
ABSTRACT
The p53 protein is a key tumor suppressor in mammals. In response to various forms of genotoxic stress p53 stimulates expression of genes whose products induce cell cycle arrest and/or apoptosis. An E3-ubiquitin ligase, Mdm2 (mouse-double-minute 2) and its human ortholog Hdm2, physically interact with the amino-terminus of p53 to mediate its ubiquitin-mediated degradation via the proteasome. Thus, pharmacological inhibition of the p53-Mdm2 interaction leads to overall stabilization of p53 and stimulation of its anti-tumorigenic activity. In this study we characterize the biological effects of a novel class of non-genotoxic isatin Schiff and Mannich base derivatives (ISMBDs) that stabilize p53 on the protein level. The likely mechanism behind their positive effect on p53 is mediated via the competitive interaction with Mdm2. Importantly, unlike Nutlin, these compounds selectively promoted p53-mediated cell death. These novel pharmacological activators of p53 can serve as valuable molecular tools for probing p53-positive tumors and set up the stage for development of new anti-cancer drugs.
Introduction
The major human tumor suppressor protein p53 is a transcriptional master regulator that controls cell cycle, senescence, apoptosis, autophagy, DNA repair and metabolism [Citation1]. Over half of human cancers are characterized by either mutations in the p53-coding gene or by alterations in the p53-dependent signaling pathways. p53 exerts its tumor suppressor functions via regulation of both coding and non-coding genes [Citation2–Citation5]. In the absence of stress signals, p53 is inactivated by an E3 ubiquitin ligase, Mdm2, that targets p53 for ubiquitin-dependent proteasomal degradation [Citation6–Citation8]. Importantly, to mediate efficient ubiquitinylation of p53, Mdm2 needs to complex with its partner, MdmX [Citation9,Citation10].The oncogenic protein Mdm2 and the tumor suppressor p53 form a negative feedback loop, in which p53 positively regulates Mdm2 expression [Citation11,Citation12] and the latter, in turn, negatively regulates p53 by promoting its degradation[Citation13]. Earlier studies have shown that Mdm2 and p53 interact through their amino termini [Citation14], which results in direct repression of the p53 transactivation domain [Citation8,Citation15]. Moreover, point mutations or deletions in the central domain of Mdm2 prevent p53 degradation [Citation16–Citation18] . Further studies have shown that both the amino- terminal and central domains of MDM2 act synergistically to bind p53 [Citation19].
Based on these features, MDM2 is arguably considered as an oncogene [Citation8,Citation20,Citation21]. Mdm2 is over-expressed in many tumors, which correlates with shorter survival of cancer patients [Citation22,Citation23]. While under normal conditions Mdm2 keeps p53 in check, upon various forms of cellular stress p53 undergoes multiple post-translational modifications that disrupt the interaction between p53 and Mdm2 resulting in p53 stabilization and activation [Citation24]. Mechanistically, phosphorylation of p53 on Ser 15 and Ser20 by ATM prevents the binding of Mdm2 and, at the same time, facilitates the interaction of p53 with histone acetyltransferase (HAT) CBP/p300 [Citation25]. The latter, together with lysine methylation by Set7/9 [Citation26,Citation27], protect lysine residues in the carboxyl-terminus of p53 from Mdm2-mediated ubiquitinylation, hence leading to its stabilization and transcriptional activation [Citation28–Citation30].
Perhaps not surprisingly, the p53-Mdm2 axis represents a very attractive target for pharmacological inhibition. To this end, a number of small molecules and stapled peptides targeting the p53-Mdm2 interaction (such as Nutlins (Nutlin-3a, RG7112, RG7388) [Citation31–Citation33], SJ-172,550 [Citation34], ATSP-7041 [Citation35]) have been developed and are currently in different phases of clinical trials.
Nutlin-3 is one of the first specific inhibitor of the MDM2-p53 interaction, which showed promise in anti-cancer studies [Citation36]. Nutlin-3 has been shown to exhibit a broad activity against various cancers that express wild-type p53, including gastric cancer and cell lymphoma [Citation37,Citation38]. Nutlin-3A is the most potent compound from a series of analogues of cis-imidazoline [Citation39]. It mimics the Mdm2 binding region of p53, represented by a short helix of three hydrophobic residues Phe19, Trp23, and Leu26 and thus inhibits the interaction between MDM2 and p53. However, the therapeutic potential of Nutlin-3 is limited because it mostly activates cell cycle arrest of tumor cells, rather than apoptosis [Citation40,Citation41]. Hence, these results justify the necessity of development of new therapeutic drugs to stabilize the tumour-suppressive protein p53 and enhance its function as a transcriptional factor in cancer cells.
Recently, by means of molecular docking followed by in cellulo screening we uncovered that Isatin Schiff and Mannich based derivatives (ISMBDs) were able to activate p53 [Citation42]. In line with this, Rana and colleagues have shown that isatin-derived spirocyclic a-methylene-g-butyrolactone can be used as a novel anticancer agent [Citation43]. Furthermore, a series of isatin-based heterocyclic compounds were found to inhibit proliferation of cancer cell lines resistant to apoptosis [Citation44]. In the current study, we elucidated the molecular mechanism of their function and demonstrated that these small molecule activators of p53 operated via blunting the interaction between p53 and Mdm2. In addition, we revealed that the selected compounds specifically activated one of the p53-dependent target gene, PUMA, resulting in the increased death of several p53-positive tumor cell lines. Collectively, our findings provide a rationale for development of new pharmacological tools based on these p53 activating molecules that would facilitate eradication of p53-positive tumors.
Materials and methods
In silico docking studies
Isatin Mannich and Schiff Based derivatives were identified using virtual screening approach as described in our previous paper [Citation42]. The Schrodinger Glide package was used for SM molecular docking studies in extra precision mode. Ligands 3D structures were prepared with Schrodinger LigPrep suite in OPLS_2005 force field. Number of treated rotatable bonds was limited to 10. The X-ray structure 1RV1 [Citation33] of the N-terminal MDM2 region (25–109 aa with 2.30 Å resolution) complexed with Nutlin-2 was used for docking studies. The water and co-crystallized molecules were removed from the protein model. Hydrogens were added, concerning that mean value of tumor intracellular pHi is 7.0. The grid point box, centered on the coordinates of the reference ligand, was used for energy grid maps calculations.
Cell cultures
Human osteosarcoma U2-OS (p53+and p53−), colon cancer HCT116 (p53+ and p53−), H1299 (p53−) and H460 (p53+) cell lines were cultured according to ATCC recommendations. All cell lines used were maintained at 37°C in 5% CO2. U2-OS cells with stable expression of shRNA against p53 were obtained by lentiviral infection as described previously [Citation5].
RNA extraction and quantitative real-time PCR
Total RNA extraction was performed using the TRIzol reagent (Invitrogen, CA, USA) and 1 µg of each RNA sample was reverse transcribed to cDNA with oligo d(T) using RevertAid RT Reverse Transcription Kit (Thermo Scientific, MA, USA). Real-time PCR was performed using the real-time PCR supermix (SsoFast EvaGreen supermix, Bio-Rad, CA, USA). Real-time PCR was performed on CFX96 Touch Real-Time PCR Detection System (Bio-Rad, CA, USA). The PCR reaction (20 μl) included 1 μl RT product, 1 × real-time PCR supermix, and 1 μM each primers. The amplification reactions were performed at 95°C for 10 min, followed by 40 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s. All reactions were run in triplicate. Relative quantification of gene expression was obtained using the 2-∆∆CT method normalizing to the GAPDH housekeeping gene (using CFX Manager software, Bio-Rad, Bio-Rad, CA, USA). The primers used were: p21 (+GACACCACTGGAGGGTGACT,_ CTCTTGGAGAAGATCAGCCG); PUMA (+GAC CTC AAC GCA CAG TAC GA,_ CAC CTA ATT GGG CTC CAT CT); BAX (+GGG TTG TCG CCC TTT TCT,_ CAG CCC ATG ATG GTT CTG ATC AG); GAPDH (+GAGGTCAATGAAGGGGTCAT,_ AGTCAACGGATTTGGTCGTA).
Western blotting analysis
Proteins from cells treated or non-treated with compounds were extracted with RIPA buffer (150 mM NaCl, 1.0% Triton X-100, 50 mM Tris, pH 7.5) supplemented with protease inhibitor cocktail (Complete Mini, Roche, Basel, Switzerland). Protein samples were separated by SDS-PAGE and transferred onto PVDF membrane (Hybond–C Extra, Amersham Bioscience, Sweden). The membrane was blocked and incubated with primary and secondary antibodies in PBST (PBS with Tween 20) and 5% non-fat milk. Membranes were incubated with antibodies directed against p53 (clone DO-1; OP43, Merck Millipore, Darmstadt, Germany), mdm2 (clone SMP-14; sc-965, Santa-Cruz, TX, USA), GAPDH (6C5, ab8245, Abcam, UK). Blots were visualized by enhanced chemiluminescence (Merck Millipore, Darmstadt, Germany).
Colony formation assay
Cell lines were plated at 500 cells per well in standard six-well plates. Cells were treated for10-14 days with 3 μM ISMBDs and incubated at 37 °C with 5% CO2 for two weeks to form colonies. Colonies were fixed in 80% methanol and stained with 0.1% crystal violet solution.
Cellular screening
For the screening of small molecules U2-OS-pLV (p53+ and p53-) reporter cell lines were generated [Citation42]. Briefly, episomal EBNA1-based vector that contains minimal HSP70 promoter with three minimal p53 binding sites inserted upstream of the start site (Supplementary Fig. S3, S4) were transfected into U2-OS and HCT116 cells and positive clones were selected with 200 µg/ml of hygromycin. Cells was were plated in 96-well black plates (Greiner Bio-One, Germany) in DMEM supplemented with 10% FBS in concentration of 2 × 104 cells/well. Incubation of cells with the compounds was performed in serum-free Opti-MEM (Thermo Scientific, MA, USA) media for 48 hours. Nutlin-3A (Sigma Aldrich, MO, USA) and DMSO (Sigma Aldrich, MO, USA) were used as positive and negative controls, respectively. After incubation period, cells were fixed with 4% paraformaldehyde PBS solution and stained with Hoechst 33,342 for 10 min in concentration 4 μg/mL. The total number of cells and the percentage of EGFP-positive cells were measured by the automated fluorescent imaging system Operetta (Perkin Elmer, MA, USA) and analyzed with the Perkin Elmer Harmony 3.1 software.
Calculation of GI50 and ED50
Growth Inhibition (GI) was calculated as percentage ratio of the cell number in control (DMSO) versus numbers of survived cells at different concentrations of the compound. Effective Dose (ED) was calculated as percentages of GFP-positive cells at different concentrations of the compound. The data were based on three independent experiments.
Statistical analysis
The potency of compounds was calculated as a ratio between percentages of EGFP-positive cells generated in response to the treatment with compounds versus Nutlin-3 to the total number of cells. %EGFP = (NEGFP/Nt)∙100%, where Nt is a total number of cells in all analyzed fields and NEGFP is a number of cells expressing EGFP with intensity level more than 290 counts/pixel (background intensity level).
γ-H2Ax evaluation after treatment with compounds
The nonspecific genotoxic effect of potential MDM2 inhibitors was evaluated by immunostaining to γ-H2Ax and estimation of DNA repair foci number. The test was performed in U2-OS p53+ and U2-OS p53- cell lines. The cells were seeded on 96-wells plates in concentration of 3 × 103 cell per well and grown in DMEM with 10% FBS overnight. The media was then replaced with Opti-MEM (ThermoScientific, MA, USA) with tested substances in a final concentration of 3 μM. Nutlin-3A (Sigma-Aldrich, MO, USA) in final concentration of 3 μM was included in the evaluations as known MDM2 inhibitor with no recorded genotoxic activity. Doxorubicin in final concentration of 0.5 μM was used for a positive control. For the negative control, cells were incubated in 0.5% DMSO. Following the 24 hours incubation, the cells were washed with PBS and fixed in 4% paraformaldehyde at RT for 15 minutes. Then the cells were permebealized in 0.5% TritonX-100 in PBS. To avoid the non-specific binding of antibodies, the cells were incubated with blocking buffer for 30 minutes. The DNA repair foci were stained with anti-γ-H2Ax antibody (Cell Signaling Technology, MA, USA) and appropriate secondary antibodies (AlexaFluor 546 conjugated anti-rabbit IgG) (ThermoScientific, MA, USA). The nuclei were visualized by Hoechst 33,342 dye (Sigma-Aldrich, MO, USA). The analysis of DNA repair foci number was made using high throughput screening system Operetta (Perkin Elmer, MA, USA) at 40x magnification by Harmony 3.1 software (Perkin Elmer, MA, USA). Mean number of foci per nucleus was calculated in 20 visual fields from each well. The final data was based on two independent experiments with three replicas for each molecule studied.
In vitro interactions
cDNAs of human TP53 and HDM2 were obtained by RT-PCR using RNA from MCF7 cell line. cDNA coding p53 (1–40 aa) and MDM2 (1–112 aa) were inserted in pGEX-5X-1 and pRSET-6His vectors, respectively. Proteins were expressed in BL21-Gold E. coli using 0.4 mM IPTG for 4 hours and were subsequently purified according to the respective manufacturer’s protocol. The GST-p53 and GST proteins bound to glutathion-sepharose beads (ThermoScientific, MA, USA) were incubated with 6His-MDM2 (1–112) in the binding buffer (150 mM NaCl, 25 mM Tris pH 8.0, 0.1% Triton X100) in the presence or absence of compounds (6 mM) at 4°C for 2 hours. The affinity resins were washed extensively three times with the binding buffer and the remained proteins were eluted by boiling in SDS loading buffer. The amounts of bound Mdm2 were analyzed by Western blotting with anti-6His antibodies (ab137839, Abcam, UK).
Luciferase assay
For luciferase assay, 1 × 106 of HCT116 p53+ and HCT116 p53- cells, respectively, were seeded in 12-well plates 24 h before transfection. Cells in each well were transfected with 0.5 μg of the luciferase reporter plasmid and 0.5 μg of the pRL Renilla Luciferase Control Reporter Vector as an internal control for transfection efficiency using the TurboFect (ThermoScientific, MA, USA) transfection reagent according to the manufacturer’s instructions. After 24 h post-transfection, cells were treated with 9 μM ISMBDs (Au, Av, Ap, Ar, and D) and Nutlin-3A (positive control). Following the 24 h treatment with ISMBDs, cells were assayed using luciferase assay kits (Promega, WI, USA) and normalized by Renilla luciferase activity in accordance with the Dual-Glo Luciferase Assay System protocol.
FACS analysis
U2-OS and U2-OS p53KD cells were treated with synthesized compounds and control reagents for 17 hours, harvested, washed once with PBS, and fixed in 70% ethanol overnight at −20°C. Staining for DNA content was performed with 50 µg/ml propidium iodide (Sigma-Aldrich, MO, USA) mixed with 1 µg/ml RNase A for 30 min. Analysis was performed on a FACS Canto II flow cytometer (BD Biosciences, NJ, USA) with Cell Quest Pro software.
Flow cytometric determination of apoptosis by Annexin V staining
For measuring the effect of compounds on apoptosis cells were treated with 3 μM of ISMBDs following the staining with Alexa Fluor 488 Annexin V Kit (Invitrogen, CA, USA) according to the manufacturer’s instructions. Briefly, treated cells were washed with cold PBS and resuspended in the 1× Annexin-binding buffer provided by the manufacturer to obtain the density ~ 1 × 106 cells/ml. Then, 5 µl of Alexa Fluor 488 Annexin V was added to 100 μl of cell suspension and incubated for 15 min. Analysis was performed using Guava EasyCyte 8 flow cytometer (Guava EasyCyte; Millipore, Billerica, MA, USA).
Results
ISMBDs activate p53-responsive GFP-reporter via breaking the p53-Mdm2 interaction
To date, more than 20 different types of chemicals are known to affect the p53-Mdm2 interaction, including benzodiazepinediones, spiro-oxindoles and cisimidazolines. All of them, despite their different structural properties, mimic the amino-terminal alpha-helix of p53 that binds Mdm2. Using this pharmacophore hypothesis ()) we carried out a molecular docking of the in-house library of 38,000 compounds available from St. Petersburg Technological Institute combined with 1800 molecules from the diversity library set obtained from Interbioscreen (Chernogolovka, Russia) for novel molecules that bind the Mdm2 cleft [Citation42].
Figure 1. Compounds identified in silico activate p53-dependent GFP reporter in cells. (a) An X-ray structure of a typical Isatin Mannich Schiff base derivative (IMSBD = Au, Av, Ap, Ar, and d) compound (scale is 17 Å) that has topological and lipophylic characteristics similar to the alpha-helical part of p53 TAD (grey ribbon) that binds MDM2. (b) Schematic diagram explains how small molecules, by dissociating the p53-Mdm2 interaction, liberate p53 that binds the DNA response elements and activates transcription of GFP; (c) representative images of U2-OS-pLV1 cell line responses to treatment with Nutlin-3A and isatin-based compounds (Au, Av, Ap, Ar, and D) by activating GFP expression; (d) To investigate effects of ISMBDs on the p53-Mdm2 interaction, the recombinant purified GST-p531-40 and 6-HIS-Mdm21-104 proteins were incubated in the presence of ISMBDs, precipitated on glutathione beads and, following washes, analyzed by western blotting for the p53-bound Mdm2 protein; (e) Western blot signals of Mdm2 used in reactions (Input), p53-bound Mdm2, and the levels of GST-p53 used in the assay are shown.
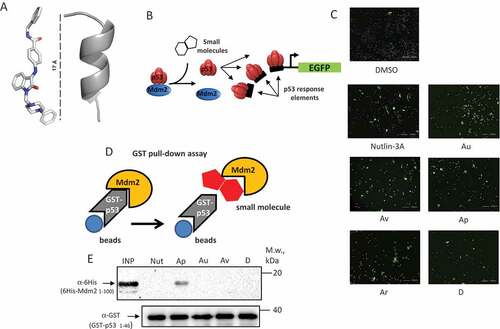
Next, we screened the top five hit compounds ( and Supplementary Fig. S1 and ) for their ability to activate expression of the p53-dependent GFP reporter plasmid, which was stably integrated into human osteosarcoma U2-OS p53+ cells ()). We reasoned that if the selected compounds successfully disrupt the p53-Mdm2 interaction, then the liberation of p53 from the Mdm2-mediated repression should result in increased number of GFP-positive cells compared to non-treated cells. As evident from ), several ISMBD showed similar levels of GFP-positive cells compared to Nutlin-3A, which was used as positive control ()). In general, we saw a good correlation between the cytotoxicity of compounds and their activity towards the p53-activated EGFP reporter (), which is also evident from the growth inhibition (GI) and efficient dose (ED) concentration curves calculated for the ISMBD compounds studied (Supplementary Fig. S6).
Table 1. Activation of p53-reporter and cytotoxicity upon treatment of U2-OS cells with ISMBD compounds.
Figure 2. ISMBDs augment expression of the PUMA-Luc reporter in p53-dependent manner. (a) HCT116 (p53+ and p53-) cells were cotransfected with the PUMA-luc reporter vector and pRL Renilla Luciferase Control Reporter Vector as an internal control for transfection efficiency. Transfected cells were treated with ISMBDs (Au, Av, Ap, Ar, and D) and Nutlin-3A (positive control) to measure luciferase activity [Citation53]; (b) Western Blot analysis of p53 protein levels was performed with the same lysates, which were used for Luciferase assay. Actin was used as a loading control.
![Figure 2. ISMBDs augment expression of the PUMA-Luc reporter in p53-dependent manner. (a) HCT116 (p53+ and p53-) cells were cotransfected with the PUMA-luc reporter vector and pRL Renilla Luciferase Control Reporter Vector as an internal control for transfection efficiency. Transfected cells were treated with ISMBDs (Au, Av, Ap, Ar, and D) and Nutlin-3A (positive control) to measure luciferase activity [Citation53]; (b) Western Blot analysis of p53 protein levels was performed with the same lysates, which were used for Luciferase assay. Actin was used as a loading control.](/cms/asset/e39eee77-a1f1-4720-87bc-5db10844f966/kccy_a_1506664_f0002_oc.jpg)
To address the question of whether activation of the p53-dependent reporter was due to disruption of the p53-Mdm2 interaction, we employed an in vitro approach whereby we directly measured the inhibitory effect of the compounds on the ability of purified amino-terminal fragments of p53 and Mdm2 proteins to form a complex (for explanation, see )). As evident from ), the Au, Av and D compounds were able to break the p53-Mdm2 interaction with the same efficiency as Nutlin-3A. Importantly, neither DMSO, nor Ap affected the formation of the p53-Mdm2 complex, suggesting that Ap activated p53 via some other, Mdm2-independent, mechanism.
We noticed that ISMBDs induced cytotoxicity in U2-OS cells (). This result posed an important question of whether this effect was p53-dependent. To address this question we measured the ISMBD-dependent activation of the pro-apoptotic p53-target gene, Puma, in HCT116 cells with different p53 status ()). Nutlin-3A and doxorubicin were used as positive controls. Importantly, all four ISMBDs tested (Au, Av, Ap, and Ar) showed p53-dependent augmentation of PUMA-luciferase expression. These results were confirmed by the western blot data showing that ISMBDs, albeit to a different extent, stabilized p53 similar to Nutlin-3A and doxorubicin ()). Summing up these data we concluded that ISMBDs not only stabilized, but also transcriptionally activated the p53 protein.
ISMBDs augment expression of the endogenous pro-apoptotic p53 target gene, PUMA
It is known that p53 exerts the anti-cancer effect mostly via regulation of its downstream target genes, whose products trigger cell cycle arrest and apoptosis [Citation1]. Therefore, we measured effects of selected compounds on expression levels of several p53 bona fide endogenous gene targets, including p21, PUMA and Bax (). Expectedly, as judged by RT-PCR, Nutlin-3A robustly activated p21 and Mdm2 (, respectively), but moderately the pro-apoptotic gene, PUMA ()). This result is consistent with previously published data that Nutlin primarily induces cell cycle arrest rather than apoptosis due to activation of the Erk anti-apoptotic pathway [Citation45]. Of note, we also detected poor activation of BAX in U2-OS cells irrespectively of the compound used ()). In contrast, we found that Au and Ar potently activated PUMA and to a much lesser extent, p21 (compare ). These results are in line with our PUMA-luciferase activation data ()). Interestingly, Ap and D activated expression of the TP53 gene itself ()), which indicates that these compounds affect the cellular level of p53 by another mechanism.
Figure 3. ISMBDs differentially affect expression of p53 target genes. U2-OS (p53+ and p53-) cells treated with ISMBDs (Au, Av, Ap, Ar, and D) were subjected to RT-PCR analysis to measure expression levels of BAX (a), TP53 (b), P21 (c), MDM2 (d) and PUMA (e) genes. Signals of non-treated p53- samples were arbitrary set as 1. Anova statistical significance values are denoted as follows: * p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001, **** p ≤ 0.0001. (3F) The corresponding samples were also examined by western blotting using antibodies against p53, Mdm2, Puma, and actin (loading control).
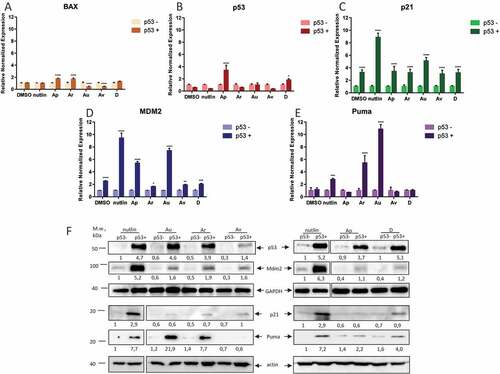
Besides Nutlin-3A, only Au and Ap were able to significantly activate transcription of another p53 target, MDM2 ()). This fact prompted us to investigate how the compounds affected the p53 targets on the protein level. To this end, we performed a series of western blotting experiments to measure the protein levels of p53, Mdm2 and Puma in U2-OS p53+ and p53- cell lines. Expectedly, all of the compounds as well as Nutlin-3A stabilized p53 on the protein level, although to a different extend ()). These results again argued that the compounds might activate p53 by different mechanisms. A heterogeneous response was also obtained for Mdm2. Surprisingly, whereas Nutlin-3A strongly induced the expression of Mdm2, the compounds showed much weaker propensity in this respect (), middle panel). In agreement with our transcription data, Au and Ar compounds strongly augmented the protein level of Puma (), bottom), highlighting the fact that these ISMBDs induce the pro-apoptotic arm of p53 activation.
ISMBDs decrease cell viability of different cancer cell lines in p53-dependent manner
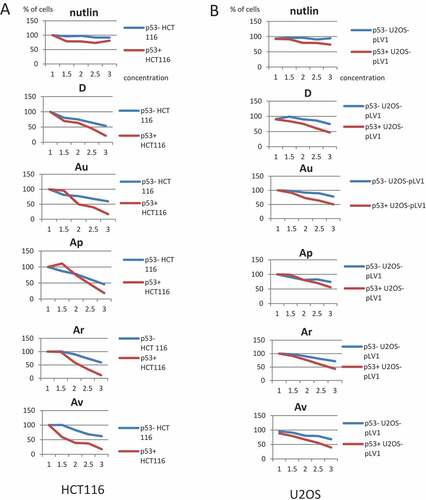
Next, we wanted to extend our observations on p53-dependent cytotoxicity conferred by ISMBDs to several cancer cell lines that differ in their p53 status. Thus, colorectal cancer cell lines HCT116 (p53+ and p53-) and osteosarcoma U2-OS (p53+ and p53-) cells were treated with increasing concentrations (from 0 to 3 μM) of D, Ar, Au, Ap, and Av (). Three compounds (Au, Ar, and Av) noticeably affected proliferation of p53-positive HCT116 cells compared to p53-negative cells ()). Further, although Nutlin-3A and the compound D did show some effect on cell viability at these concentrations, it was not as robust as after treatment with other compounds. When we examined the anti-proliferative effect of these compounds on U2-OS cells, only D and Au strongly affected the viability of p53-positive versus p53-negative cells ()). Other compounds did not have pronounced differential effect on U2-OS p53+ vs p53- cells. Collectively, these results suggest that the p53 effect of ISMBDs may be cell context-dependent.
Figure 4. ISMBDs decrease cell viability of HCT116 and U2-OS cells in p53-dependent manner. HCT116 (p53+ and p53-) (a) and U2-OS (p53+ and p53-) cells expressing pLV1-GFP reporter (b) were treated with the indicated concentrations of ISMBDs (Au, Av, Ap, Ar, and D) and counted for the number of cells using the automated microscopy system Operetta. The initial number of cells before treatment in each case was arbitrary set as 100%.
To corroborate our results, two lung tumour cell lines that differ in their p53 status (H1299 p53- and H460 p53+, respectively) were treated with 1 μM of Au to assess its cytotoxicity (Supplementary Fig. S7). As controls, three known genotoxic agents (doxorubicin, camptothecin and cisplatin) were used. Importantly, Au exhibited higher anti-proliferative effect on H460 p53+ cells compared to H1299 cells, which are p53-negative (Supplementary Fig. S7). Interestingly, while doxorubicin and cisplatin were more toxic for H460 cells, camptothecin affected both cell lines at comparable levels. Thus, we concluded that our ISMBDs were able to inhibit growth of various p53-positive cancer cell lines.
ISMBDs inhibit cell growth in p53-specific manner
Thus far, we showed that ISMBDs activated p53 resulting in selective p53-dependent enhancement of Puma expression. However, it was possible that p53 was activated by non-specific genotoxic stress elicited by these compounds. Hence, we examined the formation of γ-H2Ax foci as a universal marker of DNA damage response in cells treated with our compounds (,b and S5). Using the Operetta microscopy system, we calculated the number of γ-H2Ax foci per nucleus using Nutlin-3A and doxorubicin as positive controls. Genotoxic effect of ISMBDs in general was comparable to the effect of Nutlin-3A and was much less pronounced when compared to doxorubicin both in p53+ and p53- cells (). Collectively, these results strongly argue that our compounds stimulated p53 activity without producing strong genotoxic effects.
Figure 5. ISMBDs are non-genotoxic and attenuate proliferation of cancer cells in p53-dependent manner. U2-OS p53+ (a) and p53- (b) cells were treated with ISMBDs (Au, Av, Ap, Ar, D, An and 2), or doxorubicin (positive control) following immunostaining with antibody against γ–H2Ax. The number of nuclear foci was calculated in each case using the automated microscopy system Operetta to assess the levels of DNA damage. (c) HCT116 (p53+ and p53-) cells were treated with compounds mentioned above and examined for their proliferation potential by colony formation assay. Experiments were done in triplicates. After two weeks incubation cells were fixed, stained and counted in each case. (d) To evaluate p53-specific effects of ISMBDs on proliferation, percentage ratios of colonies formed in p53+ versus p53- cells were calculated. Anova statistical significance values are denoted as follows: * p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001, **** p ≤ 0.0001.
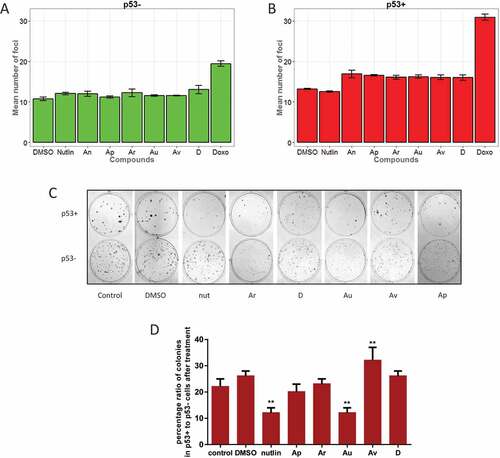
Next, we decided to explore the functional consequence of ISMBDs-mediated activation of p53 in respect to their ability to hamper proliferation of cancer cells. In line with our hypothesis, we reasoned that ISMBDs should exhibit more pronounced effect on p53-positive cells compared to the ones that lack p53.
To validate our prediction we performed the colony formation assay using HCT116 p53+ and p53- cells ()). This assay allows accurate measurement of every tumour cell in the population for its ability to undergo “unlimited” division. The results of this experiment demonstrated that the presence of p53 itself inhibited the colony formation process ()). Nutlin-3A caused further two-fold reduction in the number of p53+ colonies and had almost no effect on p53- cells (). The compounds Au and Ar also strongly inhibited colony formation of p53+ cells. However, they also negatively affected the growth of p53- cells (). When the ratio of colonies for p53+ and p53- cells was calculated for each compound it became apparent that only Au had comparable with Nutlin-3A inhibitory potency ()). Av and D had no significant effect on the colony formation process, which is probably due to their degradation in the medium as the whole process takes about two weeks. Taken together, the results of this experiment suggest that the compound Au possessed strong growth suppressive effect similar to Nutlin-3A and that this effect was likely mediated by p53-dependent activation of Puma expression.
ISMBDs induce apoptosis in p53-specific manner
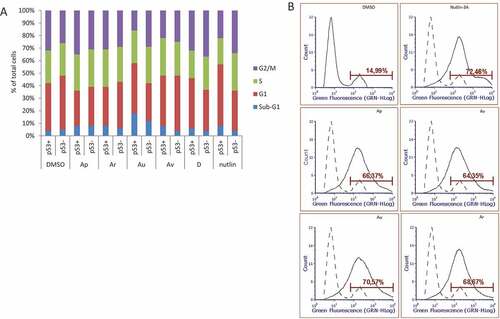
The experimental evidence suggests that ISMBDs attenuate tumour cell growth and proliferation by activating and stabilizing the p53 protein. Therefore, we decided to directly assess the effect of ISMBDs on cell cycle and the rate of apoptosis in p53-positive versus p53-negative cancer cells by FACs analysis ()). In agreement with our RT-PCR and western blotting results, we found that Au elicited the highest level of apoptosis in p53-dependent manner. However, we did not detect dramatic changes in cell cycle distribution after treatment with ISMBDs. Only Nutlin-3A caused accumulation of p53+ cells in G1 phase ()).
Figure 6. ISMBDs promote p53-dependent cell death and arrest of proliferation. (a) U2-OS (p53+ and p53-) cells treated with ISMBDs (Au, Av, Ap, Ar, and D) and Nutlin-3A (positive control) were stained with propidium iodide (PI) and analysed by FACS. Cell death was calculated by estimating the number of cells in sub-G1 phase. (b) For measuring the effect of compounds on apoptosis, cells were treated with 3 μM of ISMBDs following the staining with Alexa Fluor 488 Annexin V Kit and flow cytometry. Percentages of Annexin V-positive cells after treatment are indicated.
To corroborate our results on the effect of ISMBDs on p53-dependent cell death, we stained cells treated with ISMBDs for Annexin V, which selectively defines the fraction of apoptotic cells ()). Our results demonstrated that the treatment with ISMBDs increased the fraction of apoptotic cells ~4,5-fold over DMSO, which is comparable with the effect of Nutlin-3A.
Discussion
In the previous study [Citation42] we reported an identification of a number of isatin-like p53-activating compounds (Fig. S1). In this study, we characterized the molecular mechanisms of p53 activation by ISMBD compounds in tumor cells and described the biological consequences of this activation. Using the pharmacophore model of the amino-terminal p53 hydrophobic residues binding into the cleft of Mdm2 through molecular docking we identified a number of isatin derivatives that activated p53-dependent reporter in tumour cells. It has to be noted that only a small portion of cells responded to the treatment with compounds ()). However, this was also true for Nutlin-3A, a “gold standard” of pharmacological activation of p53. One explanation to this phenomenon may be that the promoter of p53-GFP reporter is susceptible to DNA methylation, which accumulates with continuous passaging of cells, thereby limiting the effect of the molecules. Also, to avoid false-positive effects on activation of the GFP reporter we tried to minimize the amounts of compounds to treat cells. Therefore, the highest concentration of Nutlin-3A used in screening was 3 μM, which is still in the range of acceptable concentrations for Nutlin [Citation46].
Importantly, unlike Nutlin-3A, the Au and Ar compounds strongly activated transcription of Puma, the key regulator of apoptosis. Yet, they were less potent in activating the p21 and Mdm2 genes. This result is striking as the Nutlin-3A derivatives mostly induce cell cycle arrest. Therefore, once the treatment is stopped the tumor cells resume their growth and acquire drug resistance. One explanation to specific activation of p53 would be that our compounds are genotoxic and therefore elicit DNA damage response hence stabilizing p53 via post-translational modifications and through the inhibition of 26S proteasome [Citation47,Citation48]. However, our results on staining of cells after treatment with ISMBDs for γ-H2A.X, a known marker of DNA damage, strongly argue against this possibility (). The likely explanation comes from the results of biochemical analysis which revealed that the compounds efficiently interfered with the p53 binding to Mdm2 ()).
Because all the molecules tested in cellulo were derivatives of the same Isatin Mannich and Schiff base scaffold it was somewhat puzzling that only the Au compound exhibited a significant biological effect, while others were less potent. This question clearly requires further investigation. However, it can be argued that Au is more stable compared to other isatin derivatives or may be imported to the nucleus with different efficiency. In line with this is the fact that our compounds have different numbers of predicted metabolic conversions (Fig. S2). That Au was one of the most active molecules may be explained by the fact that it has only two metabolic conversions (Fig. S2). The genome-wide study on dynamics of activation of p53 targets [Citation49] showed that the dissociation of Mdm2 from p53 by Nutlin-3A resulted in an immediate activation of hundreds of p53-dependent genomic loci before any detectable increase in total p53 and Mdm2 levels. Given that the pro-apoptotic p53 gene targets require longer time for their activation, it is possible that differential effects of the compounds correlate with their intracellular stability, i.e. the short-lived ones activate only p21 and Mdm2 and the more stable ones are also able to activate Puma. In this respect, it should be noted that the Au compound undergoes only two possible metabolic reactions (Table S1, Supplementary Fig. S2) and hence is assumed to be more stable. Also, it is more soluble than other ISMBDs tested due to the fluorine atom present in its structure (Supplementary Fig. S1). However, this hypothesis is challenged by the fact that other compounds, except Au, failed to efficiently activate the early-response p21 gene ()). Another explanation to different effects of structurally similar ISMBDs is that there are additional cell type-specific yet unknown targets for these compounds, which may confer specificity to cellular response. As an indirect support to our speculations is the fact that Nutlin-3A was shown to bind, in addition to Mdm2, the anti-apoptotic Bcl-2 family protein [Citation50,Citation51]. On a similar note, a close ISMBD analogue, 4-hydroxybenzylideneindolin-2-one has been shown to inhibit Polo-like kinase 4 (Plk4), a known regulator of p53 [Citation52].
Clearly, additional experiments are required to definitively address the question of target specificity for p53-activating ISMBD compounds studied in this work. However, the fact that these molecules potently induce p53-dependent apoptosis make them valuable tools for dissecting p53-positive tumors and set up the stage for development of new anti-cancer drugs.
Supplemental Material
Download MS Power Point (13 MB)Acknowledgments
The authors acknowledge the support from RCF grant 14-15-00816 O.F. carried out proliferation and apoptosis assays and acknowledges the support from RFBR grant 16-34-60228.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137(3):413–431.
- Barlev N, Sayan B, Candi E, et al. The microRNA and p53 families join forces against cancer. Cell Death Differ. 2010;17(2):373.
- Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010;2(8):a000935.
- Grossi E, Sánchez Y, Huarte M. Expanding the p53 regulatory network: lncRNAs take up the challenge. Biochimica Et Biophysica Acta (Bba)-Gene Regulatory Mechanisms. 2016;1859(1):200–208.
- Lezina L, Purmessur N, Antonov A, et al. miR-16 and miR-26a target checkpoint kinases Wee1 and Chk1 in response to p53 activation by genotoxic stress. Cell Death Dis. 2013;4(12):e953.
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387(6630):299–303.
- Momand J, Zambetti GP, Olson DC, et al. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69(7):1237–1245.
- Oliner JD, Pietenpol JA, Thiagalingam S, et al. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. 1993.
- Shvarts A, Steegenga W, Riteco N, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996;15(19):5349.
- Stad R, Little NA, Xirodimas DP, et al. Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Reports. 2001;2(11):1029–1034.
- Barak Y, Juven T, Haffner R, et al. mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12(2):461.
- Wu X, Bayle JH, Olson D, et al. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7(7a):1126–1132.
- Haupt Y, Maya R, Kazaz A, et al. Mdm2 promotes therapid degradation of p53. Nature. 1997;387(6630):296–299.
- Kussie PH, Gorina S, Marechal V, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274(5289):948.
- Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol. 1993;13(7):4107–4114.
- Argentini M, Barboule N, Wasylyk B. The contribution of the acidic domain of MDM2 to p53 and MDM2 stability. Oncogene. 2001;20(11):1267–1275.
- Blattner C, Hay T, Meek DW, et al. Hypophosphorylation of Mdm2 augments p53 stability. Mol Cell Biol. 2002;22(17):6170–6182.
- Zhu Q, Yao J, Wani G, et al. Mdm2 mutant defective in binding p300 promotes ubiquitination but not degradation of p53 EVIDENCE FOR THE ROLE OF p300 In Integrating Ubiquitination and Proteolysis. J Biol Chem. 2001;276(32):29695–29701.
- Kulikov R, Winter M, Blattner C. Binding of p53 to the central domain of Mdm2 is regulated by phosphorylation. J Biol Chem. 2006;281(39):28575–28583.
- Chène P. Inhibiting the p53–MDM2 interaction: an important target for cancer therapy. Nature Rev Cancer. 2003;3(2):102–109.
- Wade M, Li Y-C, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13(2):83–96.
- Haitel A, Wiener HG, Baethge U, et al. mdm2 expression as a prognostic indicator in clear cell renal cell carcinoma: comparison with p53 overexpression and clinicopathological parameters. Clin Cancer Res. 2000;6(5):1840–1844.
- Rayburn E, Zhang R, He J, et al. MDM2 and human malignancies: expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr Cancer Drug Targets. 2005;5(1):27–41.
- Marouco D, Garabadgiu AV, Melino G, et al. Lysine-specific modifications of p53: a matter of life and death? Oncotarget. 2013;4(10):1556.
- Sakaguchi K, Herrera JE, Saito S, et al. DNA damage activates p53 through a phosphorylation–acetylation cascade. Genes Dev. 1998;12(18):2831–2841.
- Lezina L, Aksenova V, Fedorova O, et al. KMT set7/9 affects genotoxic stress response via the Mdm2 axis. Oncotarget. 2015;6(28):25843.
- Morgunkova A, Barlev NA. Lysine methylation goes global. Cell Cycle. 2006;5(12):1308–1312.
- Barlev NA, Liu L, Chehab NH, et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8(6):1243–1254.
- Espinosa JM, Emerson BM. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol Cell. 2001;8(1):57–69.
- Ivanov GS, Ivanova T, Kurash J, et al. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol Cell Biol. 2007;27(19):6756–6769.
- Ding Q, Zhang Z, J-J L, et al. Discovery of RG7388, a potent and selective p53–MDM2 inhibitor in clinical development. J Med Chem. 2013;56(14):5979–5983.
- Saha MN, Qiu L, Chang H. Targeting p53 by small molecules in hematological malignancies. J Hematol Oncol. 2013;6(1):1.
- Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848.
- Reed D, Shen Y, Shelat AA, et al. Identification and characterization of the first small molecule inhibitor of MDMX. J Biol Chem. 2010;285(14):10786–10796.
- Chang YS, Graves B, Guerlavais V, et al. Stapled α− helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci. 2013;110(36):E3445–E3454.
- Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin Cancer Res. 2008;14(17):5318–5324.
- Endo S, Yamato K, Hirai S, et al. Potent in vitro and in vivo antitumor effects of MDM2 inhibitor nutlin‐3 in gastric cancer cells. Cancer Sci. 2011;102(3):605–613.
- Tabe Y, Sebasigari D, Jin L, et al. MDM2 antagonist nutlin-3 displays antiproliferative and proapoptotic activity in mantle cell lymphoma. Clin Cancer Res. 2009;15(3):933–942.
- Zhang B, Golding BT, Hardcastle IR. Small-molecule MDM2-p53 inhibitors: recent advances. Future Med Chem. 2015;7(5):631–645.
- Hu B, Gilkes DM, Farooqi B, et al. MDMX overexpression prevents p53 activation by the MDM2 inhibitor Nutlin. J Biol Chem. 2006;281(44):33030–33035.
- Michaelis M, Rothweiler F, Klassert D, et al. Reversal of P-glycoprotein–mediated multidrug resistance by the murine double minute 2 antagonist nutlin-3. Cancer Res. 2009;69(2):416–421.
- Davidovich P, Aksenova V, Petrova V, et al. Discovery of novel isatin-based p53 inducers. ACS Med Chem Lett. 2015;6(8):856–860.
- Rana S, Blowers EC, Tebbe C, et al. Isatin derived spirocyclic analogues with α-methylene-γ-butyrolactone as anticancer agents: a structure–activity relationship study. J Med Chem. 2016;59(10):5121–5127.
- Evdokimov NM, Magedov IV, McBrayer D, et al. Isatin derivatives with activity against apoptosis-resistant cancer cells. Bioorg Med Chem Lett. 2016;26(6):1558–1560.
- Lee S-Y, Ko K-W, Choe Y-J, et al. Nutlin-3, a MDM2 antagonist, induces Erk1/2 activation in U2OS cells. FASEB J. 2010;24(1 Supplement):485.16–485.16.
- Vassilev LT. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle. 2004;3(4):417–419.
- Fedorova OA, Moiseeva TN, Nikiforov AA, et al. Proteomic analysis of the 20S proteasome (PSMA3)-interacting proteins reveals a functional link between the proteasome and mRNA metabolism. Biochem Biophys Res Commun. 2011;416(3):258–265.
- Moiseeva TN, Bottrill A, Melino G, et al. DNA damage-induced ubiquitylation of proteasome controls its proteolytic activity. Oncotarget. 2013;4(9):1338–1348.
- Allen MA, Andrysik Z, Dengler VL, et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife. 2014;3:e02200.
- Ha J-H, Won E-Y, Shin J-S, et al. Molecular mimicry-based repositioning of nutlin-3 to anti-apoptotic Bcl-2 family proteins. J Am Chem Soc. 2011;133(5):1244–1247.
- Shin J-S, Ha J-H, He F, et al. Structural insights into the dual-targeting mechanism of Nutlin-3. Biochem Biophys Res Commun. 2012;420(1):48–53.
- Yu B, Yu Z, Qi P-P, et al. Discovery of orally active anticancer candidate CFI-400945 derived from biologically promising spirooxindoles: success and challenges. Eur J Med Chem. 2015;95:35–40.
- Lauriola L, Granone P, Ramella S, et al. Expression of the RNA-binding protein HuR and its clinical significance in human stage I and II lung adenocarcinoma. Histol Histopathol. 2012;27(5):617–626.