
ABSTRACT
Inflammation or dysbacteriosis-derived lipopolysaccharides (LPS) adversely influence the embryonic development of respiratory system. However, the precise pathological mechanisms still remain to be elucidated. In this study, we demonstrated that LPS exposure caused lung maldevelopment in chick embryos, including higher embryo mortality, increased thickness of alveolar gas exchange zone, and accumulation of PAS+ immature pulmonary cells, accompanied with reduced expression of alveolar epithelial cell markers and lamellar body count. Upon LPS exposure, pulmonary cell proliferation was significantly altered and cell apoptosis was inhibited as well, indicating a delayed progress of pulmonary development. LPS treatment also resulted in reduced CAV-1 expression and up-regulation of Collagen I, suggesting increased lung fibrosis, which was verified by Masson staining. Moreover, LPS induced enhanced Nrf2 expression in E18 lungs, and the increased reactive oxygen species (ROS) production was confirmed in MLE-12 cells in vitro. Antioxidant vitamin C restored the LPS induced down-regulation of ABCA3, SP-C and GATA-6 in MLE-12 cells. Furthermore, LPS induced activation of NF-κB signaling in MLE-12 cells, and the LPS-induced decrease in SP-C expression was partially abrogated by blocking NF-κB signaling with Bay-11–7082. Bay-11–7082 also inhibited LPS-induced increases of ROS and Nrf2 expression. Taken together, we have demonstrated that oxidative stress and NF-κB signaling are involved in LPS induced disruption of pulmonary cell development in chick embryos.
Introduction
Lipopolysaccharide(s) (LPS), also known as endotoxins, are components of the cell wall in Gram-negative bacteria. The recognition of LPS mediates the rapid activation of the intracellular TLR4/MD2 signaling pathway, leading to the release of pro-inflammatory mediators and other factors crucial for keeping infection under control in physiological conditions [Citation1]. Yet these signaling molecules can also elicit an acute and uncontrolled inflammatory response in the animal host that triggers cell death. As the major virulence factor of Gram-negative bacteria, LPS is known to activate leukocytes in the blood when it is released into the peripheral circulation during endotoxaemia [Citation2]. In the lung, alveolar macrophages, alveolar epithelial cells, and the TLR4 receptor pathway implement the regulatory mechanisms of the defence system. Pulmonary surfactant formation is deemed to be affected by Gram-negative bacteria and LPS [Citation3].
The composition of the intestinal microbiota is closely connected to inflammation, so any changes to the microbiota have the potential to affect inflammatory responses [Citation4]. Gram-negative bacterium-contaminated food and various dietary components can alter the gut microbiota. For example, a high-fat diet increased the proportion of Gram-negative bacteria present and the LPS leakage through the intestinal barrier in the gut [Citation5]. Furthermore, Poroyko et al. studied the reaction of the microbial community to LPS-induced lung inflammation and barrier dysfunction, and proposed that the morbid transformation of the microbiota during acute lung injury was mediated predominantly by endogenous opportunistic pathogens rather than by external infectious agents [Citation6]. The accumulation of LPS in circulating blood contributed to cell death in different tissues [Citation7], and such blood-borne LPS can be quickly removed under normal physiological conditions. However, the continuing, low-grade inflammation or dysbacteriosis can invoke the continuous production of LPS and replenishment from dormant bacterial reservoirs [Citation7]. In pregnant women, developing embryos are especially vulnerable. Unfortunately, very little has thus far been documented regarding the negative effects of LPS at this aspect. Yun et al demonstrated that differences in bacterial composition in mouse lungs could affect lung development and the alveolar structure [Citation8]. Infant respiratory distress syndrome is deemed to be due to deficient pulmonary surfactant production, which may result from structural immaturity in the lung induced by various pathogenic factors. In this study, we would like to investigate the connection between the toxicological effects of bacterial component LPS and malformation of the lungs at gestation.
Lungs, derived from embryonic endoderm at the gastrula stage, contain lower respiratory tracts and pulmonary alveoli, which are covered by pulmonary epithelial cells. The development of lungs begins from outpouching of the embryonic foregut. In the developmental process of lung branching morphogenesis, the lung endoderm starts to differentiate into epithelial cell lineages along the proximal-distal axis. The proximal progenitor cells develop into the secretory cells, mucosal cells and ciliated cells of the lung airway, while the distal progenitor cells develop into type I alveolar epithelial (AECI) and type II alveolar epithelial (AECII) cells. Squamous AECI cells cover nearly 95% of the alveolar surface and are responsible for gas exchange, while AECII cells secrete pulmonary surfactant that reduces alveolar surface tension. AECII cells can self-replicate and serve as adult stem cells to differentiate into AECI cells during normal homeostatic turnover, or when AECI cells become depleted due to injury [Citation9,Citation10]. Pulmonary reticular and elastic fibres form the bulk of the connective tissue in the walls of the alveoli.
The FGF and Wnt signaling pathways are considered to control lung specification, branching and patterning during lung development [Citation11]. FGF signaling, especially that involving Fgf9, Fgf10 and Fgfr2 genes, plays an important role in the development of endoderm and morphogenesis of the lungs [Citation12–Citation14]. Wnt functions as an important signaling molecule during early morphogenesis. GATA binding protein 6 (GATA-6) is a member of the zinc finger GATA protein family, which is mainly expressed in the developing endoderm and the vascular smooth muscle of the lung [Citation15]. It is speculated that GATA-6 plays an indispensable role in maintaining the balance between the proliferation and differentiation of pulmonary epithelial progenitor cells during lung development, through modulating Wnt signaling [Citation16]. Deficiency of GATA-6 leads to epithelial dysplasia due to the aberrant differentiation of epithelial progenitor cells during lung development.
In this study, we determined that LPS exposure can dramatically affect the development of the embryonic chick lungs. Furthermore, the possible pathological mechanisms of LPS-induced embryonic lung malformation were explored.
Materials and methods
Chick embryos and manipulation
Fertilized chicken eggs were obtained from the Avian Farm of the South China Agriculture University. The eggs were incubated until the required HH stage in a humidified incubator (Yiheng Instrument, Shanghai, China) at 38°C and 70% humidity. Starting from E7, either 100µg/ml LPS (Sigma, USA) or simple saline (control) was directly injected into the air chamber at the blunt end of the eggs every other day, and the embryos were harvested at the desired time points. All embryos were photographed using a stereomicroscope (Olympus MVX10, Japan) prior to further analysis.
Morphological and immunofluorescent staining analysis of embryonic chick lungs
The fresh lungs were weighted for wet mass right after harvest or dried overnight at 75°C for dry mass measurement[Citation17]. The fresh lungs were then fixed in 4% paraformaldehyde, dehydrated, embedded in paraffin wax and serially sectioned at 5 µm. Haematoxylin and eosin (H&E), periodic acid–Schiff (PAS) and Masson stainings were employed to observe histological pulmonary structure and detect glycogen and fibrous tissue. For immunofluorescent staining, lung transverse sections were de-waxed in xylene, rehydrated and heated for antigen retrieval. After being immersed in 3% hydrogen peroxide for 10 min to block endogenous peroxidase, the sections were blocked with 5% inactivated goat serum for 30 min at room temperature and incubated with primary antibodies against PCNA (1:300; GeneTex), pHIS3 (1:200; Santa Cruz), CAV-1 (1:200; Thermo Scientific), AQP5 (1:100; Abcam), Collagen І (1:200; Abcam), P65 (1:200; Sigma), Nrf2 (1:20; DSHB) and GATA-6 (1:300; Abcam), respectively, at 4°C overnight with shaking. The sections were then incubated with the corresponding Alexa Fluor 555 or 488 labelled secondary antibodies (1:1000; Invitrogen) at room temperature for 2 hours in a dark box. Alexa Fluor 488: excitation, 488 nm; emission, 505 to 525 nm; Alexa Fluor 555: excitation, 543 nm; emission, 560 to 620 nm. All the excitation and gain adjustment were kept constant during image acquisition in control and LPS treatment, and the sections were later counterstained with DAPI (1:1000; Invitrogen) at room temperature for 30 min. All the sections were photographed using a fluorescence microscope (Olympus IX50, Japan) linked to the NIS-Elements F3.2 software and the objective of all the Immunofluorescent staining pictures were 40X.A minimum of 3 randomly-selected images from 8 samples were assessed per group per assigned time point. We established negative control and background fluorescence to exclude false positives(suplementary1).
Transmission electron microscopy (TEM)
Embryonic chick lung specimens were fixed with 2.5% glutaraldehyde in 0.1 M PBS for 2 hours. The anterior primitive streaks were then dissected and stained with osmium tetroxide. The specimens were then embedded in resin, ultrathin-sectioned, and examined using a Tecnai G2 Spirit TWIN Transmission Electron Microscope (FEI, USA).
Cell line and manipulation
MLE-12 cell line was attained from ATCC (American Type Culture Collection, CRL-2110, USA) and cultured according to the protocol provided by ATCC. DMEM medium and penicillin/streptomycin were purchased from Gibco (NY, USA). Insulin, transferrin, sodium selenite, hydrocortisone, β-estradiol, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), L-glutamine 2 mM (in addition to that in the base medium) were purchased from Sigma (MO, USA). The cells were seeded in six-well plates (1 × 106 cells/ml) in DMEM supplemented with 2% fetal bovine serum (Gibco, Gaithersburg, MD, USA) and cultured in a humidified incubator with 5% CO2 at 37°C overnight. Cells were then exposed to 10µg/ml LPS using blank medium as a control. When needed, Bay-11–7082 and Vitamin C were added to the medium at final concentrations of 10µg/ml, respectively. Cells were harvested 48 hours later for further analysis. Immunofluorescent staining was performed 48 hours later using antibodies against GATA-6 (1:300, Abcam), Nrf2 (1:200, DSHB), P65 (1:200, Sigma) and SP-C (1:1000, Abcam), respectively. A minimum of 8 images per treatment group were assayed. The cells were photographed using an inverted fluorescence microscope (Nikon, Ti-U, Japan) linked to the NIS-Elements F3.2 software.
Quantitation of apoptotic cells
Annexin V-FITC (BD Bioscience, USA) and PI double staining were used to identify and quantify apoptotic cells present in the MLE-12 cells and chick embryo lungs. Briefly, the lung tissue was digested in 10% fetal bovine serum in Dulbecco’s modified Eagle medium, 6.5 mg/ml DNase I, and 12 mg/ml collagenase I (Roche, Indianapolis, IN) (30 min; shaking 200 rpm; 37°C). The cell suspension was strained through a 70-mm cell strainer (Fisher Scientific, Fair Lawn, NJ) and cells were collected by centrifugation (500 × g; 5 min; 4°C). Cells were resuspended in Geyes solution, centrifuged as before, and collected in PBS. the cells were collected and resuspended in cold PBS at a density of 1 × 106 cells/ml and then introduced into 200 μl of the Annexin V-binding buffer. The samples were then incubated with 2μl fluorescein isothiocyanate (FITC)-labeled Annexin V and 2μl PI at room temperature for 15min and immediately analyzed by a FACS Calibur flow cytometer (BD, NJ, USA). The acquired data were evaluated using FCS-Express software version 3.0 (De Novo).
Cell cycle assay
Tissues were digested in the same way as quantitation of apoptotic cells .Cultured cells were washed twice by centrifugation in phosphate-buffered saline at 4°C, 1300rpm for 3min. After vortexing gently, the cell suspension was slowly added to 1ml of 70% ethanol in a 1.5-ml e-tube and stored at −20°C for 1h. Following centrifugation at 1300rpm, 4°C for 3min, the pellets were resuspended in 1-ml cold phosphate-buffered saline containing RNase A (50μgml−1) and incubated at 37°C, 5% CO2 for 40min. PI was diluted to 50μgml−1 with distilled water, and transferred to a glass tube. The tube was placed on ice and analyzed using a flow cytometer (BD FACS Aria USA).
Measurement of intracellular reactive oxygen species (ROS)
Intracellular ROS was determined using a non-fluorescent dye DCF-DA (2′7-dichlorodihydrofluorescein diacetate) (Sigma-aldrich, USA), which could be oxidized by ROS to the fluorescent dye DCF (2,7-dichlorofluorescin). MLE-12 cells were seeded and incubated at 37°C and 5% CO2 overnight. The cells were then exposed to simple saline or 10µg/ml LPS. When needed, Bay-11–7082 and Vitamin C were added to the medium at final concentrations of 10µg/ml, respectively. At the end of treatment, the cells were incubated in 10μM DCF-DA for 20 min. The intensity of the fluorescence was measured using a BD FACSAria (USA).
TUNEL analysis
TUNEL staining was performed using an In Situ Cell Death Detection Kit (Roche, USA) according to the manufacturer’s instructions. Briefly, sections (4 µm) were deparaffinized in xylene, rehydrated in decreasing concentrations of ethanol, rehydrated and heated for antigen retrieval. Endogenous peroxidase was blocked in 3% hydrogen peroxide. Three different dilutions (1: 7, 1: 11, and 1: 16) of terminal deoxynucleotidyl transferase (TdT) in reaction buffer (containing a fixed concentration of digoxigenin‐labelled nucleotides) were applied to serial sections at 37°C for 1 hr before the slides were placed in Stop/Wash buffer for 10 min. Following intensive washing, a pre-diluted anti‐digoxigenin peroxidase‐conjugated antibody was applied for 30 min. Apoptotic cells were detected after incubation in the 3,3′‐diaminobenzidine (DAB) chromogen (DAKO, Carpinteria, CA, USA) for 6 min and slides were counterstained with Methyl Green (Sigma, St Louis, MO, USA). The presence of TUNEL+ cells was determined using the Image Analysis Software (Olympus, Japan). We evaluated the percentage of TUNEL+ cells relative to the total cells in the same area between the control and experimental groups (N = 8 lungs for each group).
Western blotting
Western blotting was performed in accordance with a standard procedure. Protein samples were extracted from lung tissue homogenate or MLE-12 cell lysate using a radio-immuno-precipitation assay buffer (RIPA, Sigma, USA) supplemented with protease and phosphatase inhibitors, and the protein concentrations were quantified using BCA assay. The protein samples were separated by 10% SDS-PAGE and transferred onto a PVDF membrane (Millipore, MA, USA). The membrane was blocked with 5% non-fat milk and then was incubated with primary antibodies at 4°C overnight against P65 (1:1000; Bioworld), SP-C (1:1000; Abcam), AQP5 (1:1000; Abcam), GATA-6 (1:1000; Abcam) or ABCA3 (1:1000; Abcam) in TBS buffer, respectively. β-actin was used as a loading control (1:3000; Proteintech). After incubation with HRP conjugated secondary antibodies (1:3000; EarthOx), the blots were developed with the SuperSignalTM West Femto Chemiluminescent Substrate (ThermoFisher, Rockford, USA) and Gel Doc™ XR+ System (BIO-RAD, CA, USA). The intensity of the bands was analyzed using the Quantity One software (BIO-RAD, CA, USA) according to the manufacturer’s instructions. The Western blotting results are representative of three independent experiments.
RNA isolation and qpcr
Total RNA was isolated from fresh embryonic chick lung tissue or MLE-12 cells using the E.Z.N.A® Total RNA Kit (OMEGA, Georgia, USA) according to the manufacturer’s instructions. First-strand cDNA synthesis and the SYBR® Green qPCR assay were performed using the PrimeScriptTM RT Reagent Kit (Takara, Japan). All the specific primers used are described in . The reverse transcription reactions were performed in the Bio-Rad S1000TM thermocycler (Bio-Rad, USA). qPCR mixture was then incubated at 95°C for a 3 min initial denaturation step followed by 40 PCR cycles (95°C for 5s, 60°C for 20 s, and 72°C for 20 s), using the ABI 7000 Real Time PCR machines. Corresponding relative mRNA expression was calculated by the 2−ΔΔCq method and normalized to β-actin [Citation18]. The qPCR results are representative of three independent experiments.
Table 1. Sequences of qPCR primers.
Data analyses
Data analyses were performed using Graphpad Prism 5 software package. The results are presented as Mean ± SEM. Statistical analyses were performed using one-way ANOVA and Student’s t-test. P < 0.05 was considered to be statistically significant.
Results
LPS exposure restricted the morphogenesis of embryonic chick lungs
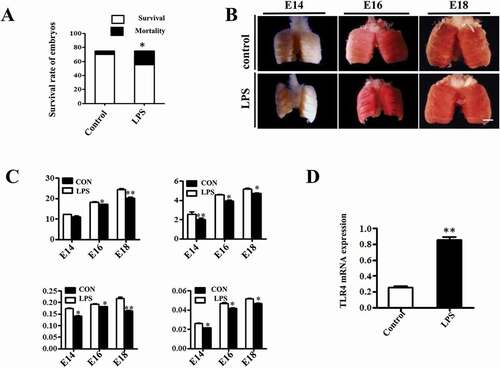
In chick embryos, lung morphogenesis takes place before birth in five stages. Bronchial epithelial cells start differentiation at E7, and emergence of AECII starts at E14 and ends at E18, with the final alveolar maturation occurring postnatally [Citation19]. Therefore, starting from E7, chick embryos were exposed to 100µg/ml LPS or simple saline (control) every other day via injection into air chambers. LPS treatment significantly reduced the survival rate of E18 chick embryos (), and LPS-treated embryonic chick lungs appeared smaller (), with significantly reduced mass compared to that of control (). The pathological action of LPS is facilitated via the TLR4/MD2 complex, which is sequentially regulated by a series of adaptor proteins [Citation3]. Hence, we sought to evaluate the TLR4 expression with qPCR, and found that LPS exposure dramatically increased TLR4 expression in E18 lungs ().
Figure 1. LPS induced macroscopic alteration in chick embryonic lungs. (A) LPS exposure significantly reduced survival rate of E18 chick embryos compared to controls. (B) The morphological appearance of embryonic chick lungs at E14, E16 and E18, and LPS induced smaller sized changes. (C) LPS exposure significantly reduced the wet and dry mass of embryos and lungs. (D) The qPCR data show LPS induced significantly up-regulated TLR4 expression at mRNA level in E18 chick lungs. *P < 0.05. **P < 0.01. Scale bar = 200μm.
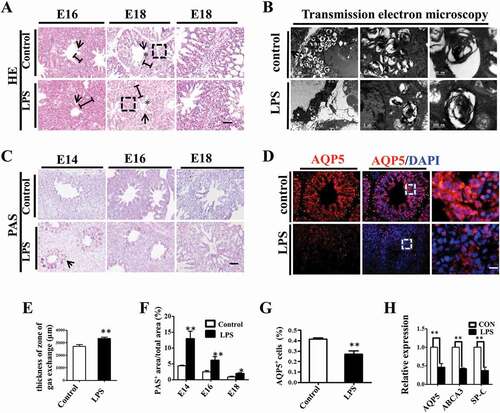
Next, we examined the histological characteristics of embryonic lungs with H&E staining. Atrial and infundibular (star) dilation, interatrial septal (arrow) thinning and gas exchange zone (bar) narrowing were seen in control lungs, which displayed a spongy lung phenotype at E18, and were well prepared for gas exchange at hatching. LPS exposure significantly enhanced the thickness of the gas exchange zone and induced a less mature and solid lung phenotype (). Immature alveolar epithelial cells contain abundant glycogen store, which will be converted into surfactant phospholipids and disappear upon maturation [Citation20]. PAS staining showed that glycogen were gradually reduced during the lung development, but LPS significantly increased the PAS+ area compared to control (), indicating that LPS induced delayed differentiation of pulmonary cells. We further examined the lamellar body count, an indicator of AECII development and fetal lung maturity [Citation21], using TEM and found that lamellar body count was remarkably decreased upon LPS exposure, suggesting an impaired AECII differentiation and maturation (). Meanwhile, we demonstrated a dramatically suppressed expression of AQP5, an AECI marker [Citation22], with immunofluorescent staining in E18 chick lungs (). Finally, qPCR confirmed that the expressions of AQP5, ABCA3 and SP-C were all reduced at the mRNA level in E18 chick lungs upon LPS exposure (). These data indicate that exposure to LPS restricted the development of the embryonic lungs.
Figure 2. LPS induced microscopic alteration in chick embryonic lungs. (A, E): H&E staining was performed on transverse sections of chick lungs. The alveolar wall and gas exchange zone are indicated with arrows and line segments, respectively. LPS increased gas exchange zone thickness, and control lungs display a more mature spongy phenotype at E18. (B): The representative TEM images of E18 chick lungs shows lamellar bodies at multiple spherical particles, ranging from 500nm to 5 μm. LPS significantly reduced LB count. (C, F): PAS staining was performed on transverse sections of embryonic chick lungs. LPS induced more PAS+ areas relative to total areas in embryonic chick lungs. (D, G): In immunofluorescent staining of E18 chick lungs, red color shows AQP5 and blue color shows DAPI staining, respectively. The far right panel indicates the dotted squares with higher magnification. LPS significantly reduced AQP5+ cells in E18 chick lungs. (H): qPCR data show LPS significantly induced down-regulation of AQP5, ABCA3 and SP-C expression at mRNA level in E18 lungs. *P < 0.05. **P < 0.01 .Scale bars = 50μm.
LPS exposure promoted fibrosis in embryonic chick lungs
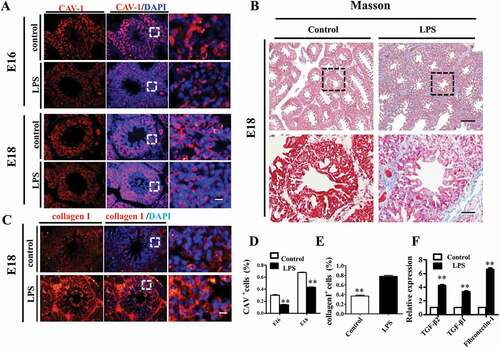
One possible cause of hyperglycaemia-induced thickening of the pulmonary septal wall is the activation of fibroblasts and overproduction of extracellular matrix in interstitial tissue. To explore this possibility, we carried out immunofluorescent staining of CAV-1, a critical regulator of lung fibrosis [Citation23], in E16 and E18 chick lungs. The results showed that the expression of CAV-1 was significantly reduced by LPS treatment compared to controls in both E16 and E18 lungs (). Masson staining is routinely used to assess tissue fibrosis, and we observed increased Masson staining of E18 chicken lung in the LPS-treated group compared to control (). Since the overexpression of type I collagen is a possible hallmark of organ fibrosis [Citation24], we also implemented immunofluorescent staining of collagen I in E18 chick lungs, and the results showed that collagen I was highly expressed more in the LPS-treated lung than in the control (). Furthermore, by using qPCR, we demonstrated that the expression levels of fibrosis related genes, such as TGF-β1, TGF-β2 and Fibronectin 1 were dramatically up-regulated upon LPS exposure (). All these data indicate that LPS promoted pulmonary fibrosis in the chick lungs.
Figure 3. LPS promoted fibrosis in embryonic chick lungs. (A, D): In immunofluorescent staining of chick lungs, red color shows CAV-1 and blue color shows DAPI staining, respectively. The far right panel indicates the dotted squares of the merged images with higher magnification. LPS induced down-regulation of CAV-1 in E16 and E18 chick lungs. (B): LPS increased Masson staining in E18 chick lungs. The lower panel indicates the dotted squares with higher magnification. (C, E): In immunofluorescent staining of E18 chick lungs, red color shows Collagen I and blue color shows DAPI staining, respectively. The far right panel indicates the dotted squares of the merged images with higher magnification. LPS induced more expression of Collagen I in E18 chick lungs. (F): qPCR data show LPS-induced up-regulation of TGF-β2, TGF-β1 and Fibronectin-1 gene expression in E18 chick lungs. *P < 0.05. **P < 0.01. Scale bars = 50 μm.
LPS exposure modulated cell proliferation and apoptosis in embryonic chick lungs
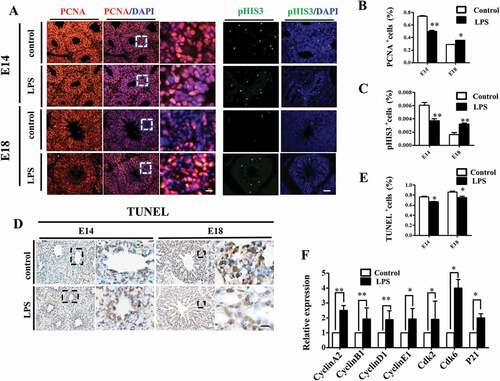
Proper cell differentiation and apoptosis are crucial for alveologenesis in the developing lungs. To evaluate the proliferation of pulmonary cells, we assessed the expression levels of S-phase marker PCNA and M-phase marker pHIS3 in E14 and E18 chick lungs. The results showed that, compared to control, both PCNA and pHIS3 were reduced upon LPS exposure at E14, but turned to more expression at E18 (), which suggests that LPS retarded the proliferation progress of embryonic lungs. Meanwhile, LPS treatment significantly reduced the numbers of TUNEL+ cells in both E14 and E18 chick lungs (), indicating that LPS inhibited apoptosis in embryonic pulmonary cells.FCM (flow cytometry) shows the same results (supplymentary figure 4). To elucidate the mechanism of growth inhibition of LPS on MLE-12 cells and chick embryo lungs, we analyzed the cell cycle. The results demonstrated that LPS exposure resulted in a significant increase in the percentage of cells in the G1 phase indicating the G1 cell cycle arrest (Supplementary figure 7A, 7A1). And the same result on E14 chick embryo lungs was observed (Supplementary figure 7B, 7B1). These data reveal that LPS exposure inhibited the growth of cells by inhibiting cell cycle process. Furthermore,we assayed the expression levels of cell cycle related genes in E18 lungs and found that expressions of Cyclin A2, Cyclin B1, Cyclin D1, Cyclin E1, Cdk2, Cdk6 and P21 were all increased in the LPS-treated lungs (). This indicates that LPS-induced alterations might be partially attributable to altered expression of cell cycle genes.
Figure 4. LPS modulated cell proliferation and apoptosis in embryonic chick lungs. (A, B, C): In immunofluorescent staining of chick lungs, red color shows PCNA, green color shows pHIS3 and blue color shows DAPI staining, respectively. The far right panel indicates the dotted squares of the merged images with higher magnification. Compared to control, PCNA and pHIS3 expression were down-regulated in E14, while up-regulated in E18 chick lungs upon LPS exposure, respectively. (D, E): TUNEL staining of E14 and E18 chick lungs, respectively. The right panels indicate the dotted squares with higher magnification. LPS reduced TUNEL+ cells in E14 and E18 chick lungs, respectively. (F): qPCR data show LPS induced up-regulation of cell cycle related gene expressions in E18 lungs. *P < 0.05. **P < 0.01. Scale bars = 50 μm.
Oxidative stress involved in the lps-induced abnormal development of chick lungs
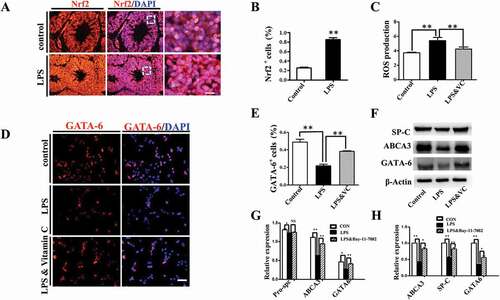
We then explored the potential involvement of oxidative stress in LPS-induced embryonic lung malformation. The immunofluorescent staining revealed that Nrf2, the master regulator of antioxidant responses [Citation25], was significantly up-regulated upon LPS exposure in E18 chick lung (). MLE-12 is a commonly used murine cell line expressing some features of normal AECII [Citation26], and we used it as an in vitro model to further verify the connection between oxidative stress and LPS-induced abnormal pulmonary cell differentiation. LPS induced an intracellular ROS production enhancement, which was partially blunted by the addition of vitamin C to the culture medium (). GATA-6 plays a crucial role in regulating pulmonary epithelial differentiation during lung development[Citation16], and LPS significantly inhibited GATA-6 expression in MLE-12 cells, while it was partially restored by vitamin C (). Western blotting and qPCR data further confirmed that LPS induced down-regulation of SP-C, ABCA3 and GATA-6 expressions, which could be restored by vitamin C (). These data demonstrated that LPS could induce the oxidative stress, which subsequently led to the restriction to embryonic lung development.
Figure 5. Oxidative stress involved in LPS-induced abnormal development of chick lungs. (A, B): In immunofluorescent staining of E18 chick lungs, red color shows Nrf2 and blue color shows DAPI staining, respectively. The far right panel indicates the dotted squares of the merged images with higher magnification. LPS induced a dramatic increase in Nrf2 expression in E18 chick lungs. (C): Flow cytometry data show intracellular ROS production in MLE-12 cells was significantly increased by LPS. (D, E): In immunofluorescent staining of MLE-12 cells, red color shows GATA-6 and blue color shows DAPI staining, respectively. LPS significantly down-regulated GATA-6 expression. (F, G): Western blot shows LPS-induced decrease in protein expression of SP-C, ABCA3 and GATA-6 in MLE-12 cells. (H): qPCR data show LPS down-regulated mRNA expression of ABCA3, SP-C and GATA-6 in MLE-12 cells. All these effects were reversed with vitamin C. *P < 0.05. **P < 0.01. Scale bars = 50 μm.
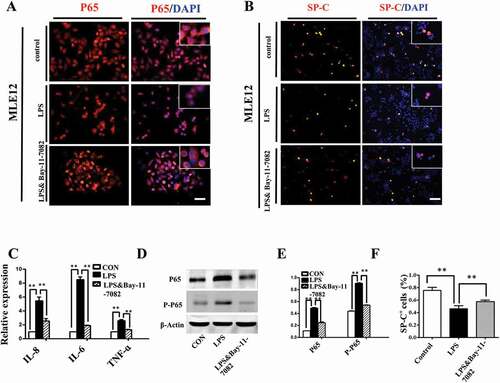
Activation of nf-κb pathway contributed to lps-induced abnormal differentiation of pulmonary cells
It has been known that the activation of NF-κB is of central importance in inflammation and immunity, cell development, growth and survival[Citation27], we demonstrated that LPS induced activation of NF-κB signaling in MLE-12 cells. Upon LPS exposure, up-regulation of IL6, IL8 and TNF-α expression was observed and this effect was abrogated by the addition of IκB/IKK inhibitor Bay-11–7082 (). Meanwhile, LPS induced P65 translocation from cytoplasm into nucleus, which was also suppressed by Bay-11–7082 (). Western blot further confirmed that LPS significantly increased both p65 and phosphorylated p65 levels, and this effect was abolished upon the application of Bay-11–7082 (). Moreover, LPS induced reduction of SP-C in MLE-12 cells, which could be partially restored by blocking NF-κB signaling with Bay-11–7082 (). These data demonstrated that NF-κB signaling involved in LPS-induced disruption of pulmonary cell differentiation.
Figure 6. Activation of NF-κB pathway contributed to LPS-induced abnormal differentiation of pulmonary cells. (A): In immunofluorescent staining of MLE-12 cells, red color shows P65 and blue color shows DAPI staining, respectively. The higher magnification images are shown at right upper corners. LPS induced P65 nuclear translocation. (B, F): In immunofluorescent staining of MLE-12 cells, red color shows SP-C and blue color shows DAPI staining, respectively. LPS induced down-regulation of SP-C expression. All these effects were restored with NF-κB inhibitor Bay-11–7082. (C): qPCR data show LPS-induced expression of IL-8, IL-6 and TNF-ɑ at mRNA level in MLE-12 cells. (D, E): Western blotting shows increased protein expression of P65, phospho-P65 in MLE-12 cells upon LPS exposure. *P < 0.05. **P < 0.01. Scale bars = 50 μm.
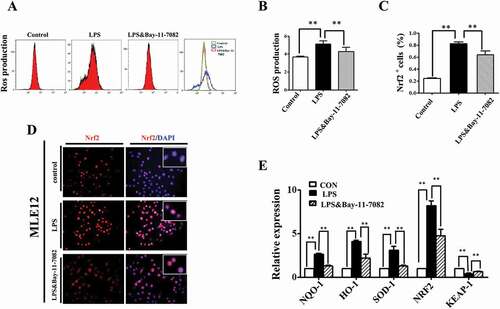
Although the exact mechanisms still under debate, numerous reports indicate that crosstalk between NF-κB and oxidative stress plays inhibitory or stimulatory roles in various cellular processes [Citation28]. In our study, we found that LPS induced ROS elevation in MLE-12 cells could be partially abrogated by blocking NF-κB signaling with Bay-11–7082 (). And the LPS induced Nrf2 expression enhancement was partially abrogated by Bay-11–7082 as well (). Furthermore, qPCR data showed that the expressions of Nrf2 and its target genes, such as NQO-1, HO-1, and SOD-1, rose with LPS exposure, and fell in the presence of Bay-11–7082. Meanwhile, Keap-1, an Nrf2 repressor protein, showed an inverse pattern ().
Figure 7. NF-kB signaling modulated ROS in LPS induced alveolar epithelial mal-differentiation. (A, B): Flow cytometry data and merged image show LPS-induced ROS elevation in MLE-12 cells was abolished with Bay-11–7082. (D, C): In immunofluorescent staining of MLE-12 cells, red colour stands for Nrf2 and blue colour stands for DAPI staining, respectively. The higher magnification images are shown at the right upper corners. LPS-induced increase in Nrf2 expression was partially blocked with Bay-11–7082. (E): qPCR data show LPS induced alteration of oxidative stress related genes in MLE-12 cells, which were revered with Bay-11–7083. *P < 0.05. **P < 0.01. Scale bars = 50 μm.
Taken together, our results imply that LPS activated NF-κB signaling could affect cellular oxidative stress level, which suppresses GATA-6 expression and interferes with the differentiation of pulmonary epithelial cells.
Discussion
The microbial infection of placental tissues frequently causes unpleasant outcomes in pregnancy, including preterm birth, neonatal morbidity and mortality [Citation29]. Clinical observations indicate that fetal inflammation is beneficial to preterm lungs and reduces incidence of RDS, while it may increase lung BPD on the other hand [Citation30]. Yun et al demonstrated that differences in bacterial composition in mouse lungs could affect lung development and the alveolar structure [Citation8]. Gao et al. reported that maternal exposure to LPS postponed the morphological maturation of the rat lung, through altering the expression of genes implicated in alveologenesis [Citation31]. Actually, very little has been documented regarding the adverse effects of LPS exposure on embryonic lung development, especially from early embryonic stages. Here, we found a disruptive influence of LPS exposure on lung development in early stage chick embryos, which manifested as higher mortality rates of E18 embryos () and thicker zones of pulmonary gas exchange (). Glycogen is the substrate for surfactant phosphatidylcholine synthesis in AECII [Citation32], and it is abundant in immature alveolar epithelial cells and disappears upon cell maturation. The increased PAS staining following LPS exposure () might indicate the misguidance of pulmonary development and/or dysfunction of AECII.
Proliferation and apoptosis participate in lung development during both the prenatal and postnatal periods [Citation33]. At the embryonic stage, branching morphogenesis begins with extensive proliferation of epithelial and mesenchymal cells, but no epithelial apoptosis [Citation34]. At the pseudoglandular stage, the bronchial airway tree is established by repeated dichotomous branching. Pulmonary progenitor cells start to differentiate, while epithelial apoptosis still does not occur [Citation35,Citation36]. At the canalicular stage, respiratory bronchioli appear, interstitial tissue decreases, AECII cells differentiate into AECI cells, and the apoptosis of interstitial tissue contributes to mesenchymal involution and thinning of the alveolar septa. Meanwhile, apoptosis of epithelial cells is increased [Citation35–Citation39]. At the saccular stage, terminal airways widen to form saccules, the interstitium between airspaces thins, the vascular network expands, and epithelial cell apoptosis increases further [Citation35–Citation39]. At the alveolar stage, there is extensive alveolar septation, an increase in the number of alveoli, further thinning of the epithelial-endothelial barrier, and then a transient increase in apoptosis at birth followed by continuous proliferation of epithelial cells [Citation40,Citation41]. In this study, we revealed that cell proliferation was inhibited upon LPS exposure at E14. However, even though continuing decreasing within the same group at E18, the proliferation level in LPS treated lungs was significantly higher than that in control, which was opposite to that at E14 (), implying differential susceptibilities of pulmonary progenitor cells towards LPS at different stages of development. The LPS-induced inhibition of cell apoptosis () disrupted apoptosis-modulated remodeling alveologenesis in the developing lung. The increased expression of cell cycle-related genes further verified our initial speculations (). Cell apoptosis during lung development might be primarily regulated by the apoptosis factor TGF-β and the anti-apoptotic factors IGF-1 and NO, in addition to other apoptosis-related proteins [Citation33].
LPS-induced inflammatory responses often lead to fibrosis. Besides, when AECII cells proliferate and differentiate into AECI cells, many fibrosis-associated cytokines tend to be secreted. Meanwhile, the transdifferentiation of myofibroblasts proceeds, ultimately further promoting lung fibrosis [Citation42]. It has also been reported that pulmonary epithelial cell transformation to mesenchymal cells (EMT) promotes fibrosis, because the transformed cells eventually differentiate into fibrous connective tissue, leading to heavy interstitial fibrous tissue deposition [Citation43]. CAV-1 is an endothelial marker and can be used as an indicator for fetal lung development [Citation44]. In this study, we found that LPS exposure inhibited CAV-1-labelled cell maturation in embryonic chick lung (), indicating a suppression of pulmonary cell differentiation. Meanwhile, LPS enhanced the expression of Collagen I as well as fibrosis related gene expression (), supporting the notion of LPS-mediated induction of fibrosis in the developing lung.
Nrf2-Keap-1 signaling pathway is of great importance in protection against oxidative damage triggered by inflammation and injury. LPS induced significant increase in Nrf2 expression in E18 lungs (), and we utilized MLE-12 cells to further investigate its possible role in LPS induced lung alteration in vitro. LPS stimulated the elevation of ROS production in MLE-12 cells (), and the expressions of ABCA3 and SP-C were inhibited at the same time (). Meanwhile, scavenging ROS with vitamin C alleviated this inhibition, indicating oxidative stress may involve in LPS induced lung mal-development. Zhang et al. reported that GATA-6 and Wnt signaling co-ordinately regulated the expansion of stem/progenitor cells and epithelial cell differentiation during both lung development and regeneration [Citation16], and LPS significantly inhibited GATA-6 expression in MLE-12 cells, which was also reversed by vitamin C (). It’s possible that LPS induced the oxidative stress, which subsequently led to the restriction to embryonic lung development.
NF-kB signaling is involved in the pathogenesis of lung fibrosis, and is frequently activated in response to LPS [Citation45,Citation46]. P65 is one of the five components of NF-κB and it enters into nucleus upon the activation of NF-κB signaling [Citation47]. Here, we observed the nuclear translocation and phosphorylation of P65 in MLE-12 cells in response to LPS exposure, and this effect was abrogated by the NF-κB signaling inhibitor, Bay-11–7082 (). Moreover, LPS exposure inhibited SP-C expression in MLE-12 cells, and Bay-11–7082 could appreciably alleviate this suppressive effect (). These data indicate the involvement of NF-κB signaling in LPS-induced pulmonary fibrosis.
It is reported that crosstalk between NF-κB and oxidative stress involves in the regulation of various cellular processes [Citation28]. LPS can generate ROS through the activation of pro-inflammatory cytokines, such as TNF-α, IFN-γ, IL-6 and IL-12 [Citation48,Citation49], which in turn can result in significant cell damage or apoptosis in embryos [Citation50,Citation51].Likewise, we also found the excessive production of pro-inflammatory cytokines () and ROS from MLE-12 cells exposed to LPS (), and again the enhancement of ROS in presence of LPS was partially repressed by blocking NF-κB signaling (), further indicating the involvement of NF-κB signaling. Furthermore, the blocking of NF-κB signaling revered the LPS induced alteration of Nrf2-Keap-1 signaling pathway, indicating the crosstalk between these two pathways in LPS induced lung mal-development.
There indeed are some reports which demonstrated the present of microbial flora in the mother’s vagina, intestines, amniotic fluid and placenta. LPS disrupts the mother’s bacterial composition which can cause inflammatory reactions and affects fetus’s development, and bacterial colonization is most common in fetus’s membrane infection, amniotic fluid and/or premature delivery. However, the mechanism of LPS developmental toxicity is still not clear. In this study, we demonstrated that LPS induced disruption of pulmonary cell development in chick embryos with the involvement of oxidative stress and NF-κB signalling, which may provide some information to solve the problem mentioned-above.We demonstrated the LPS-induced lung malformation in embryonic chick and it is attributable to at least several mechanisms. Firstly, LPS exposure interferes with the cell cycle of pulmonary precursor cells, and inhibits apoptosis. Secondly, LPS exposure clearly promotes lung fibrosis. NF-κB signaling plays an indispensable role in LPS-induced disruption of lung development via mutual interactions with oxidative stress. All of these phenomena adversely affect the differentiation of pulmonary epithelial cells through the alteration of GATA-6 expression. As a consequence, the dysfunction of the signaling pathways described above leads to aberrant alveolar development in the chick embryo. However, the precise molecular biological mechanisms remain to be further elucidated.
Competing interests
The authors declare no competing financial interests.
Supplemental Material
Download Zip (8.3 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary material can be accessed here.
Additional information
Funding
Related Research Data
References
- Heumann D, Roger T. Initial responses to endotoxins and Gram-negative bacteria. Clin Chim Acta. 2002;323(1–2):59–72.
- Dagleish MP, Brazil TJ, Scudamore CL. Potentiation of the extracellular release of equine neutrophil elastase and alpha-1-proteinase inhibitor by a combination of two bacterial cell wall components: fMLP and LPS. Equine Vet J. 2003;35(1):35–39.
- Sender V, Stamme C. Lung cell-specific modulation of LPS-induced TLR4 receptor and adaptor localization. Commun Integr Biol. 2014;7:e29053.
- Hakansson A, Molin G. Gut microbiota and inflammation. Nutrients. 2011;3(6):637–682.
- Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007 Jul;56(7):1761-72. Epub 2007 Apr 24.
- Poroyko V, Meng F, Meliton A, et al. Alterations of lung microbiota in a mouse model of LPS-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2015 Jul 1;309(1):L76-83.
- Kell DB, Pretorius E. On the translocation of bacteria and their lipopolysaccharides between blood and peripheral locations in chronic, inflammatory diseases: the central roles of LPS and LPS-induced cell death. Integr Biol (Camb). 2015 Nov; 7(11):1339–1377.
- Yun, Y, Srinivas G, Kuenzel S, et al. Environmentally determined differences in the murine lung microbiota and their relation to alveolar architecture. PLoS One. 2014;9(12):e113466.
- Evans MJ, Cabral LJ, Stephens RJ, et al. Renewal of alveolar epithelium in the rat following exposure to NO2. Am J Pathol. 1973;70(2):175–198.
- Barkauskas CE, Cronce MJ, Rackley CR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123(7):3025–3036.
- Herriges M, Morrisey EE. Lung development: orchestrating the generation and regeneration of a complex organ. Development. 2014;141(3):502–513.
- Sekine K, Ohuchi H, Fujiwara M, et al. Fgf10 is essential for limb and lung formation. Nat Genet. 1999;21(1):138–141.
- Ohuchi H, Hori Y, Yamasaki M, et al. FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multi-organ development. Biochem Biophys Res Commun. 2000;277(3):643–649.
- Yin Y, Wang F, Ornitz DM. Mesothelial- and epithelial-derived FGF9 have distinct functions in the regulation of lung development. Development. 2011;138(15):3169–3177.
- Morrisey EE, Ip HS, Lu MM, et al. GATA-6: a zinc finger transcription factor that is expressed in multiple cell lineages derived from lateral mesoderm. Dev Biol. 1996;177(1):309–322.
- Zhang Y, Goss AM, Cohen ED, et al. A gata6-Wnt pathway required for epithelial stem cell development and airway regeneration. Nat Genet. 2008;40(7):862–870.
- Plosa EJ, Young LR, Gulleman PM, et al. Epithelial beta1 integrin is required for lung branching morphogenesis and alveolarization. Development. 2014;141(24):4751–4762.
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108.
- Leibel S, Post M. Endogenous and exogenous stem/progenitor cells in the lung and their role in the pathogenesis and treatment of pediatric lung disease. Front Pediatr. 2016;4:36.
- Compernolle V, Brusselmans K, Acker T, et al. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med. 2002;8(7):702–710.
- DeRoche ME, Ingardia CJ, Guerette PJ, et al. The use of lamellar body counts to predict fetal lung maturity in pregnancies complicated by diabetes mellitus. Am J Obstet Gynecol. 2002;187(4):908–912.
- Ramírez-Lorca R, Muñoz-Cabello AM, Toledo-Aral JJ, et al. Aquaporins in chicken: localization of ck-AQP5 along the small and large intestine. Comparative biochemistry and physiology. Part A. Molecular & Integrative Physiology. 2006;143(2):269–277.
- Wang XM, Zhang Y, Kim HP, et al. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med. 2006;203(13):2895–2906.
- Désogère P, Tapias LF, Hariri LP, et al. Type I collagen-targeted PET probe for pulmonary fibrosis detection and staging in preclinical models. Sci Transl Med. 2017 Apr;9(384).
- Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 12(12):931–947.
- Mir-Kasimov M, Sturrock A, McManus M, et al. Effect of alveolar epithelial cell plasticity on the regulation of GM-CSF expression. Am J Physiol Lung Cell Mol Physiol. 302(6):L504–11.
- Wang T, Zhang X, Li JJ. The role of NF-kappaB in the regulation of cell stress responses. Int Immunopharmacol. 2002;2(11):1509–1520.
- Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21(1):103–115.
- Kim HS, Cho JH, Park HW, et al. Endotoxin-neutralizing antimicrobial proteins of the human placenta. J Immunol. 2002;168(5):2356–2364.
- Kramer BW, Kallapur S, Newnham J, et al. Prenatal inflammation and lung development. Semin Fetal Neonatal Med. 2009;14(1):2–7.
- Cao L, Wang J, Tseu I, et al. Maternal exposure to endotoxin delays alveolarization during postnatal rat lung development. Am J Physiol Lung Cell Mol Physiol. 2009;296(5):L726–37.
- Ridsdale R, Tseu I, Roth-Kleiner M, et al. Increased phosphatidylcholine production but disrupted glycogen metabolism in fetal type II cells of mice that overexpress CTP: phosphocholinecytidylyltransferase. J Biol Chem. 2004;279(53):55946–55957.
- Del Riccio V, van Tuyl M, Post M. Apoptosis in lung development and neonatal lung injury. Pediatr Res. 2004;55(2):183–189.
- Levesque BM, Vosatka RJ, Nielsen HC. Dihydrotesto-sterone stimulates branching morphogenesis, cell proliferation, and programmed cell death in mouse embryonic lung explants. Pediatr Res. 2000;47(4 Pt 1):481–491.
- Scavo LM, Ertsey R, Chapin CJ, et al. Apoptosis in the development of rat and human fetal lungs. Am J Respir Cell Mol Biol. 1998;18(1):21–31.
- De Paepe ME, Sardesai MP, Johnson BD, et al. The role of apoptosis in normal and accelerated lung development in fetal rabbits. J Pediatr Surg. 1999;34(5):863–870. discussion 870-1
- Kresch MJ, Christian C, Wu F, et al. Ontogeny of apoptosis during lung development. Pediatr Res. 1998;43(3):426–431.
- De Paepe ME, Johnson BD, Papadakis K, et al. Lung growth response after tracheal occlusion in fetal rabbits is gestational age-dependent. Am J Respir Cell Mol Biol. 1999;21(1):65–76.
- De Paepe ME, Johnson BD, Papadakis K, et al. Temporal pattern of accelerated lung growth after tracheal occlusion in the fetal rabbit. Am J Pathol. 1998;152(1):179–190.
- Schittny JC, Djonov V, Fine A, et al. Programmed cell death contributes to postnatal lung development. Am J Respir Cell Mol Biol. 1998;18(6):786–793.
- Bruce MC, Honaker CE, Cross RJ. Lung fibroblasts undergo apoptosis following alveolarization. Am J Respir Cell Mol Biol. 1999;20(2):228–236.
- Kim KK, Kugler MC, Wolters PJ, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci. 2006;103(35):13180–13185.
- Borchers AT, Chang C, Keen CL, et al. Idiopathic pulmonary fibrosis-an epidemiological and pathological review. Clin Rev Allergy Immunol. 2011;40(2):117–134.
- Amatya VJ, Takeshima Y, Kohno H, et al. Caveolin-1 is a novel immunohistochemical marker to differentiate epithelioid mesothelioma from lung adenocarcinoma. Histopathology. 2009;55(1):10–19.
- Morin C, Cantin AM, Rousseau É, et al. Proresolving action of docosahexaenoic acid monoglyceride in lung inflammatory models related to cystic fibrosis. Am J Respir Cell Mol Biol. 53(4):574–583.
- Cigana C, Curcurù L, Leone MR, et al. Pseudomonas aeruginosa exploits lipid A and muropeptides modification as a strategy to lower innate immunity during cystic fibrosis lung infection. PLoS One. 2009;4(12):e8439.
- Yoo K, Kim J, Moon J, et al. Evaluation of a volatile aroma preference of commercial red wines in Korea: sensory and gas chromatography characterization. Food Sci Biotechnol. 2010;19(1):43–49.
- Anderson CF, Mosser DM. A novel phenotype for an activated macrophage: the type 2 activated macrophage. J Leukoc Biol. 2002;72(1):101–106.
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35.
- Eriksson UJ, Borg LA. Diabetes and embryonic malformations. Role of substrate-induced free-oxygen radical production for dysmorphogenesis in cultured rat embryos. Diabetes. 1993;42(3):411–419.
- Zangen SW, Yaffe P, Shechtman S, et al. The role of reactive oxygen species in diabetes-induced anomalies in embryos of cohen diabetic rats. Int J Exp Diabetes Res. 2002;3(4):247–255.