SWI/SNF’s subunits are linked to cancer predisposition through frequent deletions and loss of heterozygosity, including BAF47/SNF5 (SNR1), a gene frequently mutated in pediatric rhabdoid tumors [Citation14–Citation19]. Other core subunits that are mutated in cancer include the ATPases BRM and BRG1 [Citation8,Citation20,Citation21].
BRM and BRG1 contain an LxCxE motif, necessary for their binding to the tumor suppressor Retinoblastoma (pRb) family of proteins [Citation22,Citation23]. Importantly, BRG1 interacts only with the hypo-phosphorylated form of pRb, and its over-expression in the mammalian carcinoma SW13 and C33a cells, which lack endogenous BRM or BRG1, results in G1 arrest [Citation22]. Additionally, BRM enhances the transcriptional repression of E2F/DP by the pRb/Histone Deacetylase Complex (HDAC) [Citation23,Citation24]. Therefore, the SWI/SNF complex functions cooperatively with the pRb/HDAC tumor suppressor complex to inhibit E2F/DP transcriptional activities and cell cycle progression. Upon mitogenic stimulation, G1-S phase regulatory cyclins (Cyclins D, E and A) are expressed, bind to, and activate the CDK4/6 or CDK2 protein kinases, which phosphorylate and inactivate pRb, thereby triggering E2F/DP target gene expression (reviewed in [Citation25]). In addition to pRb, other substrates of cyclin/CDKs are important for cell cycle progression [Citation26]. Importantly, Cyclin E/CDK2 binds to and phosphorylates components of the SWI/SNF complex, and cyclin E overexpression suppresses the ability of the BRG1-complex to induce cell cycle arrest in mammalian cells [Citation27,Citation28]. However, it is unknown whether the alleviation of BRG1-mediated cell cycle arrest by Cyclin E is a direct consequence of Cyclin E/CDK2-mediated phosphorylation of BRG1. Consistent with the results in mammalian cells, in Drosophila, Cyclin E/CDK2 also binds to SWI/SNF, and mutants of the SWI/SNF complex (Brm, Moira (BAP155) and Snr1) increase the number of S phase cells and suppress the small and disorganized eye phenotype of a hypomorphic Cyclin E mutant [Citation29,Citation30]. These data indicate that the SWI/SNF complex is an evolutionary conserved negative regulator of cell cycle progression that is regulated by Cyclin E/CDK2. However, whether cyclin/CDK-mediated phosphorylation of Brm directly controls cell cycle regulation during development is not known.
In this study, we show that Drosophila Brm can be phosphorylated by both Cyclin E/CDK2 and Cyclin A/CDK2 in vitro. To evaluate the functional role of Brm phosphorylation, we performed genetic studies in Drosophila using a CDK-site phospho-mimic mutant, brm-ASP, and a CDK-site phospho-blocking mutant, brm-ALA. We show that expression of the brm-ASP mutant overrides a pRb-induced G1-arrest within the zone of non-proliferating cells (ZNC) in the developing wing epithelium. brm-ASP expression also results in increased number of S phase cells in regions of normal cycling cells within the wing epithelium. Brm-ASP phenocopies an ATPase-dead dominant-negative Brm transgene (brm-DN) in G1-S phase regulation and tissue development. Conversely, the CDK-site phospho-blocking mutant of Brm, brm-ALA, does not dominantly affect the cell cycle, although reducing endogenous Brm complex function enables brm-ALA’s effect on inhibiting S phases to be revealed. Moreover, we show that the brm phospho-site mutants affect differentiation. Our phenotypic studies are consistent with the notion that direct phosphorylation of Brm by CDK inactivates the SWI/SNF complex, and reveal for the first time that this control is important for cell cycle regulation and differentiation during tissue development in Drosophila.
Results
Brm is phosphorylated by CDKs in vitro
Since Drosophila Brm can physically interact with Cyclin E/CDK2 [Citation29,Citation30], it is possible that it is phosphorylated by Cyclin E/A-CDK2, as occurs with mammalian BRG1 [Citation27]. Consistent with this possibility, an analysis of the Drosophila Brm protein sequence revealed 15 putative CDK phosphorylation sites (Ser/Thr Pro) ()). To determine whether Brm is phosphorylated by Cyclin E/A-CDK2, we conducted in vitro phosphorylation assays.
Figure 1. Brm is phosphorylated by CDKs in vitro.
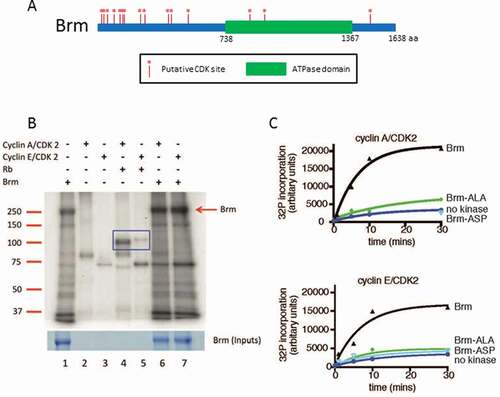
Recombinant full-length wild-type Brm (Brm-WT) with a C-terminal FLAG tag was expressed using the Baculoviral expression system and purified by immunoprecipitation (IP) using anti-FLAG antibody beads. The Brm protein was incubated with purified catalytically active recombinant human GST-tagged Cyclin A/CDK2 or GST-Cyclin E1/CDK2 (prepared in the Baculoviral expression system) in the presence of [γ-32P] ATP under CDK phosphorylation conditions, as described previously [Citation31]. As a positive control for the activity of the CDK complexes, purified mammalian MBP-pRb279-928 (prepared in E .coli as previously described [Citation31]) was used.
In the absence of added CDKs, background phosphorylation of Brm (~ 250 kDa), and various associated proteins was detected (), Lane 1). The CDK complexes alone resulted in phosphorylated bands of ~90 kDa and ~80 kDa, which respectively correspond to the molecular weights of the GST-Cyclin A and GST-Cyclin E [Citation32], (), Lanes 2–3). As expected, both CDK complexes phosphorylated MBP-pRb279-928 (~120 kDa) (), Lanes 4–5, blue box). Cyclin E/CDK2 and Cyclin A/CDK2 also phosphorylated Brm (), Lanes 6–7, red arrow). In addition, several other bands in the Brm IP also showed increased phosphorylation by the cyclin/CDK complexes: one example is the ~180 kDa band, which corresponds in size to that of the Brm complex subunits, BAP170, BAP180, or Moira (BAP155). To confirm that Brm was being phosphorylated in a CDK-dependent manner, we generated versions of Brm containing all 15 putative CDK phospho-sites mutated to either Alanine (Brm-ALA), which mimics constitutively non-phosphorylated Brm, or to Aspartate (Brm-ASP), which mimics constitutively phosphorylated Brm. We then expressed and purified the FLAG-tagged Brm-ASP and Brm-ALA constructs (containing all 15 CDK phospho-site mutations) as described above, and conducted time course kinase assays with Cyclin E/CDK2 and Cyclin A/CDK2 ()). This data revealed that whilst Brm-WT showed increasing phosphorylation over time with exogenously added Cyclin E/CDK2 and Cyclin A/CDK2 kinases, Brm-ALA showed only background level phosphorylation similar to the no kinase control over the time course ()). Similarly, Brm-ASP showed only background level phosphorylation with Cyclin E/CDK2 over the time course and with Cyclin A/CDK2 also showed only a background level of phosphorylation in the kinase assay after 30 min ()). The fact that Brm-ASP and Brm-ALA were phosphorylated at similar levels to the no kinase control indicates that there is a co-purifying kinase from the insect cells that phosphorylates Brm on non-CDK sites. Importantly, however, the Brm phospho-site mutants are not phosphorylated by the Cyclin A/E-CDK2 kinases. Altogether, our results show that Brm-WT is phosphorylated by Cyclin E/CDK2 and Cyclin A/CDK2 in vitro and that this phosphorylation is blocked by mutating the putative 15 Ser/Thr CDK phospho-sites.
Brm-ASP functions as a loss-of-function allele, whereas Brm-ALA functions as a gain-of-function allele
To assess the relevance of Brm CDK phosophorylation sites in vivo, transgenic flies containing GAL4(UAS) inducible constructs of brm-ALA and brm-ASP were generated. Genetic studies in Drosophila were then performed to characterize the functional relevance of Brm’s CDK phosphorylation status in vivo. We used the nub-GAL4 driver to express UAS-brm-WT and phospho-site mutant constructs in the developing wing pouch, which forms the adult wing (). Over-expression of brm-WT (via nub-GAL4) did not have a significant impact on the size of the wings (9.76x105 pixels versus control 9.72x105 pixels) and the wing appeared normal () compared with ), quantified in )). However, over-expression of brm-ASP had a severe impact on wing development ()). The wings were considerably smaller (1.89x105 pixels), with notching of the margin and ectopic vein tissue, as well as truncation of L5 and posterior cross-vein (PCV) veins (FIGURE 2C’, red arrow and blue circle, respectively). Interestingly, over-expression of brm-ALA also resulted in slightly smaller wings, with an average area of 9.27x105 pixels, and ectopic L2 wing vein tissue was consistently observed (with a frequency of 85.7%, n = 7) ()). These results suggest that the phosphorylation state of Brm on its CDK-phospho sites is crucial for normal Brm function during wing development.
Figure 2. Expression of CDK phosphorylation-site brm mutants affects wing development
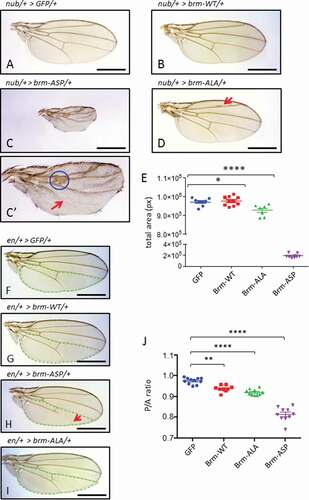
To further investigate the effect of brm phospho-site mutants on wing size, we used the engrailed-GAL4 (en-GAL4) driver to express the brm phospho-site mutant transgenes in the posterior compartment of the developing wing (-J)). Since the en-GAL4 driver is expressed in the posterior compartment of the developing wing, the developmental effects on posterior compartment development can be compared to the anterior compartment. Over-expression of brm-WT resulted in a slight reduction in Posterior/Anterior (P/A) ratio to 0.94 compared with the GFP control average of 0.97 () compared to ), quantified in )). A similar effect was also observed for the over-expression of brm-ALA, which decreased the P/A ratio of the adult wings to 0.92 () compared to ), quantified in )). Over-expression of brm-ASP strongly decreased the P/A ratio of the adult wings to 0.81 (), quantified in )). Importantly, expression of brm-ASP in the posterior region of the wing resulted in a truncation of the L5 wing vein (), red arrow), which phenocopies the wing defect seen with expression of the ATPase-dead dominant-negative brm transgene, brm-DN, via the en driver [Citation33]. Taken together, these results are consistent with the notion that brm-ASP acts as a loss-of-function mutant.
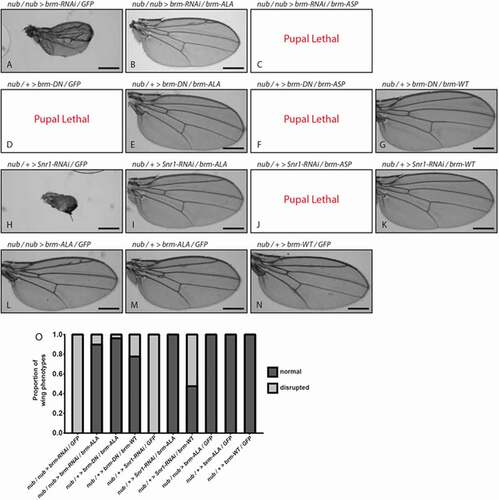
We then examined genetic interactions of brm-ASP and brm-ALA with Brm complex loss-of-function phenotypes to determine whether they were acting as loss- or gain-of-function alleles (). RNAi-mediated knockdown of Brm (brm-RNAi) in the developing wing, via the nub driver, resulted in a reduced adult wing ()), phenocopying the brm-ASP mutant. Co-expression of brm-ALA with brm-RNAi rescued the wing defect to normality, except for ectopic wing veins (), quantified in )), which were also observed with expression of brm-ALA alone in the wing (,M)). Conversely, co-expression of brm-ASP with brm-RNAi enhanced the phenotype to pupal lethality ()). Since the RNAi sequence targets brm mRNA (3649–3985 bp, transcript variant A) in the coding region, expression of the brm-ALA transgene might suppress the brm-RNAi phenotype due to titration of the brm-RNAi. Therefore, we tested the interaction of brm-ALA and brm-ASP with the dominant-negative brm transgene (UAS-brm-DN (UAS-brm-K804R)) containing a mutation in the ATPase domain, rendering it inactive without affecting its ability to associate with its partner proteins [Citation34]. nub-driven expression of brm-DN was pupal lethal ()), however co-expression of brm-ALA rescued the lethality resulting in normal wings in 95% of individauls (), quantified in )), whilst co-expression of brm-ASP did not alter the pupal lethal phenotype of brm-DN ()). Expression of UAS-brm-WT (which alone resulted in normal wings, )), was also able to rescue the nub> brm-DN lethality and somewhat restore normality to the adult wings, as expected due to titration of the Brm-DN protein away from the Brm complex, but not to the same degree as with brm-ALA (75% relative to 95% normal wings, ), quantified in )). We also examined genetic interactions of brm-ALA, brm-WT and brm-ASP with the Brm complex gene, Snr1 (-J)). RNAi-mediated knockdown of Snr1 resulted in rudimentary wings ()), which was strongly suppressed by brm-ALA co-expression, resulting in normal wings in 100% of individuals (), quantified in )), and enhanced to pupal lethality by brm-ASP co-expression ()). brm-WT expression also partially suppressed nub> Snr1-RNAi wing defects (), quantified in )), as might be expected since increased levels of Brm-WT might compensate for reduced levels of Snr1 in generating increased amounts of functional Brm complexes. However, brm-WT expression only resulted in ~ 50% normal adult wings in nub> Snr1-RNAi flies compared with 100% normal wings upon brm-ALA expression (,K), quantified in )). Thus, Brm-ALA exhibits a stronger ability than Brm-WT in suppressing the effects of a compromised Brm complex. Importantly, since brm-ALA and brm-ASP interact in opposite manners with brm-DN and Snr1-RNAi, this reveals that mutation of the 15 putative CDK-phosphorylation sites per se does not inactivate the Brm protein (for example through protein misfolding or preventing protein interactions). Indeed, the genetic interaction data indicates that brm-ASP acts as a brm loss-of-function, whilst brm-ALA acts as a brm gain-of-function allele. This is consistent with the expectation that phosphorylation of Brm inactivates it, whilst blocking Brm phosphorylation prevents its inactivation.
Figure 3. Genetic interactions reveal that brm-ASP acts as a Brm loss-of-function, whilst brm-ALA acts as a Brm gain-of-function
Brm phospho-site mutants affect the cell cycle
We then examined the effect of expression of the brm phospho-site mutant transgenes on cell cycle regulation in vivo. Since Brm forms a complex with the pRb/HDAC/E2F complex to prevent E2F target gene transcription and induce G1 phase arrest during the cell cycle [Citation35–Citation37], we expected that its phosphorylation by CDK should relieve inhibition of G1-S phase progression by the pRb/HDAC complex. To assess the role of Brm phosphorylation in G1 arrest, we focused on the Zone-of-Non-proliferating-Cells (ZNC) at the dorsal-ventral boundary within the developing wing pouch epithelium from third instar larvae, a tissue where pRb expression is known to induce a developmental G1-phase cell cycle arrest (), yellow box) [Citation38]. The ZNC comprises 3 rows of cells, the middle of which is developmentally arrested in G1 phase in the anterior compartment, surrounded by G2 phase arrested cells, whilst in the posterior compartment all 3 rows are arrested in G1 phase [Citation39].
Figure 4. Expression of CDK phosphorylation-site brm mutants affects S phase cell numbers
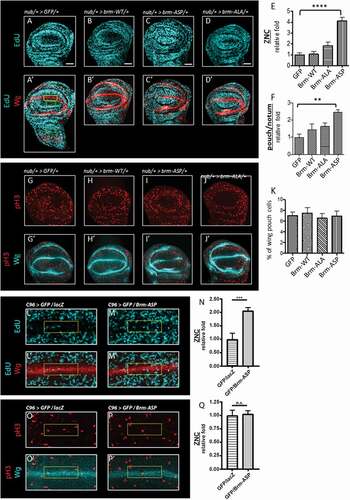
Upon expression of brm-ASP in the wing pouch using the nub-GAL4 driver (outlined by the outer ring of Wingless (Wg) staining) and monitoring S phase entry using a EdU pulse labeling experiment, we observed a 4.1 fold increase in the number of EdU+ cells in the ZNC relative to the nub-GAL4 control (), and quantified in )). We noted that brm-ASP expression not only lead to increased S phases in the G1 arrested band but also resulted in S phases occurring throughout the ZNC, suggesting that the normally G2 arrested band of cells in the anterior compartment were also not present, or was disrupted by induction of the G1 arrested cells into the cell cycle by brm-ASP. Additionally, expression of brm-ASP specifically in the G1 arrested cells using the C96-GAL4 driver, the expression of which corresponds to that of Wg in the central cells of the ZNC, also resulted in an increase in S phase cells (2-fold) relative to the control (-N)). This result indicates that CDK-phosphorylation of Brm can override the pRb-induced developmental cell cycle arrest of the G1 arrested cells in the ZNC cells. Overexpression of brm-WT did not alter S phase entry of the few cells undergoing S phases bordering the ZNC in the sampled region (), and quantified in )). It is expected that when Brm is non-phosphorylated (as mimicked by Brm-ALA), progression into S phase would be inhibited, however surprisingly, we found that ectopic expression of brm-ALA had no significant effect on S phase numbers in cells bordering the ZNC (), and quantified in )) (p = 0.16).
We also assessed whether nub-GAL4 driven brm-ASP expression could affect the proliferation of normal cycling cells within the wing pouch. The level of EdU+ cells in a non-ZNC region of the wing pouch was compared to EdU+ cells in a region within the notum, where the transgenes are not expressed (), green boxes). Similar to its effect in the ZNC, brm-ASP expression resulted in 2.5 fold increase in the number of S phase cells relative to the control within the region of normal proliferating cells in the notum (), and quantified in )). By contrast, brm-WT and brm-ALA did not significantly affect the number of S phase cells ( and quantified in )).
Next, the effect of the brm phospho-site mutant expression (via the nub-driver) on the number of mitotic cells in the entire wing pouch was assessed (-K), region outlined in yellow in 4G). In the control, only 7.01% of the pouch contained mitotic cells, as reflected by phosphorylated histone H3 (pH3) staining ()), and expression of brm-WT and brm-ALA also showed no significant effect on the number of mitotic cells (,J) and quantified in )). Surprisingly, despite an increased number of S phase cells, there was no significant increase in the fraction of mitotic cells by the expression of brm-ASP (6.93%) relative to the control (7.01%) (), and quantified in )). Additionally, expression of brm-ASP using the C96-GAL4 driver also did not significantly alter the small number of M phase cells bordering on the ZNC in the sampled region relative to the control (-Q). It is possible that there was no increase in mitotic cells due to a compensatory delay in G2 phase, as has been previously observed in the wing epithelium, when G1-S phase progression is accelerated [Citation40–Citation44]. It is also conceptually possible that brm-ASP expression might lead to S phase delay, however the fact that expression of brm-ASP using the C96 driver induces G1 cells into S phase argues that it is unlikely that Brm-ASP is resulting in prolonged S phases.
Taken together, these results show that mimicking the CDK-mediated phosphorylated state of Brm leads to an increase in S phase cell numbers in the Rbf induced G1 arrested band in the ZNC as well as in the cycling cells of the wing pouch, without affecting the number of mitotic cells. However, blocking CDK-mediated phosphorylation of Brm, using the brm-ALA transgene, did not significantly affect cell cycle progression, suggesting that this mutant might be unable to function in a dominant manner.
To test whether Brm-ALA could inhibit S phases when the activity of the endogenous Brm complex was compromised, we expressed a UAS-Snr1-RNAi transgene via the nub driver in the developing wing pouch and assessed the effect of brm-ALA or brm-WT expression on S phases (). As expected, Snr1-RNAi expression resulted in increased S phase numbers in the pouch relative to the notum regions, and in the ZNC compared with the control () compared with ), quantified ). Co-expression of brm-ALA with Snr1-RNAi resulted in a normalization of the S phase numbers in the pouch relative to the notum and, strikingly, resulted in a decreased number of S phases in the ZNC () compared with , quantified ). In comparison, expression of brm-WT together with Snr1-RNAi did not significantly reduce the increased S phases observed for Snr1-RNAi in the pouch relative to the notum or in the ZNC relative to the control or to brm-WT alone () compared with , quantified . Thus, when endogenous Brm complex function is reduced, Brm-ALA can inhibit S phases, and its greater ability to inhibit S phases in the ZNC region, where Rbf1 functions to induce the developmental G1 arrest, suggests that de-phosphorylated Brm might function with Rbf1 in inducing this developmental cell cycle arrest.
Figure 5. Snr1 knockdown induced S phase inhibition is suppressed by expression of brm-ALA, but not brm-WT. (A-E’) nub-GAL4 was used to drive the expression of GFP, GFP and Snr1-RNAi, and brm-ALA and Snr1-RNAi transgenes in the wing pouch. S phase cells were quantified by counting the number of EdU+ cells within the wing pouch (non-ZNC region) relative to EdU+ cells in the middle of the notum (white boxes, 60 by 30 pixels), and within the ZNC (yellow box, 120 by 40 pixels). (A, A’) Control wing disc. (B, B’) Snr1-RNAi expression leads to an increase in the number of S phase cells observed. (C, C’) Expression of brm-ALA strongly suppresses the Snr1-RNAi expression induced increase in S phases. (D, D’) brm-WT expression has no discernable effect on S phases. (E, E’) Expression of brm-WT does not suppress the Snr1-RNAi expression induced increase in S phases. (F, G) Histograms represent the relative fold change in EdU+ cells within the wing pouch relative to the notum, compared with control wing discs, and within the ZNC, from 8 or more samples; ± SEM. Statistical analyzes were performed using a one-way ANOVA with post-TUKEY test, with the significance set at p ≤ 0.05. * indicates p ≤ 0.05, ** indicates p ≤ 0.01. Scale bars = 50μm
The Brm dominant-negative mutant phenocopies the cell cycle defects of Brm-ASP phospho-mimic mutant
The prevailing evidence suggests that Brm acts in a complex with pRb/HDAC to inhibit S phase entry by repressing E2F/DP activity, and Cyclin/CDK mediated phosphorylation of pRb and Brm results in its inactivation to promote G1-S phase progression [Citation22–Citation24]. To assess whether the Brm-phospho-mimic mutation, brm-ASP, resulted in dominant cell cycle phenotypes similar to a loss-of-function allele, we compared its effects on S phases in the wing pouch to that of the ATPase-dead dominant-negative mutant of Brm (brm-DN) [Citation34].
As expected, expression of brm-DN led to the loss of inhibition on G1-S phase progression, as indicated by the increased amount of EdU+ cells in both the ZNC and normal cycling cells, 2.7 fold and 2.2 fold, respectively (), quantified in compared with brm-WT and the GFP controls (, quantified in ). Similar to brm-ASP, the expression of brm-DN did not have any significant effects on the progression into mitosis; 7.04% of wing pouch cells compared to 7.01% of GFP control and 7.50% of brm-WT (, quantified in )). Thus, brm-DN phenocopies the effect of brm-ASP on inducing S phase without affecting mitoses (), indicating that brm-ASP behaves as a loss-of-function mutant in regard to its effect on cell cycle regulation.
Figure 6. brm-DN shows similar effects on the cell cycle as brm-ASP
The effects of the Brm phospho-site mutants on cell death
Since, expression of brm-ASP increases the amount of S phase cells, without affecting entry into mitosis, we postulated that this deregulation of cell cycle progression might lead to apoptosis, resulting in small wings. Accordingly, we conducted TUNEL-labeling experiments to detect apoptotic cells in the wing epithelium of the brm phospho-site mutant transgenic flies (). Relative to the control wing epithelium, over-expression of brm-WT did not induce apoptosis within the wing pouch () compared with ), quantified in )). By contrast, there was 65-fold increase in TUNEL-positive cells in brm-ASP expressing tissue (7.51% versus 0.12% control) (), quantified in )). Interestingly, these apoptotic cells were localized at the anterior-posterior (A-P) boundary and at the periphery of the wing pouch ((C’)). Overexpression of brm-ALA also resulted in a slight, but statistically insignificant (p = 0.99), increase in the number of apoptotic cells, however these were scattered throughout the nub domain in the wing epithelium (), quantified in )). Thus, perturbation of CDK-mediated phosphorylation of Brm results in cell death, suggesting that appropriate phosphorylation and regulation of Brm function is important for cell survival.
Figure 7. Expression of CDK phosphorylation-site brm mutants in the wing epithelium induces apoptosis, but blocking apoptosis does not rescue the wing defects
To determine whether the adult wing defects that were observed () might be due to the increased cell death observed upon brm-ASP expression, we assessed the effect of inhibiting apoptosis, via expression of the pan-caspase inhibitor, the baculovirus p35 protein [Citation45]. Co-expression of p35 and GFP under control of the nub-GAL4 driver resulted in modestly smaller wings compared to the control of nub> GFP (, quantified in )). Although brm-ALA did not significantly increase cell death, co-expression of p35 exacerbated the vein defects of brm-ALA ()). The nub> p35 + brm-ALA wings were smaller, compared to both nub> p35 + GFP and nub> brm-ALA (), quantified in )), with notching at the wing margin and a truncation of the L5 wing vein (), green and red arrows respectively). Surprisingly, instead of rescuing the brm-ASP wing phenotype, co-expression of p35 with brm-ASP also enhanced the nub> brm-ASP phenotype, resulting in lethality at the larval stage ()). Similarly, inhibiting cell death using a c-Jun N-terminal kinase (JNK) dominant-negative transgene (bsk-DN) also did not rescue, but instead exacerbated the wing defects of nub> brm-ASP and nub> brm-ALA (-N)). Therefore, inhibiting apoptosis did not rescue the phenotypic effects of either brm-ASP or brm-ALA expression, suggesting that the developmental defects observed upon perturbing the regulation of Brm by CDK-mediated phosphorylation might be due to direct effects on differentiation.
The effects of the Brm-ASP on Decapentaplegic (BMP/TGFβ) signaling
Previous analyzes have revealed that blocking Brm complex function, using the Brm dominant-negative mutant, leads to reduced Decapentaplegic (Dpp, BMP/TGFβ family) signaling [Citation46], which is an important morphogen in wing development [Citation47,Citation48]. Therefore, we reasoned that Brm-ASP might be affecting wing differentiation by blocking Dpp signaling. Accordingly, to test whether this might be the case, we analyzed the effect of nub-driven expression of the Brm phospho-site mutants on Dpp pathway activity, using an antibody to the phosphorylated and activated version of the downstream transcription factor (pMad) (). Dpp signaling is normally activated along the anterior-posterior boundary during wing development [Citation47,Citation48], and, as expected, pMad staining was observed in this region in control wing discs ()). We then compared the effect of nub-driven expression of the brm phospho-site mutants on pMad staining intensity in the pouch region relative to the notum. Expression of brm-ALA did not significantly affect pMad staining (), relative to ), quantified in )), however brm-ASP resulted in a striking decrease in pMad levels (), relative to ), quantified in )). We also analyzed the effect of brm phospho-site mutants on Dpp signaling when cell death was blocked using the dominant-negative JNK transgene, bsk-DN (-H)). Similar results were obtained in the bsk-DN background, with expression of brm-ASP, but not brm-ALA, reducing pMad levels in the pouch relative to the notum regions (-H)). Thus, similar to Brm-DN, Brm-ASP reduces Dpp signaling, which might explain its effect on wing disc growth and differentiation (see Discussion).
Figure 8. Dpp signaling is inhibited by the expression of brm-ASP
Discussion
In our study, we have demonstrated for the first time the importance of direct CDK-phosphorylation of Brm on cell proliferation and differentiation in vivo. We show that Drosophila Brm is phosphorylated by Cyclin E1/CDK2 and Cyclin A/CDK2 in vitro and our Drosophila studies indicate that CDK phosphorylation-site mutants of Brm affect S phase numbers and differentiation during wing development. Our findings are consistent with the current model that CDK-mediated phosphorylation of Brm leads to its inactivation, similar to pRb, which is supported by the similarities between the effects of brm-ASP and the brm dominant-negative mutant, brm-DN, on S phase numbers and differentiation of the adult wing. We envisage that phosphorylation of Brm might alter its conformation and/or interactions with other subunits, thereby reducing Brm complex formation and function.
We found that in the developing wing epithelium, brm-ASP expression was able to bypass the normal developmental G1 arrest in the ZNC mediated by pRb [Citation38], suggesting that the CDK-phosphorylation and inactivation of Brm induces G1-S phase cell cycle progression. A similar trend was also observed in the analysis of normal cycling cells in non-ZNC regions. Although expression of brm-ALA alone did not show dominant effects on S phases, when endogenous Brm complex function was decreased brm-ALA was able to reduce S phases to below that of wild-type levels in the ZNC region. Altogether, these data suggest that CDK-mediated phosphorylation of Brm is important in inactivating Brm to allow S phase entry in the Drosophila wing disc epithelium.
Our results are consistent with studies in mammalian cells, which show that Cyclin E1/CDK2 phosphorylates BRG1 [Citation27]. Furthermore, Cyclin E overexpression can overcome the cell cycle inhibitory effects of BRG1, although it is unknown if this effect is direct or indirect, due to Cyclin E1/CDK2 targeting other substrates. Our results show, for the first time, that direct phosphorylation of Brm by cyclin/CDKs is important in promoting G1-S phase cell cycle progression. Previous studies in mammalian cells have also shown that inhibition of the expression of the E2F/DP target, Cyclin A, by pRb is dependent on BRG1 [Citation49], however whether Cyclin A regulation is dependent on BRG1 phosphorylation status is unknown. In Drosophila, it is not known if Cyclin A expression is regulated by Brm, but deficiencies that remove Cyclin A strongly enhance the Snr1 gain-of-function mutant wing phenotype [Citation30], suggesting that Cyclin A function is important to repress the Brm complex. The mammalian studies, as well as our studies here, show that Brm is phosphorylated by Cyclin E/CDK2 and Cyclin A/CDK2. Similar to pRb regulation, it is possible that Brm is phosphorylated sequentially by several different Cyclin/CDK complexes. In mammalian cells, pRb is first phosphorylated by Cyclin D/CDK4, followed by phosphorylation by Cyclin E/CDK2, which is necessary for entry into S phase and for Cyclin A expression (reviewed in [Citation25]). During S phase, Cyclin E is replaced by Cyclin A to form a complex with CDK2 [Citation50,Citation51], which also contributes to the hyper-phosphorylation of pRb [Citation52]. Similar CDK-driven phospho-regulation may occur to incrementally inactivate Brm during the G1-S phase progression.
Deregulation of the cell cycle by brm-ASP expression is reflected in the adult wing phenotype, which are much smaller in comparison with control wings expressing GFP, coupled with aberrant wing margin and veins. However, since increased cell death was observed upon brm-ASP expression, this severe phenotype might be attributed to cell death. How cell death is induced remains to be determined, but since dying cells from brm-ASP expression were concentrated at the anterior-posterior (A-P) boundary and outside the boundaries of the wing pouch, it is possible that this is due to cell competition mechanisms, rather than DNA damage-induced cell death, where dying cells would be expected to be distributed throughout the wing pouch. Indeed, since we observed that the Dpp pathway signaling was decreased upon brm-ASP expression, the increased cell death might be due to signaling pathway perturbations (reviewed by [Citation53,Citation54]). Wing formation is dependent on gradients of signaling pathways, which originate from the A-P boundary [Citation55]. Therefore, although confined to a small region, the cell death at the A-P boundary that is observed upon brm-ASP expression would be expected to have detrimental effects on the entire adult wing. Since brm-ASP expression decreases expression of the important patterning inducer and cell survival pathway Dpp (BMP/TGFβ) in the Drosophila wing, similarly to brm-DN [Citation46], it is possible that perturbed Dpp signaling is responsible for these phenotypic defects. Similarly, in mammalian cells, knocking-down BRG1 resulted in a 12-fold decrease in the expression of TGFβ, the mammalian homologue of Dpp [Citation56]. Indeed, the TGFβ transcriptional program is dependent on BRG1 functioning as a chromatin remodeler, as it is responsible for the expression of TGFβ-responsive genes including Myc and c-Jun [Citation56,Citation57]. Furthermore, low Dpp signaling in the Drosophila wing epithelium leads to JNK-dependent apoptosis [Citation58,Citation59]. However, in our studies, blocking apoptosis by expressing p35 or a JNK dominant-negative transgene did not suppresses the wing developmental defects due to brm-ASP expression, but instead enhanced them. Perhaps by blocking cell death, the persistence of the aberrant cells results in alterations of other signaling pathways leading to the enhancement of brm-ASP induced wing developmental defects. Blocking caspase-dependent cell death (apoptosis) with p35 can result in the ‘undead’ secretory phenotype, caused by initiating but then preventing cell death [Citation45]. This is a phenomenon whereby dying cells that are unable to undergo apoptosis continually execute compensatory proliferative effects by secreting morphogens, such as Dpp and Wg, which can lead to overgrowth of the surrounding normal tissue [Citation60–Citation62]. This effect is JNK-dependent [Citation62], and since we saw similar phenotypes with p35 and when JNK signaling was blocked by bsk-DN expression, and that Dpp signaling was still reduced in brm-ASP bsk-DN tissues, the undead effect is unlikely to be occurring here. However, it is possible that the expected effect of impaired Brm function on reducing EGFR-Ras-MAPK signaling, which we have previously observed upon brm-DN expression [Citation33], might be contributing to the developmental defects observed upon brm-ASP expression and enhanced by blocking cell death.
With nub-driven brm-ALA, although an insignificantly increased level of apoptosis was observed, co-expression with p35 or bsk-DN with brm-ALA resulted in notching of the adult wings and a truncated L5 wing vein. This phenotype is similar to defects in signaling pathways involved in the specification of L5 vein such as Dpp [Citation63] and EGFR-Ras-MAPK pathways [Citation33]. However, given that brm-ASP expression and Brm loss-of-function decreases Dpp signaling [Citation46], we would have expected brm-ALA expression to have the opposite effect on Dpp signaling. However, over-expression of brm-ALA via the nub-GAL4 driver in a bsk-DN background did not significantly affect Dpp signaling. Our previous data has shown that brm-DN expression inhibits EGFR-Ras-MAPK signaling in the developing wing [Citation33], and therefore brm-ALA expression would be expected to increase EGFR-Ras-MAPK signaling, an effect that should induce ectopic wing vein formation. Indeed, brm-ALA expression consistently induced ectopic L2 veins, suggesting that although brm-ALA expression did not have a dominant effect on the cell cycle, it was dominantly elevating EGFR-Ras-MAPK signaling. Conversely, co-expression of p35 or bsk-DN with brm-ALA resulted in L5 wing vein truncation and wing margin notching defects, suggestive of impaired EGFR-Ras-MAPK or other signaling pathways, such as Notch involved in wing vein formation and wing margin development [Citation55]. Further analysis is required to reveal how expression of the Brm phospho-site mutants with or without p35 or bsk-DN expression affect signaling pathways to induce these effects on wing vein development.
In conclusion, our analyzes have revealed that mutation of the CDK-phosphorylation sites of Brm result in substantial functional consequences on the cell cycle, cell survival and differentiation. These results indicate that CDK-mediated phosphorylation of Brm plays a direct and important role in the regulation of cell proliferation and differentiation in vivo.
Materials and methods
Expression and purification of recombinant full-length Brm
Recombinant full-length Brm with a C-terminal FLAG tag was cloned in pFL-EGFP baculoviral vector [Citation64] (kindly donated by Dr Andrew Deans, Genome Stability Unit, St Vincent’s Institute) using EcoRI and XbaI restriction enzymes (NEB) for the 5ʹ and 3ʹ ends, respectively. To generate baculoviral particles, the resulting bacmid was transfected into Sf9 cells using FuGENE HD (Roche) as previously described [Citation65]. 5 days later, supernatant containing the baculovirus was collected. For protein generation, Sf9 cells were cultured at a density of 1x106/ml and infected with recombinant baculovirus at an MOI of 2. Cells were grown for 5 days prior to harvesting. Briefly, the cells were lysed using lysis buffer (Buffer W, 1μg/ml Aprotinin, 1 μg/ml Leupeptin, 1mM DTT, 1 mM PMSF) and sonicated. Lysates obtained from above were then subjected to immunoprecipitation using anti-FLAG M2 beads (Sigma) by constant mixing at 4°C overnight. FLAG-tagged Brm was eluted using elution buffer (100 mM HEPES pH7.5, 150mM NaCl, 10% Glycerol, 1 μg/ml Leupeptin, 1 μg/ml Aprotinin, 1 mM DTT, 1 mM PMSF, 100 μg/ml FLAG peptide) 4°C overnight.
Expression and purification of recombinant full-length Brm and 15ALA/15ASP phospho-site mutants
Full-length brm or 15ALA/15ASP phospho-site mutants were cloned with a C-terminal FLAG tag into pFL-EGFP baculoviral vector [Citation64]. Bacmids were generated and baculovirus produced in Sf9 cells as previously described [Citation65]. For generation of recombinant Flag-Brm, 400mL Sf9 cells were cultured at a density of 1x106/ml in Sf900-II media and infected with recombinant baculovirus at an MOI of 3. Cells were collected after 72 hr, lysed using Buffer W (50mM HEPES pH 7.5, 150mM NaCl, 0.2% Tween-20, 0.5mM DTT, 2mM EDTA + complete protease inhibitors) and sonicated. Lysates were cleared by centrifugation (30 min, 20,000 x g, 4ʹC) and passed over 0.5mL anti-FLAG M2 beads (Sigma Aldrich), washed extensively, and eluted using elution buffer (buffer W + 125 μg/ml FLAG peptide). Peak fractions were used immediately in kinase assays.
In Vitro kinase assay
Recombinant active Cyclin E/A-CDK complexes were prepared as described previously [Citation32]. Mammalian MBP-pRb279-928 (prepared as described in [Citation31]) was used as a positive control substrate for the activity of the CDK complexes. Approximately 5 μg of FLAG-tagged Brm protein extracts were used in each in vitro kinase reaction, which was carried out as previously described [Citation31,Citation66].
Mutant and transgenes
Fly stocks used in this study are as follows;
UAS-Snr1-RNAi (#108599) (Vienna Drosophila Research Centre (VDRC)), UAS-brm-RNAi (#37720) (VDRC) [Citation67], en-GAL4, UAS-GFP (L. Johnston); nub-GAL4 (K. Harvey); C96-GAL4 (Bloomington stock 43343); (UAS-brm-DN (J. Tamkun); UAS-brm-WT, UAS-brm-ALA, and UAS-brm-ASP (generated in this study). brm-ALA and brm-ASP mutations were generated by site-directed oligonucleotide mutagenesis of a full-length brm cDNA in pBLUESCRIPT. The mutations were verified by sequencing analysis, and the constructs cloned into the Drosophila Pw+ element UAS vector, pUAST, using XbaI and NotI restriction sites. Transgenic flies were generated by injection of the pUAST plasmids along with a plasmid expressing the Δ2-3 transposase into embryo using standard methodology. Transgenic constructs on the 3rd chromosome were used for all experiments.
Immunofluorescence
To prepare both wing imaginal discs for immunohistochemistry, larvae at the third instar stage of development were dissected in PBS. Tissues were fixed with 4% PFA for 20 min, washed 3 times with PBS supplemented with 0.3% Triton-X (PBT) and blocked using PBT supplemented with 5% BSA.
Primary antibodies used are as follows;
Rabbit GFP (Invitrogen, 1:500), Mouse Wingless (4D4, DSHB, 1:500), Rabbit pH3 (Upstate, 1:40), rabbit anti-phospho Smad (pMad) 1/5 (Ser463/465) (Cell Signaling Technology, #13820, 1:40).
Secondary antibodies used are as follows:
Anti-rabbit Alexa Fluor 468, 588, anti-mouse Alexa Fluor 588, 633 (Invitrogen), donkey anti-mouse IgG, Alexa Fluor 647 (ThermoFisher Scientific, #A-31571, 1:500), and donkey anti-rabbit IgG, Alexa Fluor 594 (ThermoFisher Scientific, #A-21207, 1:500).
TUNEL staining
TdT-mediated dUTP-Nick-End-Labelling (TUNEL) in situ cell detection kit, TMR Red (Roche) was used. Briefly, larval tissues were dissected and fixed as mentioned above. Samples were then washed 3 times with PBT and incubated overnight at 4°C with PBT. Tissues were permeablized with PBT supplemented with 0.1% Sodium Citrate at 65°C for 30 min and then washed 3 times with PBT. Samples were then incubated with TUNEL reaction mix for 2h at 37°C.
EdU incorporation assay
Third instar larvae were dissected, placed in PBT containing 5-ethynyl-2′-deoxyuridine (EdU) and incubated at RT, with constant mixing, for 15 min. They were then washed with PBT and stained for Wingless using appropriate primary and secondary antibodies. EdU was activated by Click-IT reaction, as per manufacturer’s protocol (Roche), or using the modified Click-iT EdU Imaging Kit (Molecular Probes, #C10340) protocol.
Imaging of adult wings
Wings were detached from the frozen flies and mounted on coverslips using Canada Balsam/Methyl Salicylate solution. Wings were imaged against a white background using an Infinity 1 camera, or a Zeiss SteREO Discovery.V8 microscope. Wing sizes were measured using Adobe Photoshop CS3 Extended.
Statistical analyzes
The numbers of TUNEL+, EdU+ and pH3+ cells, from 7 or more wing discs, were quantified using Fiji (ImageJ ver2.0). The wing pouch and ZNC were defined by Wg staining.
For quantifying pMad levels, 60 by 60 pixel boxes were drawn in the wing pouch and notum areas. Mean pixel intensity was then determined within the boxes using the “Histogram” tool in Adobe Photoshop CC, and the ratio determined. At least 7 wing discs were examined for each sample.
Quantification of adult wing sizes, from 6 or more samples, was performed on Adobe Photoshop CS3 Extended. For the nub-GAL4 crosses, the entire wing pouch area was analyzed. In the en-GAL4 crosses, the posterior and anterior regions were defined based on the position of the L4 wing vein.
Statistical analyzes were conducted using a one-way ANOVA with a post-TUKEY test or with Student’s t-tests, and plotted ± SEM, using Graph Pad Prism ver6. Significance was set at p ≤ 0.05.
Acknowledgments
We thank all personal in our labs for helpful discussions of the results and Kasun Amaratunga and Steffi Cheung for maintaining the fly stocks. We are grateful to John Tamkun from University of California Santa Cruz, Laura Johnston from Columbia University Medical Centre, Kieran Harvey from the Peter MacCallum Cancer Centre and the Bloomington stock center [NIH P40 OD-018537] for supplying Drosophila stocks, to Flybase for its wealth of information, OzDros for Drosophila quarantine services, Sarah Ellis and the Peter MacCallum Cancer Centre Confocal Facility, and Peter Lock and the LIMS Bioimaging Facility for microscopy equipment and technical support.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Schnitzler G, Sif S, Kingston RE. Human SWI/SNF interconverts a nucleosome between its base state and a stable remodeled state. Cell. 1998;94:17–27.
- Imbalzano AN, Kwon H, Green MR, et al. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature. 1994;370:481–485.
- Shundrovsky A, Smith CL, Lis JT, et al. Probing SWI/SNF remodeling of the nucleosome by unzipping single DNA molecules. Nat Struct Mol Biol. 2006;13:549–554.
- Khavari PA, Peterson CL, Tamkun JW, et al. BRG1 contains a conserved domain of the SWI2/SNF2 family necessary for normal mitotic growth and transcription. Nature. 1993;366:170–174.
- Wang W, Cote J, Xue Y, et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 1996;15:5370–5382.
- Phelan ML, Sif S, Narlikar GJ, et al. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell. 1999;3:247–253.
- Fry CJ, Peterson CL. Chromatin remodeling enzymes: who’s on first? Curr Biol. 2001;11:R185–97.
- Roberts CW, Orkin SH. The SWI/SNF complex–chromatin and cancer. Nat Rev Cancer. 2004;4:133–142.
- Lee K, Shim JH, Kang MJ, et al. Association of BAF53 with mitotic chromosomes. Mol Cells. 2007;24:288–293.
- Lee K, Kang MJ, Kwon SJ, et al. Expansion of chromosome territories with chromatin decompaction in BAF53-depleted interphase cells. Mol Biol Cell. 2007;18:4013–4023.
- Zhang L, Chen H, Gong M, et al. The chromatin remodeling protein BRG1 modulates BRCA1 response to UV irradiation by regulating ATR/ATM activation. Front Oncol. 2013;3:7.
- Park JH, Park EJ, Lee HS, et al. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J. 2006;25:3986–3997.
- Qi W, Wang R, Chen H, et al. BRG1 promotes the repair of DNA double-strand breaks by facilitating the replacement of RPA with RAD51. J Cell Sci. 2015;128:317–330.
- Kozmik Z, Machon O, Kralova J, et al. Characterization of mammalian orthologues of the Drosophila osa gene: cDNA cloning, expression, chromosomal localization, and direct physical interaction with Brahma chromatin-remodeling complex. Genomics. 2001;73:140–148.
- Huang J, Zhao YL, Li Y, et al. Genomic and functional evidence for an ARID1A tumor suppressor role. Genes Chromosomes Cancer. 2007;46:745–750.
- Decristofaro MF, Betz BL, Rorie CJ, et al. Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J Cell Physiol. 2001;186:136–145.
- Wang X, Nagl NG Jr., Flowers S, et al. Expression of p270 (ARID1A), a component of human SWI/SNF complexes, in human tumors. Int J Cancer. 2004;112:636–642.
- Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206.
- Kohashi K, Oda Y. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Sci. 2017;108:547–552.
- St Pierre R, Mammalian KC. SWI/SNF complexes in cancer: emerging therapeutic opportunities. Curr Opin Genet Dev. 2017;42:56–67.
- Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv. 2015;1:e1500447.
- Dunaief JL, Strober BE, Guha S, et al. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell. 1994;79:119–130.
- Trouche D, Le Chalony C, Muchardt C, et al. RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci USA. 1997;94:11268–11273.
- Staehling-Hampton K, Ciampa PJ, Brook A, et al. A genetic screen for modifiers of E2F in Drosophila melanogaster. Genetics. 1999;153:275–287.
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330.
- Suryadinata R, Sadowski M, Sarcevic B. Control of cell cycle progression by phosphorylation of cyclin-dependent kinase (CDK) substrates. Biosci Rep. 2010;30:243–255.
- Shanahan F, Seghezzi W, Parry D, et al. Cyclin E associates with BAF155 and BRG1, components of the mammalian SWI-SNF complex, and alters the ability of BRG1 to induce growth arrest. Mol Cell Biol. 1999;19:1460–1469.
- Wong AK, Shanahan F, Chen Y, et al. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res. 2000;60:6171–6177.
- Brumby AM, Zraly CB, Horsfield JA, et al. Drosophila cyclin E interacts with components of the Brahma complex. EMBO J. 2002;21:3377–3389.
- Zraly CB, Marenda DR, Dingwall AK. SNR1 (INI1/SNF5) mediates important cell growth functions of the Drosophila Brahma (SWI/SNF) chromatin remodeling complex. Genetics. 2004;168:199–214.
- Suryadinata R, Sadowski M, Steel R, et al. Cyclin-dependent kinase-mediated phosphorylation of RBP1 and pRb promotes their dissociation to mediate release of the SAP30.mSin3.HDAC transcriptional repressor complex. J Biol Chem. 2011;286:5108–5118.
- Sarcevic B, Lilischkis R, Sutherland RL. Differential phosphorylation of T-47D human breast cancer cell substrates by D1-, D3-, E-, and A-type cyclin-CDK complexes. J Biol Chem. 1997;272:33327–33337.
- Herr A, McKenzie L, Suryadinata R, et al. Geminin and Brahma act antagonistically to regulate EGFR-Ras-MAPK signaling in Drosophila. Dev Biol. 2010;344:36–51.
- Elfring LK, Daniel C, Papoulas O, et al. Genetic analysis of brahma: the Drosophila homolog of the yeast chromatin remodeling factor SWI2/SNF2. Genetics. 1998;148:251–265.
- Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–473.
- Brehm A, Miska EA, McCance DJ, et al. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601.
- Magnaghi-Jaulin L, Groisman R, Naguibneva I, et al. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605.
- Duman-Scheel M, Johnston LA, Du W. Repression of dMyc expression by Wingless promotes Rbf-induced G1 arrest in the presumptive Drosophila wing margin. Proc Natl Acad Sci USA. 2004;101:3857–3862.
- Johnston LA, Edgar BA. Wingless and Notch regulate cell-cycle arrest in the developing Drosophila wing. Nature. 1998;394:82–84.
- Johnston LA, Prober DA, Edgar BA, et al. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–790.
- Prober DA, Edgar BA. Ras1 promotes cellular growth in the Drosophila wing. Cell. 2000;100:435–446.
- Neufeld TP, de la Cruz AF, Johnston LA, et al. Coordination of growth and cell division in the Drosophila wing. Cell. 1998;93:1183–1193.
- Coisy-Quivy M, Disson O, Roure V, et al. Role for Brm in cell growth control. Cancer Res. 2006;66:5069–5076.
- Reis T, Edgar BA. Negative regulation of dE2F1 by cyclin-dependent kinases controls cell cycle timing. Cell. 2004;117:253–264.
- Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1994;120:2121–2129.
- Marenda DR, Zraly CB, Dingwall AK. The Drosophila Brahma (SWI/SNF) chromatin remodeling complex exhibits cell-type specific activation and repression functions. Dev Biol. 2004;267:279–293.
- Teleman AA, Cohen SM. Dpp gradient formation in the Drosophila wing imaginal disc. Cell. 2000;103:971–980.
- Restrepo S, Zartman JJ, Basler K. Coordination of patterning and growth by the morphogen DPP. Curr Biol. 2014;24:R245–55.
- Zhang HS, Gavin M, Dahiya A, et al. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell. 2000;101:79–89.
- Pagano M, Pepperkok R, Verde F, et al. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992;11:961–971.
- Schulman BA, Lindstrom DL, Harlow E. Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc Natl Acad Sci USA. 1998;95:10453–10458.
- Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem. 1997;272:12738–12746.
- Amoyel M, Bach EA. Cell competition: how to eliminate your neighbours. Development. 2014;141:988–1000.
- Moreno E. Is cell competition relevant to cancer? Nat Rev Cancer. 2008;8:141–147.
- Blair SS. Wing vein patterning in Drosophila and the analysis of intercellular signaling. Annu Rev Cell Dev Biol. 2007;23:293–319.
- Ondrusova L, Vachtenheim J, Reda J, et al. MITF-independent pro-survival role of BRG1-containing SWI/SNF complex in melanoma cells. PLoS One. 2013;8:e54110.
- Xi Q, He W, Zhang XH, et al. Genome-wide impact of the BRG1 SWI/SNF chromatin remodeler on the transforming growth factor beta transcriptional program. J Biol Chem. 2008;283:1146–1155.
- Moreno E, Basler K, Morata G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature. 2002;416:755–759.
- Adachi-Yamada T, Fujimura-Kamada K, Nishida Y, et al. Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature. 1999;400:166–169.
- Perez-Garijo A, Martin FA, Struhl G, et al. Dpp signaling and the induction of neoplastic tumors by caspase-inhibited apoptotic cells in Drosophila. Proc Natl Acad Sci USA. 2005;102:17664–17669.
- Perez-Garijo A, Martin FA, Morata G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development. 2004;131:5591–5598.
- Perez-Garijo A, Shlevkov E, Morata G. The role of Dpp and Wg in compensatory proliferation and in the formation of hyperplastic overgrowths caused by apoptotic cells in the Drosophila wing disc. Development. 2009;136:1169–1177.
- Cook O, Biehs B, Bier E. brinker and optomotor-blind act coordinately to initiate development of the L5 wing vein primordium in Drosophila. Development. 2004;131:2113–2124.
- van Twest S, Murphy VJ, Hodson C, et al. Mechanism of Ubiquitination and Deubiquitination in the Fanconi Anemia Pathway. Mol Cell. 2017;65:247–259.
- Coulthard R, Deans A, Swuec P, et al. Architecture and DNA recognition elements of the Fanconi anemia FANCM-FAAP24 complex. Structure. 2013;21:1648–1658.
- Roesley SN, Suryadinata R, Morrish E, et al. Cyclin-dependent kinase-mediated phosphorylation of breast cancer metastasis suppressor 1 (BRMS1) affects cell migration. Cell Cycle. 2016;15:137–151.
- Dietzl G, Chen D, Schnorrer F, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156.