
ABSTRACT
We recently reported that primary acute lymphoblastic leukemia (ALL) cells are susceptible to the microtubule depolymerizing agent vincristine (VCR) in G1 phase. This finding prompted testing another G1 phase-active compound, palbociclib (PCB), a highly selective inhibitor of cyclin-dependent kinases 4/6 (CDK4/6), alone and in combination with VCR. PCB used alone caused G1 arrest in ALL cells with no effect on cell viability, and similar results were obtained for the retinoblastoma (RB)-proficient T98G glioblastoma cell line. In contrast, HeLa cells failed to arrest in the presence of PCB, consistent with their lack of dependence on the CDK4/6-RB pathway. When ALL cells were pretreated with PCB, they became refractory to death in G1 phase induced by VCR treatment, whereas HeLa cells retained VCR sensitivity after PCB pretreatment. Immunofluorescence microscopy showed that PCB did not disrupt the microtubule network nor prevent VCR from doing so. Furthermore, ALL cells pretreated with PCB retained susceptibility to the Bcl-2/Bcl-xL inhibitor ABT-263, indicating that downstream apoptotic signaling was unaffected. When released from PCB-enforced arrest, ALL cells reinitiated cycling and regained sensitivity to VCR. ALL cells treated with cycloheximide also arrested in G1 phase and became insensitive to VCR, independently reinforcing conclusions derived from PCB-imposed arrest. Thus, primary ALL cells advancing through G1 phase are strictly dependent on functional microtubules for survival whereas microtubules are dispensable for G1-arrested cells. These findings provide novel insight into interphase microtubule function and, from a therapy standpoint, strongly caution against combining microtubule targeting agents and CDK4/6 inhibitors for ALL.
Introduction
Acute lymphoblastic leukemia (ALLFootnote1) is a hematological cancer characterized by accumulation of undifferentiated progenitor blasts and occurs in both children and adults [Citation1,Citation2]. While cure rates are relatively high for pediatric ALL, cure rates are significantly lower for adults, with long-term disease-free survival rates of about 30–40%, emphasizing the need for new treatment strategies [Citation1–Citation3]. Currently, microtubule targeting agents (MTAs) such as vincristine (VCR) are widely used in combination with other drugs for the treatment of ALL [Citation4]. While MTAs are clinically relevant they have limitations including varied patient response, side effects such as myelosuppression and neurotoxicity, and development of resistance or relapse [Citation5–Citation7].
In order to reduce side effects and increase the effectiveness of MTAs, identify potential new targeted therapies, and design novel drug combinations, it is critical to determine how MTAs exert their cytotoxicity. In laboratory models, such as cell lines and xenografts models, it is well established that MTAs are able to induce cell death through disruption of spindle microtubules, leading to mitotic arrest and subsequent cell death via intrinsic apoptosis [Citation8–Citation13]. However, the extent to which mitotic or post-mitotic death contributes to clinical efficacy is uncertain and under debate. The low mitotic index and low proliferation rate of many types of tumor that are responsive to MTAs [Citation14], as well the failure of targeted antimitotic agents to elicit patient response in clinical trials, has led to the suggestion that MTAs may also, and perhaps primarily, target interphase microtubules [Citation15–Citation20].
Recently, we discovered that VCR acts in two different cell cycle phases in primary adult ALL cells [Citation21]. If a cell encounters VCR in G1 phase, death ensues directly from G1 without further cell cycle advance. However, if a cell encounters VCR beyond G1 phase, it advances to M phase and undergoes mitotic arrest and apoptosis. Thus, primary ALL cells are susceptible to VCR in G1 and M phases, but not in S or G2 phases, indicating essential roles for microtubules not only in M phase as expected but also in G1 phase. This is in contrast to established cancer cell lines which are characteristically only susceptible to MTAs in M phase [Citation22,Citation23]. Thus, primary ALL cells represent a powerful model system to investigate novel bimodal mechanisms of MTA action with potential clinical relevance.
The susceptibility of primary ALL cells to VCR in G1 phase prompted testing other G1 phase-active compounds, either alone or in combination with VCR, to determine if additive or synergistic effects were achieved. A leading candidate is the highly selective CDK4/6 inhibitor palbociclib (PCB) [Citation24]. PCB received accelerated approval for use in combination with the aromatase inhibitor letrozole or the estrogen receptor antagonist fulvestrant for patients with metastatic HR-positive and HER2-negative breast cancer, and has shown activity in combination with radiotherapy for glioblastoma [Citation25–Citation29]. In this study, we show that PCB has a cytostatic effect on ALL cells, causing G1 phase arrest without reducing viability. PCB pretreatment of either asynchronous cells or cells isolated in G1 phase completely blocked VCR-induced death, and this effect was reversible after PCB removal. Furthermore, induction of G1 arrest through an independent approach using cycloheximide treatment also rendered cells resistant to VCR. These results provide novel insight into interphase microtubule function, showing they play an essential role in the survival of primary ALL cells advancing through G1 phase but are dispensable for cells arrested in G1. Mechanistic and therapeutic implications of these findings are discussed.
Results
PCB induces a quiescent-like state in primary ALL and T98G cells
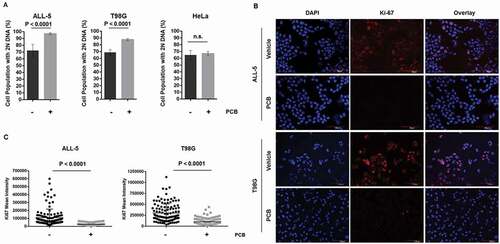
Previously, we have shown that primary ALL cells are susceptible to vincristine-induced death de novo in G1 phase [Citation21]. To exploit this property, we investigated the use of the CDK4/6 inhibitor PCB, which exerts its effects in G1 phase and has shown promising antitumor activity [Citation29]. PCB reduced viability of ALL-5 cells in a dose-dependent manner with maximum effects at around 1 µM (Fig. S1), and this concentration was therefore selected for subsequent experiments. Note that ALL cell viability was maintained above 50% even with higher concentrations of PCB (Fig. S1), suggesting that PCB was exerting a cytostatic rather than cytotoxic effect. Analysis of DNA content by flow cytometry showed that treatment of ALL-5 cells with PCB increased the population of cells with 2N DNA content from about 70% to 95–99% (), left). Previous studies have shown that RB-proficient glioblastoma cell lines undergo G1 arrest in response to PCB [Citation30]. Therefore, as a positive control, we also examined RB-proficient T98G glioblastoma cells, and obtained similar results, with PCB significantly increasing the proportion of cells with 2N DNA (), center). HeLa cells are not dependent on the RB pathway for proliferation due to expression of the E7 papilloma virus protein which inhibits and degrades RB protein [Citation31] and, as expected, the percentage of HeLa cells with 2N DNA was unaffected by PCB (), right). PCB treatment also led to a decrease in the expression of the proliferation marker, Ki-67, in both ALL-5 and T98G cells; representative immunofluorescent images are shown in ), and quantification of Ki-67 staining, averaged over 120 cells/condition, is shown in ). Essentially identical results were obtained when an independent culture of primary ALL cells, ALL-2 [Citation32], was used (data not shown). Under these conditions, there was no induction of cell death in any of the cell types examined (see below), consistent with maintenance of viability. Taken together, these results indicate that PCB causes primary ALL and T98G cells to enter into a quiescent-like G1 arrested state.
Figure 1. PCB causes G1 phase arrest in ALL-5 and T98G but not HeLa cells. A. Cells were treated with vehicle (0.1% DMSO) or 1 µM PCB for 72 h (ALL-5) or 48 h (T98G and HeLa) and DNA content determined by propidium iodide staining and flow cytometry. Data shown are mean ± S.D. (n = 4). B. ALL-5 or T98G cells were treated with 1 μM PCB for 72 h or 48 h, respectively. Cells were fixed and stained for Ki-67 (red) or with DAPI (blue) as a nuclear marker. The scale bar in ALL-5 images is 60 μm while that in T98G images is 120 μm. Images are representative of six fields of vision. C. Mean intensity fluorescence of Ki-67 was quantified for 20 cells per six fields of vision using ImageJ. Cells were selected using DAPI stain while blind to Ki-67 intensity. Each point represents a single cell while horizontal bars represent the mean ± S.D. of all data points (n = 120).
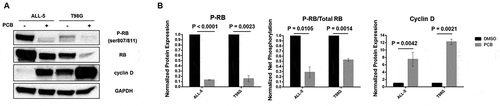
To confirm that PCB inhibited CDK4/6 under these conditions, the phosphorylation status of RB was evaluated by immunoblotting using a phospho-specific (Ser 807/811) antibody which recognizes two well characterized CDK4/6 phosphorylation sites within RB [Citation33]. There was a decrease in phospho-RB and an increase in cyclin D expression after PCB treatment in both ALL-5 and T98G cells (( and )). There was also a decrease in the level of total RB after PCB treatment. While the basis for this observation is not clear, it has been reported by others [Citation28,Citation34], and it may reflect diminished levels of RB in G1-arrested cells or be a technical issue with RB phosphorylation influencing antibody affinity. To account for this, the relative levels of RB and phospho-RB were determined by scanning blots from multiple independent experiments. As shown in ) (middle panel), phospho-RB normalized to total RB was significantly lower after PCB treatment.
Figure 2. PCB inhibits RB phosphorylation and increases cyclin D expression. A. Whole cell extracts were prepared from ALL-5 or T98G cells treated with 0.1% DMSO or 1 µM PCB for 72 h or 48 h, respectively, and subjected to immunoblot analysis for RB, phospho-RB (Ser 807/811), cyclin D, or GAPDH as a loading control. B. Protein expression was quantified by determining band intensity using ImageJ. Data shown are mean ± S.D. (n = 3).
PCB pretreatment renders cells refractory to microtubule depolymerizing agents
Since PCB treatment caused G1 arrest we sought to determine if it could be combined with VCR to modulate VCR-induced G1 phase death. When asynchronous ALL-5 cells were treated with VCR alone, high levels of cell death were observed as indicated by fragmented (sub-G1) DNA and the presence of cleaved poly(ADP-ribose) polymerase (PARP) (( and )). Importantly, we have previously and rigorously demonstrated that a large proportion (60–70%) of dead cells observed under these conditions is derived from those undergoing death directly from G1 phase [Citation21]. When ALL-5 cells were treated with PCB alone, only a slight increase in cells with sub-G1 DNA was observed and PARP remained uncleaved (( and )), confirming that PCB was cytostatic not cytotoxic. Unexpectedly however, when ALL-5 cells were pretreated with PCB to induce G1 phase arrest and then exposed to VCR, they became almost completely refractory to VCR cytotoxicity (( and )). T98G cells were also refractory to VCR after PCB pretreatment (( and )); however, this was anticipated because, unlike ALL cells, T98G cells undergo M phase arrest in response to VCR, as shown in Fig. S2, and PCB pre-treatment prevents advance to M phase. Finally, PCB pretreatment had no effect on VCR-induced cell death in HeLa cells (( and )). Of note, and in contrast to ALL and HeLa cells, intact PARP levels were much higher in asynchronous versus PCB-treated T98G cells ()), suggesting that PARP expression is reduced in arrested cells.
Figure 3. ALL-5 and T98G cells are refractory to VCR after PCB pretreatment while HeLa cells retain their sensitivity. A. ALL-5, T98G, or HeLa cells were treated with 0.1% DMSO or 100 nM (ALL-5 and HeLa) or 1 µM (T98G cells) VCR for 48 h with or without pretreatment with 1 µM PCB for 72 h (ALL-5) or 48 h (T98G and HeLa). Cells were subjected to propidium iodide staining and flow cytometry to determine the percentage of cells with < 2N (sub-G1) DNA. Data shown are mean ± S.D. (n = 3). B. Extracts were made under the same conditions and subjected to immunoblot analysis for poly(ADP-ribose) polymerase (PARP) (detecting intact and cleaved PARP), cleaved PARP (detecting cleaved PARP only), or GAPDH as a loading control. C. ALL-5 cells in G1 phase were isolated using centrifugal elutriation as described in Experimental Procedures and incubated with either vehicle or 100 nM VCR for 48 h, without or with pretreatment with 1 µM PCB for 72 h. Cells were fixed and stained with propidium iodide and DNA content analyzed by flow cytometry. Data shown are representative of two independent experiments of three replicates each.
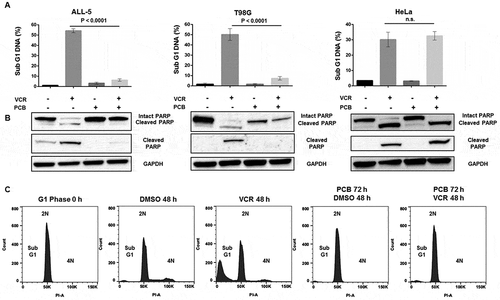
To further investigate the susceptibility of G1 phase ALL cells to VCR and protection by PCB, we isolated cells in G1 phase by centrifugal elutriation, as previously described [Citation21,Citation35]. As shown in ), cells thus isolated were highly phase-purified, with 98–99% exhibiting 2N DNA content (first panel). When treated with DMSO, cells embarked on the cell cycle, as shown by the presence of cells with > 2N DNA after 48 h (, second panel), and as shown by incorporation of 5-ethynyl-2ʹ-deoxyuridine (EdU) as reported previously [Citation21]. When treated with VCR, cells died without evidence of mitotic arrest, with 37 ± 4% (n = 6) of the cells exhibiting sub-G1 DNA content (), third panel). When treated with PCB, cells with 2N DNA were maintained as expected (), fourth panel) and furthermore, when VCR was added after PCB, cells remained arrested in G1 phase with minimal evidence of cell death (fifth panel).
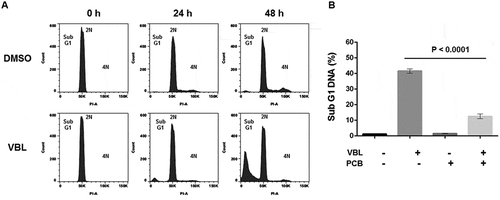
To support the conclusion that PCB blocks MTA susceptibility of primary ALL cells in G1 phase, the effects of vinblastine (VBL) were also examined (). VBL was selected as a comparative drug since, like VCR, it exerts its effects via microtubule depolymerization [Citation8]. Centrifugal elutriation was used to isolate a pure G1 phase population which was treated with vehicle or VBL and analyzed for DNA content over time (). In the presence of DMSO, cells advanced in the cycle as indicated by an increase in cells with > 2N DNA (), upper panels). In the presence of VBL, fragmented DNA was observed without evidence of significant cell cycle advance or mitotic arrest, with 37 ± 7% (n = 6) of cells exhibiting sub-G1 DNA content after 48 h (), lower panels). This is consistent with death ensuing directly from G1 phase, as found for VCR. ALL-5 cells were then pretreated with PCB prior to treatment with VBL, and sub-G1 DNA content determined similarly. As shown in ), PCB inhibited VBL-induced DNA fragmentation with high statistical significance.
Figure 4. Vinblastine induces death in G1 phase which is prevented by PCB pretreatment. A. ALL-5 cells in G1 phase were isolated using centrifugal elutriation and treated with 0.1% DMSO or 100 nM vinblastine (VBL) for the times indicated. Cells were fixed and stained with propidium iodide and DNA content analyzed by flow cytometry. Data shown are representative of two independent experiments of three replicates each. B. ALL-5 cells were treated with 0.1% DMSO or 100 nM VBL for 48 h with or without pretreatment with 1 µM PCB for 72 h. Cells were fixed and stained with propidium iodide and subjected to flow cytometry to determine the percentage of cells with < 2N DNA. Data shown are mean ± S.D. (n = 3).
PCB does not influence the microtubule network nor apoptotic signaling pathways
We considered whether PCB affected VCR action upstream, at the level of microtubule depolymerization, and therefore visualized microtubules by immunofluorescence microscopy ()). HeLa cells were used as a control, and microtubules, which were readily visualized in untreated cells, were absent after VCR treatment (left panels). Since ALL cells grow in suspension, it was necessary to first centrifuge them onto glass slides prior to processing for immunofluorescence. While this procedure caused some perturbations in cell morphology, interphase microtubules were readily visualized in untreated cells (observed against a nuclear background which is the dominating organelle) and were depolymerized after VCR treatment (middle panels). In addition, normal interphase microtubules were observed in PCB pretreated cells, and these were absent after VCR treatment (), right panels). These results confirm that vincristine caused depolymerization of interphase microtubules in ALL cells, and that PCB pretreatment did not disrupt microtubule integrity nor prevent VCR from doing so.
Figure 5. Pretreatment with PCB does not prevent VCR from disrupting the microtubule network or block downstream apoptotic signaling. A. HeLa or ALL-5 cells were treated with 0.1% DMSO or 100 nM vincristine (VCR) for 14 h, with or without pretreatment with 1 µM PCB for 72 h, as indicated. Cells were fixed and permeabilized and α-tubulin visualized by fluorescence microscopy. B. ALL-5 cells were treated with 0.1% DMSO or 100 nM ABT-263 for 16 h with or without pretreatment with 1 µM PCB for 72 h. Cells were fixed and stained with propidium iodide and subjected to flow cytometry to determine the percentage of cells with < 2N DNA. Data shown are mean ± S.D. (n = 3). C. Extracts were made under the same conditions and subjected to immunoblot analysis for poly(ADP-ribose) polymerase (PARP) (detecting intact and cleaved PARP), cleaved PARP (detecting cleaved PARP only), or GAPDH as a loading control.
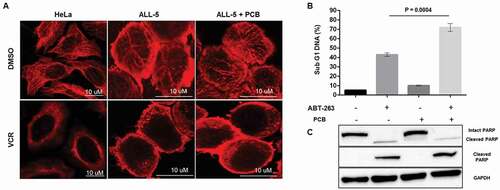
To test the possibility that PCB blocks the action of VCR downstream, at the level of cell death pathways, the Bcl-2/Bcl-xL inhibitor ABT-263 was used to activate the intrinsic apoptosis pathway without or with PCB pretreatment. We have shown previously that ABT-263 induces apoptosis in primary ALL cells which are highly dependent on Bcl-2 [Citation32]. Treatment of ALL-5 cells with ABT-263 alone induced DNA fragmentation and PARP cleavage as expected ()), and when cells were pretreated with PCB and then exposed to ABT-263, cell death was not prevented. In fact, there was a significant increase in cells with fragmented DNA after co-treatment, and this augmentation was reproducibly observed in several independent experiments. These results suggest that PCB pretreatment, or the quiescent state, sensitizes cells to Bcl-2 inhibition, and show that PCB does not block activation of the core apoptotic pathway.
PCB-induced arrest is reversible and restores vincristine susceptibility
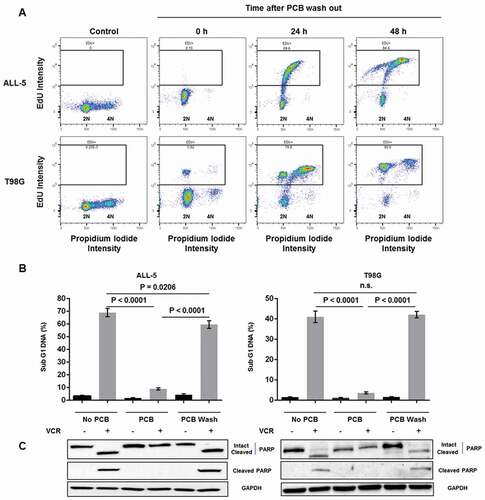
In order to determine if PCB-induced arrest was reversible, EdU incorporation assays were performed to track cell cycle reentry upon removal of PCB. First, untreated asynchronous ALL-5 and T98G cells, in the absence of EdU, were stained with propidium iodide and analyzed by two-parameter flow cytometry, in order to define a threshold distinguishing EdU-negative from EdU-positive cells (), left panels). Cells were then treated with PCB to induce G1 arrest, PCB was removed by repeated washings in PBS, EdU was added, and cells harvested at intervals up to 48 h and analyzed by flow cytometry. As shown in ), at zero time, both ALL-5 and T98G cell populations largely exhibited 2N DNA content and were negative for EdU label, as expected. With increasing times of incubation, cells shifted diagonally, simultaneously increasing DNA content and incorporating EdU, and by 48 h of incubation, 84% of ALL-5 cells and 95% of T98G cells were EdU labeled. These results indicate that after PCB removal, cells are competent to resume cycling. Because T98G cells have a much shorter doubling time than primary ALL cells (24 h versus 65 h), by 48 h of incubation, most T98G cells have divided, appearing in the cytogram as EdU-positive with 2N DNA (), lower right panel). Once it was determined that cells could reenter the cell cycle when PCB was removed, we investigated whether VCR sensitivity was regained. As shown in ( and ), after PCB removal, both ALL-5 and T98G cells regained their sensitivity to VCR, with levels of DNA fragmentation and PARP cleavage comparable to those of VCR treated cells without PCB pretreatment.
Figure 6. PCB-induced arrest is reversible and cells regain sensitivity to VCR after reentering the cell cycle. A. ALL-5 and T98G cells were stained with propidium iodide and subjected to two-parameter flow cytometry to create a gate distinguishing EdU-negative and EdU-positive cells (left panels). ALL-5 and T98G cells were then treated with 1 µM PCB for 72 h (ALL-5) or 48 h (T98G), washed thrice with PBS, and incubated in growth media containing 10 µM EdU for the times indicated. Cells were harvested, fixed, and stained for EdU incorporation and with propidium iodide and analyzed by two-parameter flow cytometry. Data shown are representative of three independent experiments. The percentage of cells positive for EdU is indicated at the top of each panel. B. ALL-5 and T98G cells were treated with 0.1% DMSO or 100 nM (ALL-5) or 1 µM (T98G) VCR for 48 h with or without pretreatment with 1 µM PCB for 72 h (ALL-5) or 48 h (T98G). A third set of cells were pretreated with 1 µM PCB for 72 h (ALL-5) or 48 h (T98G), after which cells were washed thrice with PBS and treated with 0.1% DMSO or 100 nM (ALL-5) or 1 µM (T98G) VCR for 48 h. Cells were stained with propidium iodide and subjected to flow cytometry to determine the percentage of cells with < 2N DNA. Data shown are mean ± S.D. (n = 3). C. Extracts were made under the same conditions and subjected to immunoblot analysis for poly(ADP-ribose) polymerase (PARP) (detecting intact and cleaved PARP), cleaved PARP (detecting cleaved PARP only), or GAPDH as a loading control.
Cycloheximide treated ALL cells arrest in G1 phase and become insensitive to VCR
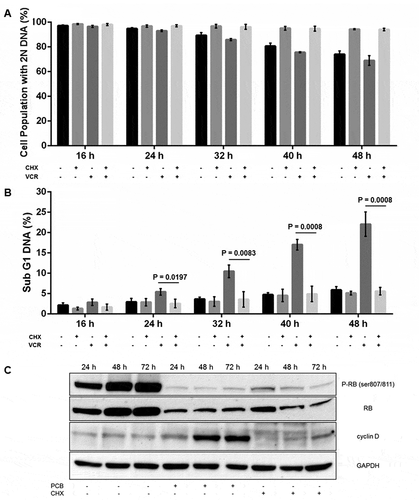
The effects of the protein synthesis inhibitor cycloheximide (CHX) were next investigated, with the initial intention of determining whether G1 phase death of primary ALL cells in response to VCR was dependent on protein synthesis. Cells in G1 phase were prepared and treated with either 1 µg/mL CHX (pre-determined to be a sub-toxic concentration, Fig. S3), or 100 nM VCR, or both, and analyzed for DNA content by propidium iodide staining. As shown in ), control cells exhibited 2N DNA content which decreased with time as cells advanced out of G1 phase. In contrast, when G1 phase cells were treated with CHX, the proportion of cells with 2N DNA remained unchanged, as did cells with sub-G1 DNA when compared with control cells ()), indicating CHX induced G1 arrest without cell death. When cells isolated in G1 phase were treated with VCR, the proportion of cells in G1 phase diminished incrementally over time in concert with an increase in cells with sub-G1 DNA, reflecting cell death from G1 phase. However, when cells were co-treated with CHX and VCR, the proportion of cells in G1 phase remained unchanged with time, as did cells with sub-G1 DNA (versus control cells), indicating protection from cell death. To gain further insight into the basis for the ability of CHX to block VCR cytotoxicity in G1 phase, phospho-RB, RB, and cyclin D levels under the same conditions were evaluated by immunoblotting. As shown in ), phospho-RB levels increased with time in control G1 phase cells, consistent with cell cycle advance. After PCB treatment, phospho-RB levels were strongly diminished and cyclin D levels elevated, as observed earlier in asynchronous cells (). After CHX treatment of G1 phase cells, phospho-RB levels were strongly reduced, similar to PCB-treated cells, but in contrast, cyclin D immunoreactive bands were atypical, with several species present, some of higher molecular weight than normally observed ()). Thus the lack of normal RB phosphorylation after CHX treatment may be due, at least in part, to impaired synthesis of cyclin D critical for CDK4/6 activation, and is consistent with the observation of G1 arrest.
Figure 7. Cycloheximide treated ALL cells arrest in G1 phase and become insensitive to VCR. A,B. ALL-5 cells isolated in G1 phase by centrifugal elutriation were treated with 1 µg/mL cycloheximide (CHX) or 100 nM VCR or both and stained with propidium iodide and analyzed for DNA content by flow cytometry. The percentage of cells with 2N DNA (G1 phase) (A) or sub-G1 DNA (B) are shown (mean ± S.D., n = 3). C. Whole cell extracts were prepared from G1 phase ALL-5 cells treated with 1 µM PCB or 1 µg/mL CHX for the times indicated and subjected to immunoblot analysis for RB, phospho-RB (Ser 807/811), cyclin D, or GAPDH as a loading control.
Discussion
Although our understanding of how MTAs exert their cytotoxicity in the clinical setting remains incomplete, several compelling lines of evidence suggest that interphase as well as spindle microtubules are targeted [Citation15,Citation17,Citation19,Citation20]. We have presented conclusive evidence previously that primary ALL cells are susceptible to vincristine and other MTAs, used alone at relatively low concentrations, not only in M phase as expected but also in G1 phase [Citation21]. Thus primary ALL cells represent a powerful model system to uncover novel mechanisms of MTA action with potential clinical relevance. Because the CDK4/6 inhibitor PCB exerts its effects in G1 phase and has been reported to sensitize cancer cells to a variety of anticancer agents [Citation36], it was of considerable interest to investigate the effects of PCB in this system. The results presented in this study show that PCB as a single agent is cytostatic, arresting primary ALL and RB-competent T98G glioblastoma cells in G1 phase, in parallel with inhibition of RB phosphorylation, while having no effect on RB pathway-defective HeLa cells. This is consistent with results using numerous other RB-competent cell lines [Citation37].
When ALL cells were pretreated with PCB to promote G1 phase arrest and then treated with VCR, we observed profound insensitivity to VCR. This was true whether asynchronous or G1 phase purified cells were used, and was also observed for VBL. The lack of effect of VCR after PCB pretreatment was not due to an inability of VCR to cause microtubule depolymerization nor to PCB-induced defects in apoptosis signaling. Conventionally, MTAs act in M phase, and the ability of PCB, through G1 arrest, to block effects of a drug acting in mitosis is not unexpected. For T98G cells, this is certainly the mechanism of protection, since they are only susceptible to VCR in M phase, and PCB precludes advance to the point in the cycle where VCR manifests its effects. For ALL cells, however, the mechanism is distinct. In this case, PCB pretreatment protected cells from susceptibility to death directly from G1 phase, which is characteristically observed in response to VCR in this cell type [Citation21]. Whereas the ability of PCB pretreatment to block a drug active in mitosis could be anticipated, the effects of PCB on a drug acting in G1 phase could not readily be predicted. Importantly, CHX also caused G1 arrest and subsequent insensitivity to VCR, providing independent validation of the findings with PCB and strengthening the notion that protection is related to a lack of active cycling through G1 rather than a specific effect of PCB.
Overall, the results show that functional microtubules are essential for the survival of primary ALL cells progressing through G1 phase, but are dispensable for cells arrested in G1 phase. Since primary ALL cells are not susceptible to MTAs when progressing through S or G2 phases, taken together, the findings suggest that the microtubules present in these cells, aside from their established role in mitosis, carry out specific processes essential for satisfactory completion of G1 phase. These observations represent a fundamental advance in our appreciation and understanding of interphase microtubule function. G1 phase is associated with an increase in cellular biomass as well as preparation for DNA synthesis, processes not occurring in arrested cells, thus it seems likely that G1 phase microtubules play crucial roles in these or related functions. In terms of potential mechanism, certain proteins have been shown to undergo intracellular trafficking via the interphase microtubule network; these include RB, p53, various kinases, transcription factors, hormone receptors, DNA repair proteins, and other classes of proteins [Citation15,Citation38–Citation41]. Interphase microtubules also play roles in organelle trafficking and regulation of signaling and stress responses [Citation42–Citation44] . In preliminary studies with primary ALL cells isolated in G1 phase, we have found that VCR impairs cytosol to nucleus trafficking of several proteins involved in RNA processing, protein degradation, and transcriptional regulation (data not shown). Further work will be required to establish the full spectrum of functions of G1 phase microtubules and how broadly they impact fundamental cellular activities, and to determine how cell survival is compromised when these processes are inhibited by MTAs. While mitotic death occurs via the intrinsic mitochondrial pathway [Citation9–Citation13], the death pathway(s) activated by VCR and other MTAs in G1 phase are currently unknown; such studies are in progress.
CDK4/6-specific inhibitors, including PCB and others such as ribociclib and amebaciclib, are being advanced clinically in several types of cancer either alone or in combination with conventional drugs or more targeted agents [Citation36,Citation37]. The ability to predict combinatorial effects through the use of relevant laboratory and preclinical models is therefore of growing significance. Our results clearly showed that PCB pretreatment causes robust G1 arrest and blocks the effects of VCR exerted either in G1 (ALL cells) or M phase (T98G cells). These findings caution against the combination of CDK4/6 inhibitors and MTAs for cancer treatment. On the other hand, PCB pretreatment reproducibly sensitized ALL cells to Bcl-2 inhibition ()), suggesting that G1-arrested ALL cells may have a lower apoptotic threshold than asynchronous cells. PCB also strongly sensitized pancreatic cancer cell lines to Bcl-2 inhibition [Citation45], suggesting that this effect may be of general significance and that such a combination merits further investigation. Studies with several different types of cancer have shown that CDK4/6 inhibitors have the ability to sensitize cells to other second agents. For example, CDK4/6 inhibitors cooperate with PI3K inhibitors in breast cancer models [Citation46] and sensitize pancreatic cancer cell lines to MEK and mTOR inhibitors [Citation34,Citation47]. Another study showed that RB-proficient sarcoma cells were sensitized to Wee1 kinase inhibition after prior transient treatment with PCB [Citation48]. This novel sequential combination treatment exploited the reversibility of PCB to synchronize the cells thus maximizing the effects of the kinase inhibitor during the ensuing S and G2 phases. The observations made here and elsewhere emphasize that G1 arrest has the potential to either oppose or augment the effects of a second agent. Given the likely expansion in the use of CDK4/6 inhibitors for cancer therapy, careful evaluation of individual combinations will be increasingly important.
Materials and methods
Materials
Palbociclib was obtained from Selleckchem (S1116); vincristine sulfate was from Santa Cruz Biotechnology, Inc. (SC-201,434); cycloheximide (C104450) and vinblastine sulfate (V1377) were from Sigma-Aldrich and ABT-263 was from ApexBio Technology (A3007).
Cell culture
Primary ALL-5 cells were derived from the bone marrow of a 37 year old patient as described previously [Citation49]. Although these cells can be cultured up to 6 months with no obvious change in their properties, in the present study they were exclusively used at low passage (up to passage 10) for all experiments and are thus referred to as primary cells. They were maintained in IMDM media containing 10 µg/mL cholesterol, 6 mg/mL human serum albumin, 2 mM L-glutamine, 2% v/v amphotericin-B/penicillin/streptomycin, 1 µg/mL insulin, 200 µg/mL APO-transferrin, and 50 μM β-mercaptoethanol, as described previously [Citation32,Citation49]. T98G cells were maintained in EMEM with 1% penicillin/streptomycin and 10% FBS. HeLa cells were maintained in DMEM (5 g/L glucose), 1% penicillin/streptomycin/glutamine and 10% FBS.
Cell viability assay
Cell viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) as described [Citation50]. Cells (105 per well) were seeded in 96-well plates, and agents added in a fixed final concentration of 0.1% DMSO. After treatment for time periods specified in individual experiments, MTT reagent (50 µg/10 µL/well) was added and incubated overnight at 37°C. The following day, 0.1 mL of 10% SDS in 0.01 M HCl was added, and after overnight incubation, absorbance readings were taken at 540 nm.
DNA content analysis
Cells were harvested and fixed in 70% ethanol. DNA content was evaluated by propidium iodide staining according to the manufacturer’s instructions (BD Pharmigen) and flow cytometry was carried out by the UAMS Flow Cytometry Core Facility using a FACSCalibur (Becton Dickinson, Mountain View, CA). The data were analyzed using the FlowJo (FlowJo, LLC) software.
Immunofluorescence microscopy
ALL cells were collected and suspended in Cytospin buffer composed of Hank’s buffered salt solution with 30% FBS. Cells were centrifuged onto a glass slide using a Cytospin centrifuge at 800 rpm for 3 min at a concentration of 200,000 cells/200 µL. T98G and HeLa cells were grown directly onto coverslips. Cells were fixed with 100% cold methanol and permeabilized with 0.2% Triton X-100/0.01% sodium azide in PBS. To visualize nuclei, cells were stained with 4,6-diamidino-2-phenylindole (DAPI). Proliferating cells were visualized by staining with Ki-67 antibody (1:300; Cell Signaling 9449) and microtubules by staining with α-tubulin antibody (1:300; Abcam ab18251), with Cy-5 conjugated AffiniPure Donkey anti-rabbit as secondary antibody (1:100; Jackson Laboratories). Cells were visualized using a Zeiss LSM410 confocal microscope. Quantification of the mean intensity of Ki-67 was determined using ImageJ for 20 cells per six fields of vision per condition.
Immunoblot analysis
Whole cell extracts were prepared in the presence of protease and phosphatase inhibitors as previously described [Citation21] and samples (25 µg protein per lane) analyzed by SDS-PAGE using 12% acrylamide gels followed by transfer to PVDF membrane. Membranes were probed with the following antibodies: cyclin D (1:2000; Santa Cruz sc20044), RB (1:2000; BD Pharmigen 554,138), phospho-(Ser 807/811) RB (1:2000; Cell Signaling 8516), PARP (1:2500; Cell Signaling 9532s), cleaved PARP (1:2500; Abcam ab32561) and GAPDH (1:10,000; Cell Signaling 2118s). Secondary antibodies against either mouse IgG (1:5000; BioRad 170–6516) or rabbit IgG (1:5000; BioRad 170–6515) were used dependent on the origin species of the primary antibodies. Apparent molecular weights derived from SDS-PAGE/immunoblotting of the proteins examined are as follows: phospho-RB, 110 kDa; RB, 110 kDa; cyclin D, 37 kDa; GAPDH, 37 kDa; PARP, 110 kDa; cleaved PARP, 85 kDa.
Centrifugal elutriation
Centrifugal elutriation was performed using a Beckman JE-5.0 elutriation rotor as previously described [Citation21,Citation35] with slight modifications. Briefly, ALL cells (4–6 x 108) were suspended in 25 mL of elutriation buffer (Hank’s buffered salt solution containing 1.6 g/L 2-naphthol-6,8-disulfonic acid dipotassium salt and 2% FBS), passed through a 25G needle twice, and introduced into the elutriation chamber at a flow rate of 25 mL/min with a rotor speed of 3,000 rpm. Rotor speed was reduced to 2920 rpm to collect the F1 wash fraction and then to 2620 rpm to collect the F2 fraction containing cells in G1 phase, verified by DNA content analysis. Cells were resuspended in fresh growth medium.
EdU incorporation assays
These were performed as described previously [Citation21]. Briefly, ALL-5 or T98G cells (1.5 x 106) were suspended in growth medium containing 10 µM EdU for periods up to 48 h, washed with PBS, fixed with 70% ethanol overnight, and then washed with 3% BSA in PBS. Cells were incubated with permeabilization buffer (0.5% Triton X-100 in PBS) for 20 min, washed in PBS, resuspended in 200 µL of Click-iT reaction cocktail with Alexa Fluor® 488 prepared as per manufacturer’s instructions (Click-iT® EdU Imaging Kit by Life Technologies), and incubated for 30 min in the dark. Cells were washed, incubated in propidium iodide for 2 h at room temperature, and analyzed by two-parameter flow cytometry at the UAMS Flow Cytometry Core Facility.
Statistical analysiss
Data are expressed as mean ± S.D. Student t-tests were performed in GraphPad to determine p values; p values of < 0.05 were considered statistically significant. GraphPad Version 6 software was used to plot viability curves. Experiments were conducted with replicates of at least three and repeated at least twice, unless stated otherwise.
Supplemental Material
Download MS Word (417.5 KB)Acknowledgments
We thank Dr. Robert Eoff for assistance with T98G cell culture and Dr. Anisha Kothari for helpful advice and discussions.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemental data for this article can be accessed here.
Additional information
Funding
Notes
1. The abbreviations used are: ALL, acute lymphoblastic leukemia; PCB, palbociclib; VCR, vincristine; VBL, vinblastine; MTA, microtubule targeting agent; CDK, cyclin-dependent kinase; RB, retinoblastoma protein; PARP, poly(ADP-ribose) polymerase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; EdU, 5-ethynyl-2ʹ-deoxyuridine; CHX, cycloheximide.
Related Research Data
References
- Dores GM, Devesa SS, Curtis RE, et al. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood. 2012;119:34–43.
- Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood. 2015;125:3977–3987.
- Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013;381:1943–1955.
- Douer D. Efficacy and safety of vincristine sulfate liposome injection in the treatment of adult acute lymphocytic leukemia. Oncologist. 2016;21:840–847.
- Moore A, Pinkerton R. Vincristine: can its therapeutic index be enhanced? Pediatr Blood Cancer. 2009;53:1180–1187.
- McGrogan BT, Gilmartin B, Carney DN, et al. Taxanes, microtubules and chemoresistant breast cancer. Biochim Biophys Acta. 2008;1785:96–132.
- Miltenburg NC, Boogerd W. Chemotherapy-induced neuropathy: A comprehensive survey. Cancer Treat Rev. 2014;40:872–882.
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265.
- Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7:637–651.
- Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8:7–12.
- Yamada HY, Gorbsky GJ. Spindle checkpoint function and cellular sensitivity to antimitotic drugs. Mol Cancer Ther. 2006;5:2963–2969.
- Manchado E, Guillamot M, Malumbres M. Killing cells by targeting mitosis. Cell Death Differ. 2012;19:369–377.
- Topham CH, Taylor SS. Mitosis and apoptosis: how is the balance set? Curr Opin Cell Biol. 2013;25:780–785.
- Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell. 2012;23:1–6.
- Komlodi-Pasztor E, Sackett D, Wilkerson J, et al. Mitosis is not a key target of microtubule agents in patient tumors. Nat Reviews Clin Oncol. 2011;8:244–250.
- Komlodi-Pasztor E, Sackett DL, Fojo AT. Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res. 2012;18:51–63.
- Furst R, Vollmar AM. A new perspective on old drugs: non-mitotic actions of tubulin- binding drugs play a major role in cancer treatment. Die Pharmazie. 2013;68:478–483.
- Salerni BL, Bates DJ, Albershardt TC, et al. Vinblastine induces acute, cell cycle phase-independent apoptosis in some leukemias and lymphomas and can induce acute apoptosis in others when Mcl-1 is suppressed. Mol Cancer Ther. 2010;9:791–802.
- Field JJ, Kanakkanthara A, Miller JH. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg Med Chem. 2014;22:5050–5059.
- Ogden A, Rida PC, Reid MD, et al. Interphase microtubules: chief casualties in the war on cancer? Drug Discov Today. 2014;19:824–829.
- Kothari A, Hittelman WN, Chambers TC. Cell cycle-dependent mechanisms underlie vincristine-induced death of primary acute lymphoblastic leukemia cells. Cancer Res. 2016;76:3553–3561.
- Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–122.
- Sakurikar N, Eichhorn JM, Alford SE, et al. Identification of a mitotic death signature in cancer cell lines. Cancer Lett. 2014;343:232–238.
- Toogood PL, Harvey PJ, Repine JT, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48:2388–2406.
- Beaver JA, Amiri-Kordestani L, Charlab R, et al. FDA approval: palbociclib for the treatment of postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res. 2015;21:4760–4766.
- Walker AJ, Wedam S, Amiri-Kordestani L, et al. FDA approval of palbociclib in combination with fulvestrant for the treatment of hormone receptor-positive, HER2- negative metastatic breast cancer. Clin Cancer Res. 2016;22:4968–4972.
- Rocca A, Schirone A, Maltoni R, et al. Progress with palbociclib in breast cancer: latest evidence and clinical considerations. Ther Adv Med Oncol. 2017;9:83–105.
- Whittaker S, Madani D, Joshi S, Chung SA, Johns T, Day B, Khasraw M, McDonald KL. Combination of palbociclib and radiotherapy for glioblastoma. Cell Death Discov. 2017;3:17033.
- Kwapisz D. Cyclin-dependent kinase 4/6 inhibitors in breast cancer: palbociclib, ribociclib, and abemaciclib. Breast Cancer Res Treat. 2017;166:41–54.
- Michaud K, Solomon DA, Oermann E, et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010;70:3228–3238.
- Matlashewski G. The cell biology of human papillomavirus transformed cells. Anticancer Res. 1989;9:1447–1456.
- Alford SE, Kothari A, Loeff FC, et al. BH3 inhibitor sensitivity and Bcl-2 dependence in primary acute lymphoblastic leukemia cells. Cancer Res. 2015;75:1366–1375.
- Knudsen ES, Wang JY. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol. 1997;17:5771–5783.
- Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014;5:6512–6525.
- Delgado M, Kothari A, Hittelman WN, et al. Preparation of primary acute lymphoblastic leukemia cells in different cell cycle phases by centrifugal elutriation. J Vis Exp. 2017.
- Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev. 2016;45:129–138.
- VanArsdale T, Boshoff C, Arndt KT, et al. Molecular pathways: targeting the cyclin D-CDK4/6 axis for cancer treatment. Clin Cancer Res. 2015;21:2905.
- Roth DM, Moseley GW, Glover D, et al. A microtubule-facilitated nuclear import pathway for cancer regulatory proteins. Traffic. 2007;8:673–686.
- Thadani-Mulero M, Nanus DM, Giannakakou P. Androgen receptor on the move: boarding the microtubule expressway to the nucleus. Cancer Res. 2012;72:4611–4615.
- Giannakakou P, Sackett DL, Ward Y, et al. p53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat Cell Biol. 2000;2:709–717.
- Poruchynsky MS, Komlodi-Pasztor E, Trostel S, et al. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc Natl Acad Sci USA. 2015;112:1571–1576.
- Melkov A, Abdu U. Regulation of long-distance transport of mitochondria along microtubules. Cell Mol Life Sci. 2018;75:163–176.
- Maizels Y, Gerlitz G. Shaping of interphase chromosomes by the microtubule network. FEBS J. 2015;282:3500–3524.
- Parker AL, Kavallaris M, McCarroll JA. Microtubules and their role in cellular stress in cancer. Front Oncol. 2014;4:153.
- Franco J, Balaji U, Freinkman E, et al. Metabolic reprogramming of pancreatic cancer mediated by CDK4/6 inhibition elicits unique vulnerabilities. Cell Rep. 2016;14:979–990.
- Vora SR, Juric D, Kim N, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26:136–149.
- Heilmann AM, Perera RM, Ecker V, et al. CDK4/6 and IGF1 receptor inhibitors synergize to suppress the growth of p16INK4A- deficient pancreatic cancers. Cancer Res. 2014;74:3947–3958.
- Francis AM, Alexander A, Liu Y, et al. CDK4/6 inhibitors sensitize Rb-positive sarcoma cells to Wee1 kinase inhibition through reversible cell-cycle arrest. Mol Cancer Ther. 2017;16:1751–1764.
- Nijmeijer BA, Szuhai K, Goselink HM, et al. Exp Hematol. 2009;37:376–385.
- Eichhorn JM, Alford SE, Sakurikar N, et al. Molecular analysis of functional redundancy among anti-apoptotic Bcl-2 proteins and its role in cancer cell survival. Exp Cell Res. 2014;322:415–424.