
ABSTRACT
Deregulation of the cell cycle is a hallmark of cancer that enables limitless cell division. To support this malignant phenotype, cells acquire molecular alterations that abrogate or bypass control mechanisms in signaling pathways and cellular checkpoints that normally function to prevent genomic instability and uncontrolled cell proliferation. Consequently, therapeutic targeting of the cell cycle has long been viewed as a promising anti-cancer strategy. Until recently, attempts to target the cell cycle for cancer therapy using selective inhibitors have proven unsuccessful due to intolerable toxicities and a lack of target specificity. However, improvements in our understanding of malignant cell-specific vulnerabilities has revealed a therapeutic window for preferential targeting of the cell cycle in cancer cells, and has led to the development of agents now in the clinic. In this review, we discuss the latest generation of cell cycle targeting anti-cancer agents for breast cancer, including approved CDK4/6 inhibitors, and investigational TTK and PLK4 inhibitors that are currently in clinical trials. In recognition of the emerging population of ER+ breast cancers with acquired resistance to CDK4/6 inhibitors we suggest new therapeutic avenues to treat these patients. We also offer our perspective on the direction of future research to address the problem of drug resistance, and discuss the mechanistic insights required for the successful implementation of these strategies.
Breast cancer is a heterogeneous disease with multiple subtypes classified based on the expression of estrogen (ER) and progesterone (PR) receptors, and the presence of human epidermal growth factor receptor 2 (HER2) amplification or overexpression. This characterization segregates tumours into major clinical subgroups: those that are hormone receptor positive (HR+ i.e. ER+, PR+) and HER2-negative; HER2-positive (HER2+); or triple-negative (TNBC; ER-, PR-, and HER2-). These receptor-defined subtypes of breast cancer exhibit differential clinical behaviours and prognoses, and this classification guides the use of therapies targeting these receptors [Citation1–Citation5]. For instance, HER2+ tumours are treated with drugs that specifically target the HER2 receptor (e.g. trastuzumab, pertuzumab, lapatinib), while hormone receptor-positive patients are treated with endocrine therapies aimed at disrupting hormone receptor signaling or systemic estrogen production. These drugs include selective estrogen receptor modulators (SERMs, e.g. tamoxifen), selective estrogen receptor degraders (SERDs, e.g. fulvestrant), and aromatase inhibitors (e.g. letrozole, anastrozole, exemestane). While cytotoxic chemotherapy has a role in all of these subgroups, it is the mainstay of treatment for TNBC where the ER and HER2 targets are absent.
Advancements in early detection and the use of systemic therapies described above for early stage breast cancer have led to improved rates of survival and cure. However, despite this significant progress, disease relapse, metastatic dissemination and the subsequent development of drug resistance remain significant clinical challenges that limit the survival of patients with all breast cancer subtypes, particularly endocrine-therapy resistant cancers and chemotherapy-resistant TNBCs [Citation6,Citation7]. Recognition of these clinical hurdles has spurred major efforts to elucidate the biological mechanisms underlying drug resistant phenotypes, and to develop new treatment strategies to overcome them.
In this review, we discuss both recently approved and investigational cell cycle targeting agents for the treatment of breast cancer, including cyclin dependent kinase 4/6 (CDK4/6) inhibitors in ER+ breast cancers and TTK protein kinase (TTK) and polo-like kinase 4 (PLK4) inhibitors in TNBCs. These new anti-cancer agents represent examples of next generation cell-cycle inhibitors which have been rationally developed to target tumour-specific vulnerabilities in order to improve their therapeutic index. We briefly describe mechanisms of drug action and discuss potential biomarkers predictive of response. As the number of patients treated with recently approved CDK4/6 inhibitors continues to increase, we foresee that breast cancers with acquired resistance to this class of treatment will constitute an emerging and distinct clinical disease entity. Thus, it is increasingly important to understand the selective pressures these drugs impose on tumour cells and their impact on tumour evolution. Recognizing this, we propose potential strategies to target this new group of drug resistant breast cancers, which will be important to enable the optimal development and application of cell cycle targeting agents in the clinic.
The cell cycle
Progression of cells through the cell cycle is an essential process governing genome duplication and cell division and has been extensively reviewed elsewhere [Citation8]. Briefly, the cycle itself is comprised of four ordered phases, denoted G1 (Gap 1), S (DNA synthesis), G2 (Gap 2), and M (Mitosis) and contains multiple checkpoints throughout to ensure the faithful replication and segregation of chromosomes into daughter cells (). These checkpoints serve to prevent genomic instability, which can drive or potentiate tumorigenesis. Cyclins and cyclin-dependent kinases (CDKs) are the orchestrators of the cell cycle, and their expression and activities fluctuate across specific phases [Citation8,Citation9]. Formation of cyclin-CDK complexes enable control of cell cycle progression via phosphorylation of targets such as Rb. The cyclin-CDK complexes themselves are regulated by cyclin-dependent kinase inhibitors (CKIs) including the INK4s: p16INK4A/CDKN2A, p15INK4B/CDKN2B, p18INK4C/CDKN2C, and p19INK4D/CDKN2D; and the CDK-interacting protein/kinase inhibitory proteins (CIP/KIPs): p21CIP1/WAF1/CDKN1A, p27KIP1/CDKN1B, and p57KIP2/CDKN1C. Additionally, E3 ubiquitin ligases are essential for regulating expression of various mitotic proteins to control cell cycle transitions, including the Skp1–Cul1–F-box-protein (SCF) complex and anaphase promoting complex/cyclosome (APC/C) [Citation10–Citation12].
Figure 1. The mammalian cell cycle is governed by CDKs, cyclins, and ubiquitin ligases. Genomic integrity is guarded by multiple checkpoints and inhibitors (red) that halt cell cycle progression in the absence of appropriate environmental stimuli, in the presence of DNA damage, or when chromosomes are not sufficiently aligned in mitosis. The centrosome cycle, depicted in green, is important for generating the bipolar spindle apparatus to enable proper chromosome segregation.
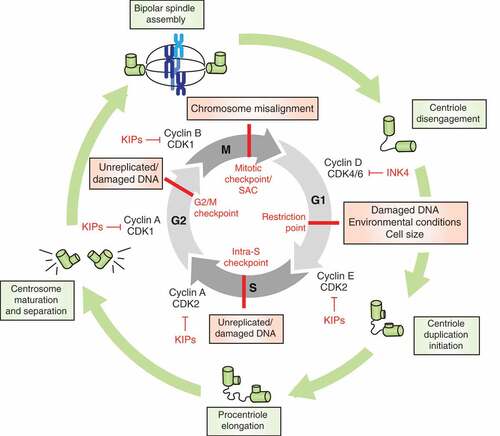
Progression of cells through G1 phase of the cell cycle is normally regulated by mitogens. In the case of hormone-receptor positive breast cancer, estrogen drives cell cycle progression by binding to the ER, leading to its dimerization, translocation to the nucleus, and transcriptional activity at estrogen response elements (EREs) [Citation13]. In the nucleus, activated ER directs expression of genes involved in promoting cell cycle progression and cell division, including cyclin D1 [Citation14,Citation15]. Production of cyclin D1 leads to the formation of cyclin D-CDK4/6 complexes that phosphorylate and partially inactivate Rb, releasing its inhibition of E2F transcription factors [Citation16]. E2F-mediated transcription drives the expression of targets including cyclins E and A. Cyclin E-CDK2 complexes hyper-phosphorylate and completely inactivate Rb, which enables cells to overcome the restriction point in G1, such that mitogen is no longer required for cell cycle progression. At this point, E2F activity leads to transcription of genes required for G1-S transition and DNA replication [Citation16]. Near the end of S phase, cyclin A-CDK2 drives the transition from S to G2, and subsequent activation of CDK1 by cyclin A causes cells to enter mitosis. Upon cyclin A inactivation, CDK1 complexes with cyclin B to push cells through mitosis.
Both G1/S and G2/M transitions are dependent on the satisfaction of cell cycle checkpoints that monitor the integrity of DNA [Citation17]. In the presence of DNA damage, cell cycle progression is halted via inhibition of CDKs to enable time for error correction or if damage is too extensive, cells undergo cell death or senescence. The spindle assembly checkpoint (SAC), also known as the mitotic checkpoint, is regulated by the kinase MPS1/TTK, and functions in mitosis to prevent progression of cells to anaphase until all chromosomes are properly attached to the mitotic spindle [Citation18,Citation19]. In this way, the SAC protects dividing cells from genomic instability resulting from chromosome segregation errors. Once the SAC is satisfied, mitotic exit is initiated by liberation of the APC/C E3 ligase complex which tags cyclin B and securin for ubiquitin-mediated degradation.
Deregulation of the cell cycle in breast cancer
Cancer cells override the many safeguards and checkpoints of the cell cycle to enable limitless proliferation in spite of their aneuploidy and other cellular defects that would prevent non-malignant cells from dividing. This phenotype is achieved through the acquisition of various genetic and epigenetic molecular alterations that hyperactivate or inactivate key components of the cell cycle, and impose specific cellular dependencies upon cancer cells to sustain aberrant proliferation. Different breast cancer subtypes exhibit different molecular alterations and dependencies on the cell cycle and its checkpoints [Citation20].
In hormone-receptor (ER+) positive/HER2-negative “luminal” breast cancers, malignant cells generally retain dependence on estrogen for its mitogenic effects. Estrogen binding to hormone receptors initiates a signal transduction pathway that culminates in receptor-driven expression of genes promoting cell growth, survival and proliferation. Cyclin D1 (CCND1) is an ER target gene that drives cell cycle progression through the restriction point by enabling cyclin D-CDK4/6 complexes as described above [Citation13]. ER+ tumours exhibit elevated expression of the gene encoding the estrogen receptor, ESR1, and its corresponding protein. Luminal cancers also have a high frequency of CCND1 DNA amplification. Activating mutations in PIK3CA are common in ER+ breast cancers, and contribute to cell cycle progression through mitogenic AKT/mTOR signaling [Citation20]. Unlike HER2+ and TNBC subtypes, ER+/luminal tumours typically have competent Rb and p53 tumour suppressor pathways, and are relatively genomically stable with a primary dependency on estrogen signaling. On the contrary, TNBCs exhibit RB1 mutations or deletions which compromise the integrity of cell cycle control by the Rb/E2F/CDK4/6 pathway, as well as frequent alterations in DNA damage response genes such as BRCA1. These tumours also tend to be highly aneuploid with near-universal loss of TP53 function, recurrent CCNE1 DNA amplifications, and PTEN loss of function [Citation6,Citation20–Citation22]. Multiple cellular dependency studies have demonstrated that TNBC tumours are reliant on the spindle assembly checkpoint and exhibit high expression levels of mitotic checkpoint genes (e.g. TTK, BUB1, MAD2, AURKB) and DNA repair proteins, presumably due to their high levels of genomic instability [Citation22–Citation26]. Copy gains of CDK4 are common across breast cancer types, with the highest frequency in HER2+ tumours. In addition to the pathognomonic amplification of ERBB2 (the gene encoding the HER2 receptor), mutations of TP53, PIK3CA, and PTEN and DNA amplification of CCND1 are also frequent in this subtype. Thus, although heterogeneity in mechanisms exist across subtypes, the presence of alterations that contribute to aberrant progression of the cell cycle is a hallmark of breast cancer cells.
Targeting the cell cycle in breast cancer
As described above, deregulation of the cell cycle through inactivation of tumour suppressors and aberrant activation of cyclins and cyclin-dependent kinases (CDKs) is a defining characteristic of breast cancer [Citation20]. Given its essentiality for sustaining cancer cell proliferation, it is not surprising that this cellular process represents a promising therapeutic target [Citation5,Citation27]. However, early generation cell cycle targeting agents performed poorly in the clinic due to a lack of target specificity and dose-limiting toxicities. Despite the disappointing clinical efficacy of past drugs, refinement of the therapeutic strategy to improve therapeutic index and the development of new potent and selective inhibitors has renewed optimism in targeting the cell cycle as an anticancer therapeutic strategy. Key to this progress has been the recognition that targeting cell cycle regulators that are specifically disrupted and essential in malignant cells provides a therapeutic window such that the vulnerabilities they impose on cancer cells may be exploited with tolerable side effects owing from normal tissue toxicity. This is the rationale behind most cell cycle inhibitors currently being investigated in breast cancer patients.
Microtubule binding agents and first-generation cell cycle/CDK inhibitors
Microtubule binding agents (MTBAs) exert anti-tumour effects in mitosis of dividing cancer cells by stabilizing (taxanes, including paclitaxel and docetaxel) or destabilizing (vinca alkaloids, eribulin) microtubules, and are a mainstay in cancer therapy [Citation28]. While not directly targeting cell cycle regulators, these agents activate the spindle assembly/mitotic checkpoint (SAC), which prevents anaphase and mitotic exit until all chromosomes have achieved a bipolar attachment to the spindle. In doing so, MTBAs induce cell cycle arrest in mitosis and prolonged mitotic arrest leads to cell death [Citation28]. Several MTBAs are approved for the treatment of breast cancer, either alone or in combination with other chemotherapies (or HER2-targeted monoclonal antibodies). Taxanes have been demonstrated to increase cure rates when used as adjuvant treatment of early breast cancer, and high rates of tumor response can be achieved in early lines of therapy for metastatic disease [Citation29,Citation30]. While MTBAs are incompletely cross resistant, responses in later lines of treatment fall considerably.
In contrast to MTBAs, the first generation cell cycle targeting inhibitors had little success in the treatment of solid tumors [Citation27]. These agents include the non-selective, multi-CDK inhibitors flavopiridol, dinaciclib and seliciclib [Citation31], as well as other mitotic kinase inhibitors such as those targeting aurora kinase B (AURKB) and polo-like kinase 1 (PLK1). The disappointing failure of these agents has been attributed in significant part to dose limiting toxicities caused by undesirable target inhibition in non-malignant cells, including neuropathy, myelosuppression, and gastrointestinal effects [Citation27,Citation31]. The lack of predictive biomarkers to inform patient selection for these agents may have also contributed to their limited success [Citation31]. While preclinical data has identified opportunities for repurposing some of these agents, including dinaciclib, the mechanisms are based on inhibition of transcriptional rather than cell cycle-related functions [Citation32].
These experiences crystallized the need to exploit cancer- and subtype-specific vulnerabilities (e.g. genomic instability and abnormalities in cell cycle proteins that are not essential in non-malignant cells) to achieve a therapeutic window for breast cancer selectivity, and has guided the development of the newest generation of cell cycle targeting agents. The targets of this new class of cell cycle inhibitors include CDK4/6, TTK (aka MPS1), and PLK4.
Targeting the cell cycle in ER+ breast cancer: CDK4/6 inhibitors
When the pathway is intact, phosphorylation-mediated inactivation of Rb by the cyclin D-CDK4/6 complex is required for progression through the restriction point in G1. Dependence of breast cancer cells on these cell cycle regulators is evident by the prevalence of genetic alterations that elevate CDK4/6 and CCND1 expression. At the mechanistic level, multiple studies have demonstrated that cyclin D1 and CDK4 are required for the development of breast tumours in mouse models [Citation33–Citation36]. Interestingly, other knockout studies have shown that CDK4/6 and cyclin D may be dispensable for non-malignant cells [Citation37,Citation38]. Together, these findings suggested the cyclin D-CDK4/6 axis could be a promising therapeutic target with a breast cancer-specific therapeutic window, and spurred the development of CDK4/6 specific inhibitors. Three ATP-competitive inhibitors (palbociclib (PD0332991), ribociclib (LEE011), and abemaciclib (LY2835219)) have recently been evaluated in clinical trials in combination with standard endocrine therapies in metastatic breast cancer, and have demonstrated significant improvements in tumor response rate and progression-free survival compared to endocrine therapy alone, which has led to their FDA approvals in combination with aromatase inhibitors or fulvestrant [Citation39–Citation41]. In addition to these approved compounds, several other CDK4/6 inhibitors are being evaluated in clinical trials as combination therapies with standard chemotherapies in breast and other malignancies (e.g. small-cell lung cancer) [Citation42] (clinicaltrials.gov; ).
Table 1. Approved and investigational CDK4/6 inhibitors in clinical development.
Based on their mechanism of action, CDK4/6 inhibitors are predicted to be effective only in tumours with a competent Rb pathway ( and ). This fact explains the preferential anti-tumour activity of these agents in luminal/ER+ breast tumours where Rb is rarely inactivated, as opposed to basal and HER2+ tumours in which RB1 is frequently inactivated and tumours have no requirement to bypass the Rb1-imposed brake on cell cycle progression in G1. This feature was initially demonstrated by testing palbociclib in a large panel of breast cancer cell lines in vitro, in which CDK4/6 inhibition resulted in G0/G1 arrest in sensitive lines [Citation43]. Given the general understanding of this signaling network, several candidate biomarkers of response to CDK4/6 inhibitors related to the cyclin D-CDK4/6 and Rb/E2F pathway have been proposed, and the Phase II study of palbociclib in ER+ breast cancer did originally include a biomarker-selected cohort requiring amplification of cyclin D1 (CCND1, using FISH ratio >1.5) or loss of p16 (INK4A or CDKN2A, using FISH ratio <0.8); however, the final analysis combined all patients based on lack of differential benefit between genomically enriched and unselected cohorts [Citation44]. Other candidate predictive biomarkers that have been considered or included in various studies include Ki67 levels; expression of p16, CDK4, CDK6, Cyclin E, Cdk2 and Rb; hormone-receptor status; as well as specific mutations (e.g. PIK3CA, ESR1, and TP53) [Citation45,Citation46]. While several of these have been reported not to predict differential benefit from the addition of CDK4/6 inhibitor, a recent report from the PALOMA-3 trial demonstrated relatively less benefit in cyclin E overexpressing (CCNE1 mRNA) tumors when palbociclib was added to fulvestrant, supporting the idea that Cyclin E-CDK2 represents a major bypass mechanism for CDK4/6 inhibitors [Citation47]. While current clinical use of CDK4/6 inhibitors is based solely on ER+/HER2- status, investigation of biomarkers predictive of CDK4/6 inhibitor response, beyond ER-positivity, is an ongoing, essential area of research [Citation45].
Figure 2. Targeting the cell cycle in breast cancer. (a) Cell cycle vulnerabilities of HR+ breast tumours. The Rb/E2F/CDK4/6 pathway is generally competent in HR+ breast cancers, making these cells dependent on CDK4/6 for progression through the restriction point. In such cells with intact Rb, CDK4/6 inhibitors induce cell cycle arrest in G0/G1. (b) Potential mechanisms of resistance to CDK4/6 inhibitors in HR+ breast cancers. To overcome resistance to CDK4/6 inhibition, cells acquire molecular alterations that enable them to bypass the restriction point and uncouple their dependency on CDK4/6 activity for cell cycle progression. (c) Cell cycle vulnerabilities of TNBC tumours. TNBC tumours are typically TP53 mutant, highly aneuploid, and often exhibit centrosome amplification. These features make TNBC cells susceptible to the induction of further, intolerable, aneuploidy and multipolar spindle formation. TTK and PLK4 inhibitors potentiate genomic instability and chromosome segregation errors in mitosis leading to cell death. (d) Potential mechanisms of resistance to TTK inhibition in TNBC. Gatekeeper mutations in TTK and delay of mitotic exit through impaired function of the APC/C have been demonstrated to confer resistance to TTK targeting drugs. Blue arrows/type indicate DNA amplification or activating mutations. Dashed lines indicate loss of activity/function. * indicates a gatekeeper mutation. Cell cycle checkpoints are indicated in red.
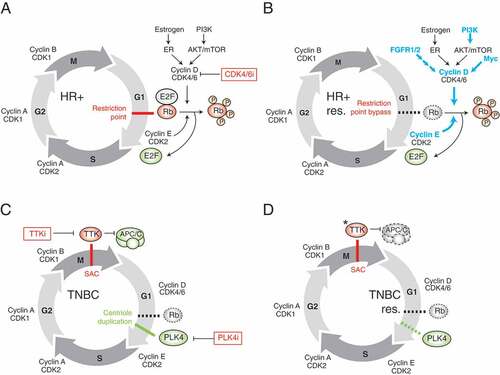
The remarkable clinical success of these compounds, and widespread adoption into standard care has prompted numerous studies to determine mechanisms of acquired resistance, since CDK4/6 inhibitors roughly double median progression-free survival in patients with metastatic disease but are not curative in this setting [Citation31]. Deciphering drug resistance in this context will be critical for understanding how to best treat the growing population of patients with CDK4/6 inhibitor-resistant breast cancer. To date, the majority of these studies have been conducted in preclinical models, although some investigations have identified resistance mechanisms in the clinical setting. A variety of approaches including functional genomics, pharmacogenomics and comparative phenotypic analyses of innately sensitive and resistant models, and characterization of breast cancer cell lines with acquired CDK4/6 inhibitor resistance have been used. Such studies have revealed a number of candidate mechanisms including: upregulated AKT signaling [Citation48–Citation50], deregulated expression of Cyclin E-CDK2 and CCNE1 DNA amplification [Citation48,Citation49,Citation51], acquired RB1 inactivating mutations [Citation48,Citation52], FGFR1 amplification or activating FGFR2 mutations [Citation53,Citation54], and upregulation of PDK1, MYC, or SKP2 [Citation49,Citation55] ()). However, it remains to be seen whether these mechanisms identified in vitro will be clinically relevant in drug-treated patients. In the clinical setting, somatic loss-of-function mutations in RB1 were acquired, or pre-existing mutations selected for, in multiple patients treated with palbociclib and ribociclib for 5–13 months [Citation56], and these mutations are suspected to confer therapeutic resistance.
Based on these findings, and not surprisingly, it seems that resistance to CDK4/6 inhibition results from loss of dependence on CDK4/6 for Rb1 inactivation and consequential activation of E2F-driven transcription for G1/S transition. Analyses of paired tumor or liquid biopsies obtained prior to treatment and following disease progression in larger clinical datasets will contribute substantially to understanding the relative contribution of different mechanisms of acquired resistance. Further insights and characterization of these mechanisms will undoubtedly lead to the identification of combination therapies and strategies aimed at preventing or overcoming CDK4/6 inhibitor resistance. For example, multiple groups have already demonstrated preclinical combination activity by combining CDK4/6 inhibitors with AKT pathway/mTOR inhibitors in breast cancer models [Citation48,Citation50,Citation51,Citation57]. Interestingly, Nikitorowicz-Buniak et al. have recently reported that endocrine therapy and palbociclib resistant ER+ breast cancers exhibit an induced dependency on the SAC kinase, TTK/MPS1 [Citation58,Citation59], proposing an alternative strategy to circumvent CDK4/6 inhibitor resistance.
Novel cell cycle inhibitors: TTK and PLK4 inhibitors
With better understanding of the cell cycle dependencies in different breast cancer subtypes, and in light of the success achieved with CDK4/6 inhibitors in ER+ breast cancer, there has been substantial renewed interest in the development of novel selective inhibitors against other cell cycle targets. In the subsequent sections, we describe the next generation of cell cycle drugs including their mechanisms of action and the current status of knowledge regarding drug resistance and clinical biomarkers for these compounds.
TTK inhibitors
TTK protein kinase (TTK), also known as monopolar spindle 1 (MPS1), is a key regulator of the spindle assembly checkpoint (SAC) [Citation18,Citation19,Citation60] ()). Through phosphorylation of its substrates, TTK recruits checkpoint proteins to kinetochores during mitosis in order to establish and maintain the mitotic checkpoint. This checkpoint is critical for maintaining genomic stability during mitosis because it prevents anaphase and the initiation of mitotic exit until all chromosomes have achieved a bipolar attachment to the mitotic spindle [Citation19]. Given its regulation of the SAC, TTK ensures adequate chromosome segregation and genomic integrity. TTK is often overexpressed in cancer cells, which may be a consequence of its involvement in mitosis, since aggressive tumours with high mitotic indexes naturally exhibit high expression levels of cell cycle genes; however, its overexpression could also reflect tumour cell dependency on the SAC to enable viable segregation of their aneuploid and unstable genomes into daughter cells [Citation22–Citation26]. A gene expression meta-analysis of TNBCs identified an “aggressiveness”-associated gene expression signature, which was enriched for genes involved in chromosomal instability, including TTK [Citation61]. This study also confirmed a dependency of TNBC cells on TTK. Regardless of whether its overexpression reflects an association with highly aggressive tumour biology or a functional dependence of cancer cells on the SAC, TTK expression distinguishes aneuploid malignant from diploid non-malignant cells that have additional competent pathways guarding genomic integrity, and provides a therapeutic window to target breast cancer cells. The principle underlying the use of TTK inhibitors to treat genomically unstable tumours such as TNBC is to abrogate the SAC and rush cells through mitosis before the chromosomes can be adequately segregated. This results in mitotic segregation errors and leads to intolerable levels of genomic instability in susceptible cancer cells, ultimately leading to cell death. Several TTK inhibitors are currently being evaluated in early phase clinical trials as single agents or in combination with taxane therapy, including a compound developed by our institution’s therapeutics group, CFI-402257 () [Citation62,Citation63]. Additional TTK inhibitors in preclinical development include CCT271850, AZ3146, NMS-P715, MPI-0479605, CCT251455, and MPS1-IN-3, demonstrating the interest and clinical potential for this class of anti-cancer compounds [Citation64–Citation71].
Table 2. TTK (MPS1) inhibitors in clinical development.
Similar to other clinical kinase inhibitors, gatekeeper mutations in the active site of TTK have been described to confer resistance to TTK inhibitors in vitro [Citation72], although the clinical relevance of such mutations must be evaluated prospectively in patients who develop resistance ()). In a preclinical setting, we recently described delay of mitotic exit as a mechanism of TNBC resistance to the clinical TTK inhibitor, CFI-402257 using CRISPR/Cas9 functional genomic screens [Citation73] ()). Our findings independently validated those of Sansregret et al. who identified the same mechanism of resistance using siRNA screens with the tool compound reversine, another TTK inhibitor [Citation74,Citation75]. Both studies identified that compromising the anaphase-promoting complex/cyclosome (APC/C) function through genetic inactivation or knockdown of individual APC/C components led to elongation of mitosis with fewer chromosome segregation errors and consequentially, less cell death in cancer cells treated with TTK inhibitors. Using a complementary approach, assessment of TTK-inhibitor resistant HCT-116 colon cancer cells revealed that 2/6 resistant clones had acquired mutations in CDC16, another APC/C component, that prolonged mitotic duration, substantiating this mechanism of TTK inhibitor resistance [Citation75].
As TTK inhibitors progress through clinical trials, patient biopsies will become available for genomic profiling and will enable pharmacogenomic studies to identify molecular correlates associated with drug response. Such findings will be critical for informing patient stratification for therapy. In the meantime, potential biomarkers of TTK inhibitor response may be identified in a preclinical setting and investigated for clinical utility once tumour genomic and drug response data are available. Zaman et al. reported that activating mutations in CTNNB1 (β-catenin) were associated with enhanced sensitivity to TTK inhibitors compared to CTNNB1 wild-type models [Citation76], although minimal evaluation of the functional impact of CTNNB1 mutation on TTK inhibitor response was conducted, and only in an in vitro setting. Furthermore, the rarity of CTNNB1 mutations in breast cancers limits the prospective application of this putative biomarker in this disease. p53 status may be associated with response to TTK inhibition as p53−/- HCT-116 colon carcinoma cells were found to be more sensitive to the broad-spectrum serine-threonine kinase inhibitor, SP600125, which inhibits TTK with an IC50 of 1.95 µM [Citation77]. However, the lack of selectivity of SP600125 complicates interpretation of the role of TTK inhibition in mediating preferential p53-deficient cell killing in this study. Finally, comparison of genetic dependencies in PTEN-mutant and wild-type breast cancer cell lines using siRNA screens revealed TTK inhibition is preferentially deleterious in PTEN-deficient cancer cells, suggesting PTEN could be a biomarker for the efficacy of TTK inhibitors [Citation26,Citation78].
Based on the APC/C-mediated biology of resistance to TTK inhibition, we investigated whether clinical tumours harbour alterations to APC/C complex components, which could be indicative of APC/C functional capacity and therefore predictive of TTK inhibitor response. These analyses revealed recurrent genetic disruption and gene expression changes in APC/C components in various tumour types [Citation73]. Furthermore, we identified a two-gene metagene (ANAPC4 and CDC20 mRNA expression signature) that was strongly correlated with in vitro response to CFI-402257 in breast and lung cancer cell line panels that we tested. Finally, analysis of The Cancer Genome Atlas tumour database revealed clusters of tumours with very low metagene scores across multiple tissue types, suggesting the potential utility of this metagene for identifying patients that could be refractory to TTK inhibition [Citation73].
PLK4 inhibitor
Centrosomes are the microtubule organizing centers in cells and consist of two barrel-like structures called centrioles. During G1, disengagement of the centrioles comprising the centrosome occurs. Coincident with the G1/S transition is the initiation of centriole duplication ()). This is followed by procentriole formation and elongation in S and G2 phases, and finally, centrosome maturation and separation results in two centrosomes that will form the mitotic spindle [Citation79,Citation80]. Polo-like kinase 4 (PLK4) is the director of centriole duplication; PLK4-mediated phosphorylation of proteins involved in centrosome biology is an upstream event in the signaling cascade responsible for centriole biogenesis that is required for centrosome generation [Citation79–Citation81]. PLK4 expression is tightly regulated to maintain centriole and centrosome numerical integrity, as depletion of PLK4 abrogates centriole duplication and overexpression leads to centriole amplification. Overexpression of PLK4 and associated centrosome abnormalities are common in cancer, including breast malignancies, and correlate with disease aggressiveness [Citation82–Citation85]. It is thought that PLK4-driven centrosome amplification may be a major contributor to the chromosomal instability associated with tumorigenesis. Although earlier work found no increased incidence of spontaneous tumour formation in p53+/- or p53-/- mice upon PLK4 overexpression and centrosome amplification [Citation86], a recent study demonstrated increased tumour formation upon PLK4-mediated centrosome amplification in an APCMin/+ model of intestinal cancer [Citation87], lending further support for the idea of exploiting PLK4 as a therapeutic target. Based on the proposed mechanism of PLK4 overactivation driving centrosome amplification, inhibition of PLK4 would potentiate aneuploidy and genomic instability and lead to cancer cell death [Citation27].
Our therapeutics group has developed the first clinical PLK4 inhibitor, CFI-400945, an ATP competitive agent which is entering phase 2 clinical trials [Citation88,Citation89]. PLK4 was identified as a promising target with particular efficacy in TNBC cell lines based on an siRNA screen targeting kinases. Interestingly, and consistent with the opposite effects described for deletion and overactivation of PLK4 in the literature, CFI-400945 exhibits different phenotypic effects at different doses, with centrosome depletion at high doses and centriole overduplication (and consequential centrosome amplification) at low doses. Low dose CFI-400945 led to partial inhibition of PLK4, preventing its autoregulation via trans-autophosphorylation of its degron leading to increased PLK4 protein expression [Citation88,Citation90,Citation91]. In either case, CFI-400945 treatment leads to chromosome segregation errors due to formation of aberrant mitotic spindles (eg. multipolar spindles), increased DNA content consistent with genomic instability, and ultimately cell death in various cancer models [Citation88,Citation92,Citation93].
To date, we have yet to identify robust mechanisms of acquired resistance to CFI-400945 using in vitro cancer models, and this remains an active area of research. Cellular resistance to centrosome depletion induced by the tool compound centrinone, a highly selective PLK4 inhibitor, was shown to be mediated by a p53-dependent 53BP1-USP28 circuit in non-malignant, diploid RPE1 cells [Citation94]. This finding of 53BP1 and USP28 inactivation driving resistance to centrosome loss was also observed in two elegant studies that generated RPE1 models in which endogenous PLK4 was replaced with an analog-sensitive mutant that is inactivated upon chemical treatment to cause centrosome loss [Citation95,Citation96]. While this resistance mechanism may occur in tumours with proficient p53 signaling, its clinical relevance remains to be answered. In cancers characterized by high levels of genomic instability, centrosomal aberrations, and prevalent TP53 mutations like TNBC, TP53-independent mechanisms will likely drive resistance [Citation85].
Initial functional characterization of CFI-400945 effects in breast cancer cell lines revealed a potential association between PTEN status and drug response, in which response was associated with PTEN deficiency [Citation88]. This finding was consistent with a report that PLK4 inactivation was synthetic lethal in PTEN-mutated breast cancers [Citation26], suggesting PTEN status could serve as a biomarker predictive for CFI-400945. Investigation of PTEN as a potential biomarker in breast tumour PDX with defined responses to CFI-400945, along with additional hypothesized biomarkers based on PLK4 biology such as chromosomal instability and centrosome copy number are ongoing. Interestingly, in support of this hypothesis, a recent comprehensive analysis of centrosome amplification in the NCI-60 cancer cell line panel demonstrated enrichment of centrosome amplification in aggressive TNBC, as well as in microsatellite stable colorectal cancer (vs MSI CRC), which are two diseases for which significant clinical or pre-clinical anti-tumor activity has been observed with CFI-400945 [Citation85]. Ultimately, the potential associations between PTEN inactivation and centrosome amplification and CFI-400945 sensitivity will be determined using clinical tumour specimens from ongoing clinical trials.
Future perspective
Based on the early success of CDK4/6 inhibitors in the clinic and the promising activity of TTK inhibitors and CFI-400945 as anticancer agents, the next logical steps in the development of these compounds are to understand drug resistance, to identify biomarkers for patient selection, and to inform treatment sequencing. Prolonged treatment of incurable metastatic cancers with these new agents will undoubtedly drive tumour evolution and provide selective pressure for the expansion of drug resistant clones. Thus, understanding this evolutionary process and how it modifies tumour genomes to generate drug resistant phenotypes will be critical for devising strategies to combat disease progression ()). Elucidating resistance mechanisms and characterizing drug-induced cellular effects also has the potential to inform potential biomarkers to predict innate tumour response.
Figure 3. Strategies for identifying clinical correlates of drug response and combination or sequential therapies to overcome drug resistance. (a) Approaches to elucidate resistance mechanisms, vulnerabilities of drug resistant cells, and biomarkers predictive of drug response. (b) Re-purposing TTK inhibitors for the treatment of CDK4/6 inhibitor-resistant breast cancer. PGx = pharmacogenomics. PDX = patient-derived xenograft. ct-DNA = circulating tumour DNA. ET = endocrine therapy. SAC = spindle assembly checkpoint.
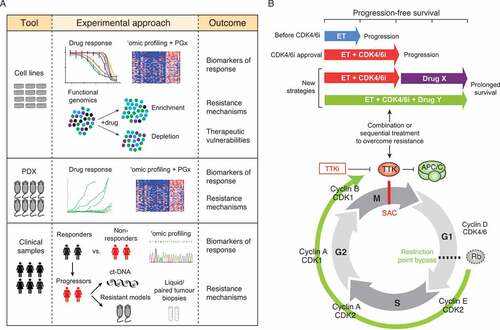
There are several complementary strategies that should be employed to answer these questions including functional genomics, genomic characterization of tumours with acquired drug resistance, and pharmacogenomic approaches. As we and others have demonstrated for TTK inhibitors [Citation73,Citation75], CRISPR/Cas9 and siRNA-based genome-wide screens are powerful tools for deciphering resistance mechanisms. With advances in gene editing technology, it is now feasible to conduct functional genomic screens with different genetic effects including gene knockout, repression and activation [Citation97], which greatly expands the potential for discovery. This approach can also be used to identify synthetic lethal interactions and provide insights into combination therapies to overcome resistance. For instance, screens to identify the dependencies of resistant cancer cells may reveal biological pathways essential for the survival of drug resistant cells or direct drug targets. Design of such studies to assess “druggable” targets could lead to re-purposing of already FDA-approved drugs to combat drug resistance with combination or sequential therapies ()).
In the case of CDK4/6 inhibitors, which are now standard of care for the treatment of metastatic ER+/HER2- breast cancer, the number of patients with acquired resistance to these drugs will continue to grow. Sampling of progressive disease will be essential for identifying resistance mechanisms, and multiple efforts are underway to define the genomic evolution of clinical resistance using either liquid (i.e. circulating tumor DNA, ctDNA) or paired tumor biopsies. The generation of pre-clinical models of acquired drug resistance, as well as the development of patient-derived models from patients who developed clinical resistance will provide the experimental tools to study the drug resistance phenotype [Citation98]. For instance, preclinical studies have demonstrated that breast cancer models with acquired resistance to CDK4/6 inhibitors exhibit a dependence on TTK [Citation58,Citation59]. This suggests that TTK-targeting drugs represent a promising sequential therapy for patients who progress on CDK4/6 inhibitors ()).
While a clinical cohort of cancer patients with acquired resistance to TTK inhibitors and CFI-400945 has not yet emerged given they are being investigated in early-phase clinical trials, pharmacogenomic analyses have great power to identify molecular correlates of drug response [Citation99,Citation100]. RNA- and exome-sequencing of cancer models including cell lines and PDX with well-defined drug response data can identify potential biomarkers predictive of innate resistance as well as foreshadow potential acquired resistance mechanisms. An emphasis on collection of tumour samples including paired biopsies in patients demonstrating initial response to therapy, and serial circulating tumour DNA from patients enrolled in these clinical trials will contribute to the important goal of identifying correlates and mechanisms of drug response and resistance [Citation98].
Undoubtedly the resurgence of basic, translational, and clinical interest in targeting the cell cycle, spurred by the success of CDK4/6 inhibitors in ER+ breast cancer, will yield important discoveries and significant clinical advances. Equipped with the genomic and experimental tools to dissect mechanisms and mediators of response and resistance, the field is well positioned to maximize the impact of cell cycle targeting therapies, and tackle the challenges and opportunities that treatment-driven disease evolution may bring.
Acknowledgements
The authors would like to thank the members of the Campbell Family Institute for Breast Cancer Research Therapeutics Group for useful discussions regarding the use and development of the compounds described in this review.
Disclosure statement
The authors’ institution, University Health Network, is developing the PLK4 and TTK inhibitors discussed in this article and holds patents for their use as treatments for breast and other cancers. DC has served as a consultant or advisor for Pfizer, Novartis and Merck.
Additional information
Funding
References
- Arteaga CL, Sliwkowski MX, Osborne CK, et al. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2012;9:16–32.
- Russnes HG, Lingjærde OC, Børresen-Dale A-L CC. Breast cancer molecular stratification: from intrinsic subtypes to integrative clusters. Am J Pathol. 2017;187:2152–2162.
- Gingras I, Gebhart G, De Azambuja E, et al. HER2-positive breast cancer is lost in translation: time for patient-centered research. Nat Rev Clin Oncol. 2017;14:669–681.
- Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–1948.
- Ignatiadis M, Sotiriou C. Luminal breast cancer: from biology to treatment. Nat Rev Clin Oncol. 2013;10:494–506.
- Bianchini G, Balko JM, Mayer IA, et al. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674–690.
- Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–247.
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166.
- Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15:122.
- Sivakumar S, Gorbsky GJ. Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat Rev Mol Cell Biol. 2015;16:82–94.
- Zhou Z, He M, Shah AA, et al. Insights into APC/C: from cellular function to diseases and therapeutics. Cell Div. 2016;11:9.
- Senft D, Qi J, Ronai ZA. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat Rev Cancer. 2018;18:69–88.
- Platet N, Cathiard AM, Gleizes M, et al. Estrogens and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion. Crit Rev Oncol Hematol. 2004;51:55–67.
- Klein EA, Assoian RK. Transcriptional regulation of the cyclin D1 gene at a glance. J Cell Sci. 2008;121:3853–3857.
- Sabbah M, Courilleau D, Mester J, et al. Estrogen induction of the cyclin D1 promoter: involvement of a cAMP response-like element. Proc Natl Acad Sci USA. 1999;96:11217–11222.
- Bertoli C, Skotheim JM, De Bruin RAM. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013;14:518–528.
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323.
- Musacchio A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol. 2015;25:R1002–18.
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol. 2012;22:R966–80.
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
- Xu H, Eirew P, Mullaly SC, et al. The omics of triple-negative breast cancers. Clin Chem. 2014;60:122–133.
- Curtis C, Shah SP, Chin S-F, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352.
- Daniel J, Coulter J, Woo J-H, et al. High levels of the Mps1 checkpoint protein are protective of aneuploidy in breast cancer cells. Proc Natl Acad Sci USA. 2011;108:5384–5389.
- Yuan B, Xu Y, Woo J-H, et al. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin Cancer Res. 2006;12:405–410.
- Patel N, Weekes D, Drosopoulos K, et al. Integrated genomics and functional validation identifies malignant cell specific dependencies in triple negative breast cancer. Nat Commun. 2018;9:1044.
- Brough R, Frankum JR, Sims D, et al. Functional viability profiles of breast cancer. Cancer Discov. 2011;1:260–273.
- Dominguez-Brauer C, Thu KL, Mason JM, et al. Targeting mitosis in cancer: emerging strategies. Mol Cell. 2015;60:524–536.
- Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9:790–803.
- De Laurentiis M, Cancello G, D’Agostino D, et al. Taxane-based combinations as adjuvant chemotherapy of early breast cancer: a meta-analysis of randomized trials. J Clin Oncol. 2008;26:44–53.
- Gradishar WJ. Taxanes for the treatment of metastatic breast cancer. Breast Cancer. 2012;6:159–171.
- Finn RS, Aleshin A, Slamon DJ. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016;18:17.
- Johnson SF, Cruz C, Greifenberg AK, et al. CDK12 inhibition reverses de novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 2016;17:2367–2381.
- Reddy HKDL, Mettus RV, Rane SG, et al. Cyclin-dependent kinase 4 expression is essential for neu-induced breast tumorigenesis. Cancer Res. 2005;65:10174–10178.
- Yu Q, Sicinska E, Geng Y, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23–32.
- Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–1021.
- Landis MW, Pawlyk BS, Li T, et al. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006;9:13–22.
- Malumbres M, Sotillo R, Santamaría D, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118:493–504.
- Kozar K, Ciemerych MA, Rebel VI, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118:477–491.
- Rocca A, Schirone A, Maltoni R, et al. Progress with palbociclib in breast cancer: latest evidence and clinical considerations. Ther Adv Med Oncol. 2016;9:83–105.
- O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13:417–430.
- Barroso-Sousa R, Shapiro GI, Tolaney SM. Clinical development of the CDK4/6 inhibitors ribociclib and abemaciclib in breast cancer. Breast Care. 2016;11:167–173.
- Klein ME, M K, Davis LE, et al. CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell. 2018. DOI:10.1016/j.ccell.2018.03.023.
- Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77.
- Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35.
- Fang H, Huang D, Yang F, et al. Potential biomarkers of CDK4/6 inhibitors in hormone receptor-positive advanced breast cancer. Breast Cancer Res Treat. 2017. DOI:10.1007/s10549-017-4612-y
- Knudsen ES, Witkiewicz AK. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer Res. 2017;3:39–55.
- Turner NC, Liu Y, Zhu Z, et al. Abstract CT039 - Cyclin E1 (CCNE1) expression associates with benefit from palbociclib in metastatic breast cancer (MBC) in the PALOMA3 trial [Internet]. American Association for Cancer Research, Annual Conference; 2018 Apr 15, Chicago, IL [cited 2018 May 1]. Available from: http://cancerres.aacrjournals.org/content/78/13_Supplement/CT039
- Mt H-A, Palafox M, Asghar U, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016;76:2301–2313.
- Jansen VM, Bhola NE, Bauer JA, et al. Kinome-wide RNA interference screen reveals a role for PDK1 in acquired resistance to CDK4/6 inhibition in ER-positive breast cancer. Cancer Res. 2017;77:2488–2499.
- Lenihan C, Bouchekioua-Bouzaghou K, Shia A, et al. Abstract P3-06-02: characterization of resistance to the selective CDK4/6 inhibitor palbociclib in ER positive breast cancer. Cancer Res. 2016;76:P3–06–02–P3–06–02.
- Asghar U, Barr A, Cutts R, et al. 44PUnravelling mechanisms of resistance to CDK4/6 inhibitors using triple negative breast cancer (TNBC). Ann Oncol. 2017 [ cited 2018 Mar 18];28. Available from: https://academic.oup.com/annonc/article-abstract/doi/10.1093/annonc/mdx145/3792864
- Dean JL, Thangavel C, McClendon AK, et al. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29:4018–4032.
- Mao P, Kusiel J, Cohen O, et al. Abstract PD4-01: the role of FGF/FGFR axis in resistance to SERDs and CDK4/6 inhibitors in ER+ breast cancer. Cancer Res. 2018;78:PD4–01–PD4–01.
- Formisano L, Lu Y, Jansen VM, et al. Abstract 1008: gain-of-function kinase library screen identifies FGFR1 amplification as a mechanism of resistance to antiestrogens and CDK4/6 inhibitors in ER+ breast cancer. Cancer Res. 2017;77:1008.
- Pan Q, Sathe A, Tong Z, et al. 37Identification of molecular mechanisms that confer therapy response to CDK4/6 inhibition using a genome-wide CRIPR-dCsa9 gain-of-function screen. Ann Oncol. 2017 [ cited 2018 Mar 18];28. Available from: https://academic.oup.com/annonc/article/doi/10.1093/annonc/mdx511.003/4469352/37Identification-of-molecular-mechanisms-that
- Condorelli R, Spring L, O’Shaughnessy J, Lacroix L, Bailleux C, Scott V, Dubois J, Nagy Rj, Lanman Rb, Lafrate Aj, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol. 2017. DOI:10.1093/annonc/mdx784
- Michaloglou C, Crafter C, Siersbæk R, et al. Combined inhibition of mTOR and CDK4/6 is required for optimal blockade of E2F function and long term growth inhibition in estrogen receptor positive breast cancer. Mol Cancer Ther. 2018. DOI:10.1158/1535-7163.MCT-17-0537
- Nikitorowicz-Buniak J, Pancholi S, Simigdala N, et al. Abstract P1-09-03: global knockdown of cellular kinases identifies MPS1 as a novel modulator of endocrine and palbociclib resistance highlighting a new role for MPS1 inhibitors. Cancer Res. 2018;78:P1–09–03–P1–09–03.
- Nikitorowicz-Buniak J, Pancholi S, Simigdala N, et al. Abstract 4950 - MPS1 as a novel target in endocrine- and palbociclib-resistant estrogen receptor positive breast cancer [Internet]. American Association for Cancer Research, Annual Conference; 2018 Apr 15, Chicago, IL [cited 2018 May 1]. Available from: http://cancerres.aacrjournals.org/content/78/13_Supplement/CT039
- Liu X, The WM. MPS1 family of protein kinases. Annu Rev Biochem. 2012;81:561–585.
- Al-Ejeh F, Simpson PT, Sanus JM, et al. Meta-analysis of the global gene expression profile of triple-negative breast cancer identifies genes for the prognostication and treatment of aggressive breast cancer. Oncogenesis. 2014;3:e100.
- Mason JM, Wei X, Fletcher GC, et al. Functional characterization of CFI-402257, a potent and selective Mps1/TTK kinase inhibitor, for the treatment of cancer. Proc Natl Acad Sci USA. 2017;114:3127–3132.
- Wengner AM, Siemeister G, Koppitz M, et al. Novel Mps1 kinase inhibitors with potent antitumor activity. Mol Cancer Ther. 2016;15:583–592.
- Tannous BA, Kerami M, Van Der Stoop PM, et al. Effects of the selective MPS1 inhibitor MPS1-IN-3 on glioblastoma sensitivity to antimitotic drugs. J Natl Cancer Inst. 2013;105:1322–1331.
- Naud S, Westwood IM, Faisal A, et al. Structure-based design of orally bioavailable 1H-pyrrolo[3,2-c]pyridine inhibitors of mitotic kinase monopolar spindle 1 (MPS1). J Med Chem. 2013;56:10045–10065.
- Faisal A, Mak GWY, Gurden MD, et al. Characterisation of CCT271850, a selective, oral and potent MPS1 inhibitor, used to directly measure in vivo MPS1 inhibition vs therapeutic efficacy. Br J Cancer. 2017;116:1166–1176.
- Tardif KD, Rogers A, Cassiano J, et al. Characterization of the cellular and antitumor effects of MPI-0479605, a small-molecule inhibitor of the mitotic kinase Mps1. Mol Cancer Ther. 2011;10:2267–2275.
- Colombo R, Caldarelli M, Mennecozzi M, et al. Targeting the mitotic checkpoint for cancer therapy with NMS-P715, an inhibitor of MPS1 kinase. Cancer Res. 2010;70:10255–10264.
- Hewitt L, Tighe A, Santaguida S, et al. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J Cell Biol. 2010;190:25–34.
- Jemaà M, Galluzzi L, Kepp O, et al. Characterization of novel MPS1 inhibitors with preclinical anticancer activity. Cell Death Differ. 2013;20:1532–1545.
- Kusakabe K-I, Ide N, Daigo Y, et al. Discovery of Imidazo[1,2-b]pyridazine derivatives: selective and orally available Mps1 (TTK) kinase inhibitors exhibiting remarkable antiproliferative activity. J Med Chem. 2015;58:1760–1775.
- Koch A, Maia A, Janssen A, et al. Molecular basis underlying resistance to Mps1/TTK inhibitors. Oncogene. 2016;35:2518–2528.
- Thu KL, Silvester J, Elliott MJ, et al. Disruption of the anaphase-promoting complex confers resistance to TTK inhibitors in triple-negative breast cancer. Proc Natl Acad Sci USA. 2018;115:E1570–7.
- Burkard ME, Weaver BA. Tuning chromosomal instability to optimize tumor fitness. Cancer Discov. 2017;7:134–136.
- Sansregret L, Patterson JO, Dewhurst S, et al. APC/C dysfunction limits excessive cancer chromosomal instability. Cancer Discov. 2017;7:218–233.
- Zaman GJR, Jadm DR, Libouban MAA, et al. Inhibitors as a targeted therapy for CTNNB1 (β-catenin) mutant cancers. Mol Cancer Ther. 2017;16:2609–2617.
- Jemaà M, Vitale I, Kepp O, et al. Selective killing of p53-deficient cancer cells by SP600125. EMBO Mol Med. 2012;4:500–514.
- Mendes-Pereira AM, Lord CJ, Ashworth A. NLK is a novel therapeutic target for PTEN deficient tumour cells. PLoS One. 2012;7:e47249.
- Ea N, Aj H. Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol. 2018. DOI:10.1038/nrm.2017.127
- Wang G, Jiang Q, Zhang C. The role of mitotic kinases in coupling the centrosome cycle with the assembly of the mitotic spindle. J Cell Sci. 2014;127:4111–4122.
- Maniswami RR, Prashanth S, Karanth AV, et al. PLK4: a link between centriole biogenesis and cancer. Expert Opin Ther Targets. 2018;22:59–73.
- Denu RA, Zasadil LM, Kanugh C, et al. Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC Cancer. 2016;16:47.
- Pannu V, Mittal K, Cantuaria G, et al. Rampant centrosome amplification underlies more aggressive disease course of triple negative breast cancers. Oncotarget. 2015;6:10487–10497.
- Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci. 2011;7:1122–1144.
- Marteil G, Guerrero A, Vieira AF, et al. Over-elongation of centrioles in cancer promotes centriole amplification and chromosome missegregation. Nat Commun. 2018;9:1258.
- Vitre B, Aj H, Kulukian A, et al. Chronic centrosome amplification without tumorigenesis. Proc Natl Acad Sci USA. 2015;112:E6321–30.
- Levine MS, Bakker B, B B, et al. Centrosome amplification is sufficient to promote spontaneous tumorigenesis in mammals. Dev Cell. 2017;40:313–322.e5.
- Mason JM, Lin D-C-C, Wei X, et al. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell. 2014;26:163–176.
- Bedard PL, Cescon DW, Fletcher G, et al. Abstract CT066: first-in-human phase I trial of the oral PLK4 inhibitor CFI-400945 in patients with advanced solid tumors. Cancer Res. 2016;76:CT066–CT066.
- Cunha-Ferreira I, Bento I, Pimenta-Marques A, et al. Regulation of autophosphorylation controls PLK4 self-destruction and centriole number. Curr Biol. 2013;23:2245–2254.
- Aj H, Fachinetti D, Zhu Q, et al. The autoregulated instability of Polo-like kinase 4 limits centrosome duplication to once per cell cycle. Genes Dev. 2012;26:2684–2689.
- Kawakami M, Mustachio LM, Zheng L, et al. Polo-like kinase 4 inhibition produces polyploidy and apoptotic death of lung cancers. Proc Natl Acad Sci USA. 2018;115:1913–1918.
- Lohse I, Mason J, Cao PM, et al. Activity of the novel polo-like kinase 4 inhibitor CFI-400945 in pancreatic cancer patient-derived xenografts. Oncotarget. 2017;8:3064–3071.
- Meitinger F, Anzola JV, Kaulich M, et al. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol. 2016;214:155–166.
- Fong CS, Mazo G, Das T, et al. 53BP1 and USP28 mediate p53-dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife 2016;5. DOI:10.7554/eLife.16270
- Bg L, Daggubati V, Uetake Y, et al. A USP28-53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol. 2016;214:143–153.
- Doench JG. Am I ready for CRISPR? A user’s guide to genetic screens. Nat Rev Genet. 2018;19:67–80.
- Cescon D, Siu LL. Cancer clinical trials: the rear-view mirror and the crystal ball. Cell. 2017;168:575–578.
- El-Hachem N, Ba-Alawi W, Smith I, et al. Integrative cancer pharmacogenomics to establish drug mechanism of action: drug repurposing. Pharmacogenomics. 2017;18:1469–1472.
- Madani Tonekaboni SA, Soltan Ghoraie L, Manem VSK, et al. Predictive approaches for drug combination discovery in cancer. Brief Bioinform. 2016. DOI:10.1093/bib/bbw104