
ABSTRACT
Systemic sclerosis (SSc) is a multisystemic fibrotic disease characterized by excessive collagen deposition and extracellular matrix synthesis. Though transforming growth factor-β (TGF-β) plays a fundamental role in the pathogenesis of SSc, the mechanism by which TGF-β signaling acts in SSc remains largely unclear. Here, we showed that TGF-β type II receptor (TGFBR2) was significantly upregulated in both human SSc dermal tissues and primary fibroblasts. In fibroblasts, siRNA-induced knockdown of TGFBR2 resulted in a reduction of p-SMAD2/3 levels and reduced production of type I collagen. Additionally, functional experiments revealed that downregulation of TGFBR2 yielded an anti-growth effect on fibroblasts through inhibiting cell cycle progression. Further studies showed that miR-3606-3p could directly target the 3′-UTR of TGFBR2 and significantly decrease the levels of both TGFBR2 mRNA and protein. Furthermore, SSc dermal tissues and primary fibroblasts contain significantly reduced amounts of miR-3606-3p, and the overexpression of miR-3606-3p in fibroblasts replicates the phenotype of TGFBR2 downregulation. Collectively, our findings demonstrated that increased TGFBR2 could be responsible for the hyperactive TGF-β signaling observed in SSc. Moreover, we identified a pivotal role for miR-3606-3p in SSc, which acts, at least partly, through the attenuation of TGF-β signaling via TGFBR2 repression, suggesting that the regulation of miR-3606-3p/TGFBR2 could be a promising therapeutic target that could improve the treatment strategy for fibrosis.
Introduction
Systemic sclerosis (SSc), or scleroderma, is a complex multisystem autoimmune disease characterized by vascular injury, inflammation, and accumulation of excessive extracellular matrix (ECM) protein, ultimately resulting in fibrosis of the dermal and internal organs, including the lungs, the gastrointestinal tract, and the heart [Citation1–Citation4]. Although the exact etiology and precise mechanism driving this disease remain largely elusive, accumulating evidence has shown that the transforming growth factor β (TGF-β) pathway may be the most important factor in the pathogenesis of SSc [Citation5–Citation7].
TGF-β is a potent inducer of collagen synthesis, fibroblast proliferation, as well as differentiation. TGF-β receptor types 1 and 2 (TGFBR1 and TGFBR2) have been implicated in the transmission of TGF-β signaling to affect multiple intracellular functions [Citation8,Citation9]. Experimental deletion of TGFBR2 has been shown to decrease BLM-induced dermal and pulmonary fibrosis both in vitro and in vivo [Citation10–Citation13]. There is also evidence to suggest that TGFBR2 might be involved in the pathogenesis and progression of fibrosis of the kidneys, liver, and heart in mice [Citation14–Citation16]. It can therefore be concluded that alterations to TGFBR2 might be a potential underlying mechanism in the pathological process of SSc. However, when comparing endogenous levels of TGFBR2 in SSc and healthy individuals, the conclusion remains controversial. For example, both Kawakami et.al and Yoshihide Asano et.al reported higher TGFBR2 expression levels in scleroderma fibroblasts than in normal fibroblasts [Citation2,Citation17]. In another study, Jaspreet Pannu showed that the TGFBR2 level was decreased by 30% in SSc fibroblasts [Citation18]. On the other hand, Dong et al. have reported that TGFBR2 protein levels did not appear to differ between SSc and control cell lines [Citation19]. Thus, while there is evidence that the TGF-β signaling pathway is altered in SSc fibroblasts, there is no consensus on TGFBR2 alterations in SSc.
Additionally, the reason for the TGFBR2 alterations observed in SSc also remains unclear, especially concerning epigenetics. MiRNAs are endogenous, single-stranded, evolutionarily conserved noncoding RNAs about 24 nucleotides in length that act as crucial posttranscriptional regulators through binding to the 3ʹ-untranslated regions (3′-UTRs) of their target genes [Citation20]. Currently, the specific signatures of SSc have been detected by examination of miRNA profiles in serum, skin tissues, and fibroblasts [Citation21–Citation24]. Moreover, some unique miRNAs have even been demonstrated to be involved in SSc [Citation22,Citation23,Citation25,Citation26]. Therefore, these miRNAs could be potential biomarkers for SSc diagnosis and a target for therapeutic manipulation. Using computational algorithms, we predicted that miR-3606-3p could directly target the 3′-UTR of TGFBR2. This inspired us to focus on the functional contribution of miR-3606-3p to TGFBR2 alteration in SSc.
In the present study, we found that TGFBR2 was increased in SSc skin tissues and fibroblasts, while miR-3606-3p attenuated TGF-β signaling through direct targeting of TGFBR2. Collectively, our results demonstrated that a downregulation of miR-3606-3p could, in part, account for the abnormal elevation of TGFBR2 in SSc.
Materials and methods
Patients
Twenty patients who fulfilled the criteria for SSc diagnosis were enrolled from the Shanghai Traditional Chinese Medicine-Integrated Hospital (2013). Laboratory tests were conducted for all patients in order to confirm the diagnosis for SSc. Twenty-one age- and gender-matched healthy individuals were enrolled as normal controls. Clinical characteristics including age, gender and disease type are showed in Supplementary Table 1. All patients and healthy controls provided written informed consent before they were entered into the study. This study was approved by the local ethical committee of Fudan University, China.
Biopsy specimens and cell culture
Dermal biopsy specimens were obtained from 13 SSc patients who fulfilled the criteria for SSc as defined by the American College of Rheumatology (formerly the American Rheumatism Association) (1937). Dermal biopsy specimens from normal controls with no history of autoimmune or other dermal disease were used as control comparators. Dermal samples were transported in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) for processing the same day as described previously [Citation27,Citation28]. The dermal samples were washed in 75% ethanol, phosphate buffered saline (PBS), and DMEM with 10% FBS. Cultured fibroblast strains were established by mincing tissues and placing them into 60-mm culture dishes secured by glass overlays. The third to fifth passage of human dermal fibroblasts were placed in 12-well culture plates at the density of 1 × 105 cells per well for gene and protein expression assays. All cells were cultured in DMEM medium supplemented with 10% FBS, 2 mM glutamine, 50 mg/ml gentamicin, and incubated at 37°C with 5% CO2.
Immunohistochemical staining
Human skin tissues were fixed with 4% paraformaldehyde and embedded in paraffin. Hematoxylin/eosin (H&E) and Masson’s trichrome staining were used to detect tissue structure. A Nikon Eclipse 80i microscope (Nikon, Badhoevedorp, the Netherlands) was used to measure dermal thickness. The maximal distance between the epidermal-dermal junction and the dermal–subcutaneous fat junction in all skin sections described above was analyzed according to the method mentioned in the previous report by Jiaying Lu et al. [Citation28]. Two independent examiners performed evaluation of the sections. For human immunohistochemistry (IHC), primary antibodies for TGFBR2 (1:50; Santa Cruz Biotechnology, https://www.scbt.com/scbt/home) and S100A4 (1:100; Abcam, http://www.abcam.cn) were used along with goat anti-mouse IgG and goat anti-rabbit IgG secondary antibody, respectively. After antibody incubation, tissue sections were examined under the microscope.
RNA extraction, cDNA synthesis, and real-time quantitative reverse transcriptase polymerase chain reaction (qRT-PCR)
Total RNA was isolated from cells using a HP Total RNA Kit (Omega Biotech, Stamford, CT, USA) according to the manufacturer’s protocol. Synthesis of cDNA with reverse transcriptase was performed using an M-MLV First Strand Kit (Life Technologies, Gaithersburg, MD, USA). Real-time qRT-PCR was conducted on a Roch-LC480 Real-Time PCR system (Roche, Switzerland) using the SYBR-Green-based method. Primer sequences for TGFBR2 mRNA, miR-3606-3p, GAPDH and U6 detection are described in . Relative expression levels of TGFBR2 mRNA and miR-3606-3p were determined following normalization to GAPDH and U6, respectively.
Table 1. Primers for reverse transcription and qRT-PCR.
Western blot analysis
Protein products from cell lysates were fractionated by 8% SDS-PAGE electrophoresis and transferred to nitrocellulose membranes (Millipore, Billerica, MA, USA). Membranes were blocked with BSA/TBST buffer for 1 h and then incubated with primary antibodies overnight at 4°C followed by incubation with HRP-conjugated secondary antibodies. Detection was performed using an ECL kit (Pierce, Rockford, IL, USA). Each experiment was done in triplicate. Protein expression was normalized against GAPDH. The following antibodies were used in the analysis: anti-TGFBR2 and anti-GAPDH from Santa Cruz Biotechnology, https://www.scbt.com/scbt/home; anti-Collagen I from merck millipore, http://www.merckmillipore.com/CN/zh; anti-SMAD2/3 and p-SMAD2/3 from Cell Signaling Technology, https://www.cst-c.com.cn. All western blots were calculated through densitometry analysis using the Image J software.
Prediction microRNAs of target genes
To scanning the potential microRNAs that target to target genes, TargetScan software (a canonical microRNA prediction program) was used. Many rules of microRNA-mRNA target binding need to be taken into account. To precisely predict the target microRNAs for genes, these features are following [Citation29]: seed sequence match, conservation, free energy of microRNA-mRNA duplex, site accessibility, contribution of multiple binding sites, local ALU content, local mRNA sequence, ribosomal shadow, position effects and 3ʹpairing.
Construction of the luciferase reporter plasmid, transient transfection, and luciferase assay
Three 407-bp fragments of the human TGFBR2 3′-UTR containing three predicted miR-3606-3p target sites (positions 7258–7265, 7329–7335 and 8163–8169) were directly synthesized with restriction enzymes NheI and Xbal (GenScript Biotech, Shanghai, China) and subcloned into the pmirGLO vector to generate the respective pmirGLO-TGFBR2-3′-UTR-wildtype. Comparably, the pmirGLO-TGFBR2-3′-UTR-mutant was constructed by introducing 7 mutational bases into the miR-3606-3p target sites to obtain six mutated fragments. Then, 50 ng of the above-described luciferase constructs were cotransfected into cells with 20 nM of either miR-3606-3p mimic (5ʹ-AAAAUUUCUUUCACUACUUAG-3ʹ) or negative control (NC). A scrambled sequence (5ʹ-UUCUCCGAACGUGUCACGUTT-3ʹ) was used as NC. All the transient transfections were done using Lipofectamine 2000 (Invitrogen). After 48 hours, cells were harvested and analyzed for luciferase activity using the Dual -Luciferase Reporter Assay System (Promega, Madison, WI, USA). Each experiment was performed in triplicate. Results are represented as relative Renilla luciferase activity, which are obtained following normalization to the firefly luciferase activity.
RNA interference
Two pre-designed short interfering RNA (siRNA) sequences, which target human TGFBR2, were directly synthesized (GenePharma, Shanghai, China). Sequences for TGFBR2 siRNAs are as follows: si-TGFBR2, 5ʹ-GATTCAAGAGTATTCTCACTT-3ʹ. A scrambled sequence (5ʹ-TTCTCCGAACGTGTCACGT-3 was used as non-targeting control siRNA (NC). Cells were transiently transfected with 100 pmol of siRNA sequences using Lipofectamine 2000 (Invitrogen). After 72 h transfection, the cells were collected for further experiments.
Collagens content measurement
Total soluble collagens content in the cell medium were quantified using the Sircol assay (Biocolor, Belfast, UK) according to the manufacturer’s protocol. The amount of collagen protein in cell medium was normalized to the total amount of protein as determined using a BCA Protein Assay kit (Beyotime, Nanjing, China).
Cell proliferation assay
Cell proliferation was determined using the xCELLigence system (Roche). Briefly, cells transfected with miR-3606-3p mimics and si-TGFBR2 or NC were diluted in complete culture medium with a grad of 200 cells and added to the E-Plates 16 (Roche, East Sussex, UK). For 48 hours, the cell density was measured as the cell index every ten minutes, a unit indicating the percentage of the well occupied by cells.
Cell cycle analysis
Propidium iodide (PI) staining flow cytometry was used to assess distribution of cell cycle phases. According to the instructions for the Cell Cycle Analysis Kit (Beyotime, Shanghai, China), cells were transfected with miR-3606-3p mimics and si-TGFBR2 or NC for 72 h in 6-well plates and then harvested and fixed in 70% ethanol at 4°C overnight followed by staining in a mixture of PI/RNase A. Finally, after being kept in the dark at 37°C for 30 min, the staining cells were analyzed using a flow cytometer (FC500 Flow Cytometer; Beckman Coulter, USA). BD Cellquest software was used to calculate the number of cells in each cell cycle phase.
Cell apoptosis assay
Cells were transfected with miR-3606-3p mimics and si-TGFBR2 or NC in 6-well plates. After 72 h, cells were harvested, rinsed with PBS, and suspended in 195 μl 1X binding buffer containing 5 μl Annexin V/FITC and 10 μl propidium iodide (Annexin V/FITC kit; Beyotime). Then cells were incubated at room temperature for 10 ~ 20 min in the dark, and the fluorescence was immediately measured using a flow cytometer.
Statistical analysis
An independent two group-test or one-way Analysis of Variance (ANOVA) with LSD multiple comparison test was used to evaluate significant differences between groups. A Pvalue less than 0.05 was considered significant.
Results
The level of TGFBR2 is increased and the level of miR-3606-3p is decreased in SSc primary dermal fibroblasts and dermal tissues
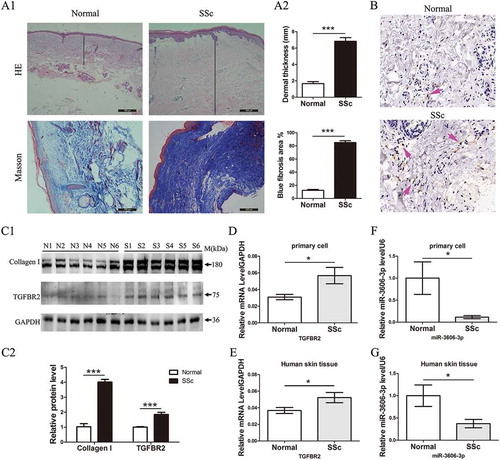
The level of TGFBR2 was detected in SSc skin tissues and primary fibroblasts from skin. H&E and Masson’s staining revealed a thicker dermis (normal vs. SSc: 0.8 ± 0.1 vs. 1.8 ± 0.2 mm, P < 0.05) and more collagen content in SSc skin than controls ()). The expression of S100A4, a widely used systemic sclerosis biomarker, was higher in SSc skin tissues by IHC assay (). Consistently with increased S100A4 level, IHC also revealed that the TGFBR2 level was about two fold higher, quantified by counting the number of TGFBR2 positive cells, in the SSc samples than in the control ()). Western blots analysis directly confirmed the increased expression of type I collagens and TGFBR2 in primary SSc dermal fibroblasts compared to those of normal controls ()). Transcriptional analysis by qRT-PCR further exhibited the higher expression of TGFBR2 in SSc skin samples and fibroblasts (). In contrast, miR-3606-3p, a predicted miRNA of TGFBR2, was downregulated in SSc fibroblasts and tissues (). These findings suggested that the decrease of miR-3606-3p and increase of TGFBR2 might contribute to the deposition of collagen associated with the SSc process.
Figure 1. Increased TGFBR2 and decreased miR-3606-3p levels in SSc primary fibroblasts and dermal tissues. (a1) H&E and Masson’s staining of SSc and normal dermal tissues. (a2) Dermal thickness was calculated by measuring the distance between the dermal-epidermal junction and the derma–subcutaneous fat junction in five randomly selected fields for each skin section. (b) Arrow indicates TGFBR2-positive staining by IHC. (c1) Western blots analysis of type I collagens and TGFBR2 protein expression in six normal and six SSc patient fibroblast cells. GAPDH was used as internal control. (c2)The fold change of type I collagens and TGFBR2 protein expression was calculated by using gray scanning software. (d) qRT-PCR analysis of TGFBR2 mRNA levels in primary fibroblast cells from normal (n = 10) and SSc (n = 13) patients. The relative TGFBR2 mRNA level are expressed after normalization to GAPDH. (e) qRT-PCR analysis of relative TGFBR2 mRNA levels in 20 SSc and 21 normal skin tissues. (f) qRT-PCR analysis of relative miR-3606-3p levels in primary fibroblast cells from normal (n = 10) and SSc (n = 13) patients. Levels of miR-3606-3p are expressed as a relative index normalized to U6. (g) qRT-PCR analysis of relative miR-3606-3p in 20 SSc and 21 normal skin tissues. *P < 0.05; **P < 0.001; ***P < 0.001.
MiR-3606-3p reduces TGFBR2 expression by targeting the TGFBR2 3′-UTR in fibroblasts
Based on the above findings, we next determined whether or not TGFBR2 is the direct target for miR-3606-3p. We predicted the potential targets of miR-3606-3p by using TargetScan software program. This software program predicted the mRNA of TGFBR2, an essential receptor at the early stage of the profibrotic TGF-β signaling pathway, and thus we further investigate how miR-3606-3p influenced the expression of TGFBR2. The seed region of miR-3606-3p all base-paired with the 3′-UTR of TGFBR2 mRNA. Bioinformatics prediction tools identified that the 3′-UTR of TGFBR2 harbors three potential binding sites (positions site1:7258–7265, site2:7329–7335 and site3:8163–8169) for miR-3606-3p, suggesting that TGFBR2 could be a potential target for miR-3606-3p. In order to confirm this, three TGFBR2 3′-UTR wild type-containing binding sites (site 1, site 2 and site 3) of miR-3606-3p were subcloned into the pmirGLO vector ()), and cotransfected with a luciferase construct containing miR-3606-3p mimics into two primary dermal fibroblasts defined as SSc-1 and SSc-2. As illustrated in , the relative luciferase activity was only significantly decreased in the TGFBR2 3′-UTR wild type site 3 group, whereas there was no significant difference in the TGFBR2 3′-UTR wild type site 1 or site 2 group, suggesting that miR-3606-3p may selectively bind to the sequence containing site 3 (8163–8169) of the TGFBR2 3′-UTR. To further confirm the binding site, the analogous clones of the TGFBR2 3′-UTR mutant site 3 were constructed, and the mutant sequence is shown in ). Results of the luciferase assay showed that when compared with a scramble control, the TGFBR2 3′-UTR mutant group did not reveal any significant difference in luciferase activity ()).
Figure 2. miR-3606-3p reduces TGFBR2 expression by directly targeting the TGFBR2 3′-UTR. (a) Schematic graph showing the subcloning of the predicted miR-3606-3p binding sites (positions site1:7258–7265, site2:7329–7335 and site3:8163–8169) of the TGFBR2 3′-UTR into the pmirGLO luciferase construct. The predicted duplex formation between miR-3606-3p and wild type binding sites is indicated. (b,c) Luciferase activities of the construct containing the wild type TGFBR2 3′-UTR reporter gene in two primary fibroblast cell types cotransfected with negative control (NC) or miR-3606-3p. Scrambled sequences were used as NC. Relative Renilla luciferase activity is determined normalization to the firefly luciferase activity. (d) The mutant miR-3606-3p binding site of TGFBR2 3′-UTR. (e) Luciferase activities of the mutant construct in two fibroblast cell types cotransfected with NC or miR-3606-3p. (f) qRT-PCR analysis of miR-3606-3p levels in the two fibroblast cell types transfected with miR-3606-3p mimics or NC for 48 h. U6 was used as internal control. (g) TGFBR2 mRNA expression in the two fibroblast cell types transfected with miR-3606-3p mimics or NC. GAPDH was used as internal control. (h-i) TGFBR2 protein levels in the two fibroblast cell types transfected with miR-3606-3p mimics or NC. All western blots assay were performed in triplicate independent experiment. The repeated bands showed in figures were represented in two individual experiment with similar experimental operation. *P < 0.05; **P < 0.01; ***P < 0.001.
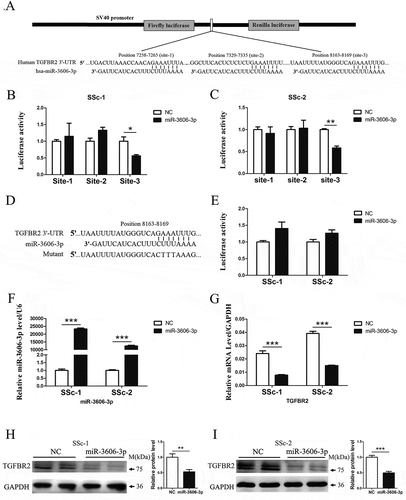
Furthermore, overexpression of miR-3606-3p ()) significantly diminished the level of TGFBR2 mRNA ()) and protein (,i)). Taken together, these results suggested that miR-3606-3p could directly target the 3′-UTR of TGFBR2. We had thus revealed a new target for TGFBR2 that had never before been reported and offered a novel clue in the regulation of diseases with abnormal high expression of TGFBR2.
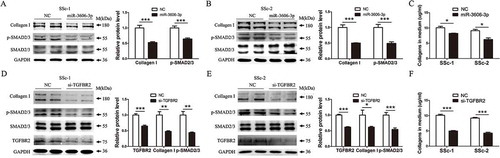
MiR-3606-3p overexpression or TGFBR2 knockdown reduces collagen deposition though the inhibition of canonical SMAD-dependent TGF-β signaling in fibroblast cells
Next, we examined whether or not the repression of TGFBR2 mediated by miR-3606-3p would disrupt TGF-β/SMAD signaling in fibroblasts. Western blots analysis showed that transfection of two types of primary fibroblasts with miR-3606-3p mimics significantly reduced the protein levels of type I collagen and p-SMAD2/3 ( and ). Additionally, the sircol assay revealed an anti-collagen-producing ability attributed to miR-3606-3p, suggesting that there is lower total ECM in the cell medium ( and ). We next set out to further analyze the expression of type I collagen and p-SMAD2/3 in response to TGFBR2 downregulation in the fibroblasts. Consistent with this, data analysis of western blots in siRNA-induced TGFBR2 downregulation revealed similar results to those obtained via miR-3606-3p overexpression in the fibroblasts ( and ). Collectively, our findings demonstrated that miR-3606-3p could serve as a novel anti-fibrotic microRNA and suppress fibroblast collagen deposition at both the cellular and extracellular levels. The anti-fibrotic role of miR-3606-3p in fibroblast cells is mainly via the reduction of TGFBR2 expression and diminishing p-SMAD2/3 phosphorylation.
Figure 3. Overexpression of miR-3606-3p or knockdown of TGFBR2 reduces collagen deposition though inhibition of the canonical SMAD-dependent TGF-β signaling in fibroblast cells. (a,b) Detection of p-SMAD2/3 and type I collagens protein levels in two fibroblast cell types overexpressing miR-3606-3p at 48 h. SMAD2/3 and GAPDH were used as loading controls. Scrambled sequences were used as NC. (c) Detection of extracellular collagens in the medium of miR-3606-3p overexpressed fibroblast cells at 48 h. Scrambled sequences were used as NC. (d,e) Detection of p-SMAD2/3 and type I collagen levels in two fibroblast cell types with TGFBR2 knockdown at 48 h. SMAD2/3 and GAPDH were used as loading controls. Scrambled sequences were used as NC. (f) Detection of extracellular collagens in the medium of TGFBR2-deficienct fibroblast cells at 48 h. Scrambled sequences were used as NC. All western blots assay were performed in triplicate independent experiment. The repeated bands showed in figures were represented in two individual experiment with similar experimental operation.*P < 0.05; **P < 0.01; ***P < 0.001.
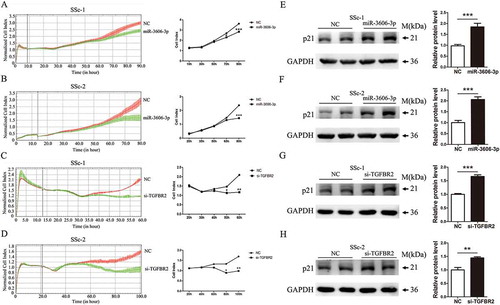
MiR-3606-3p overexpression and TGFBR2 knockdown repress growth of SSc fibroblasts
Considering the notion that TGF-β signaling has an promotional influence on cell proliferation, we were inspired to investigate the role of miR-3606-3p in TGF-β-mediated effects on the growth of fibroblast cells. As illustrated in and , the results of the RTCA assay showed that the cell index of fibroblasts overexpressing miR-3606-3p was significantly lower than that of miR-NC cells, suggesting that miR-3606-3p is a novel microRNA that can exert anti-proliferative effects on SSc cells. Accordingly, to elucidate the effect of TGFBR2 on fibroblastic growth, we first induced knockdown of TGFBR2 using specific siRNA. The RTCA results revealed that the proliferation of TGFBR2 silencing cells was significantly weakened when compared with that of control cells ( and ). We next focused on the p21 gene (cyclin dependent kinase inhibitor 1A, CDKN1A), which has been described to be a critical inhibitor of cell proliferation, and found that overexpression of miR-3606-3p led to an increase in p21, and that knockdown of TGFBR2 also resulted in enhanced p21 expression ( and . These results suggested that miR-3606-3p could suppress TGFBR2-induced growth in dermal fibroblast cells.
Figure 4. Overexpression of miR-3606-3p or knockdown of TGFBR2 represses fibroblasts growth. RTCA assay results of cell proliferation ability in two fibroblast cell types with overexpression of miR-3606-3p (a,b), or knockdown of TGFBR2 (c,d). Cell growth was persistently determined every 10 min for at least 60 h following miR-3606-3p transfection. Images of cell index analysis for cell proliferation in two fibroblast cell types with miR-3606-3p overexpression or TGFBR2 knockdown. Detection of p21 protein levels in the cells described in panel A-D with miR-3606-3p overexpression or TGFBR2 knockdown, NC, negative control (e-h). All western blots assay were performed in triplicate independent experiment. The repeated bands showed in figures were represented in two individual experiment with similar experimental operation.*P < 0.05; **P < 0.01; ***P < 0.001.
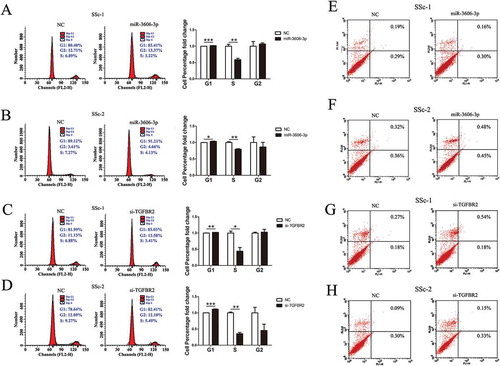
MiR-3606-3p overexpression and TGFBR2 knockdown inhibit cell cycle in SSc fibroblasts
We next aimed to examine the distribution of cell cycle phases and the cell apoptosis effect in miR-3606-3p overexpression and TGFBR2-silenced fibroblasts. We found that fibroblasts transfected with miR-3606-3p mimic or si-TGFBR2 led to a significant reduction in S phase, accompanied by an increase in G1 (, whereas no significant effect on cell apoptosis was observed (. When taken together, these results suggested that miR-3606-3p inhibited cell proliferation and that TGFBR2 promoted cell proliferation by regulating fibroblasts cycle.
Figure 5. Overexpression of miR-3606-3p or knockdown of TGFBR2 inhibited fibroblast cell cycle, but had no effect on cell apoptosis. Flow cytometric analysis of cell cycle progression of two fibroblast cell types transfected with miR-3606-3p (a,b), or si-TGFBR2 (c,d). Cells were harvested at 72 h post-transfection and stained with propidium iodide. Shown in the right of each panel is the fold-change in percentage of different cell cycle phases. Flow cytometric analysis of apoptosis of two fibroblast cell types transfected with miR-3606-3p (e,f), or si-TGFBR2 (g,h), and non-targeting control (NC). Cells were harvested at 72 h post-transfection and stained with Annexin V/FITC and propidium iodide. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
SSc is a typical fibrotic disease that is characterized by thickening and persistent fibrosis of the skin with involvement of multiple internal organs [Citation30]. The TGF-β pathway is a potent pro-fibrotic pathway that plays a critical role in the progression and development of SSc. Studies have shown that TGF-β’s principal influence on fibroblasts is based on effects on proliferation, activation, and accumulation and stimulation of extracellular matrix (ECM) [Citation31]. As we known, upon stimulation, TGFBR2 first binds to TGF-β factor and auto-phosphorylates, then activates TGFBR1, triggering a cascade response that ultimately results in gene regulation of a series of genes involved in fibrosis, such as collagens, α-SMA, CTGF, MMPs, TIMPs, fibronectin, and so on [Citation32–Citation35].
In contrast to the consistently elevated levels of TGFBR1 that have been observed in SSc, the levels of the TGFBR2 constitutive receptor remain largely controversial. So we force on the TGFBR2 alteration and its contribution in SSc. In this study, we observed an upregulation of TGFBR2 in SSc skin tissues and primary fibroblasts. Moreover, knockdown of TGFBR2 decreased p-SMAD2/3 level and inhibited fibroblast cell cycle progression, suggesting the promotion ability of TGFBR2 on TGF-β signaling and cell proliferation. Consistently, TGF-β plays an important role in fibrosis diseases through promoting fibroblast cell cycle progression, growth and differentiation. The hyperactive TGF-β signaling may attribute to higher TGFBR2 level in SSc. So we can conclude that TGFBR2 and the ligand TGF-β exhibit the similar effect on cell cycle progression, at least in fibroblasts. On the contrary, as Zhang et al showed, TGF-β exhibits potent repressive effect on cell cycle, especially in epithelial cells [Citation36]. Resistance to TGF-β-induced growth inhibition is common in NSCLC (non-small-cell lung cancer) epithelail cell lines [Citation37]. Notably, TGFRB2 level was frequently reduced, suggesting TGFBR2 reduction may be an underlying mechanism of refractoriness to TGF-β growth inhibitory signals. Therefore, we can conclude that TGFBR2 and the ligand TGF-β exhibit the similar effect on cell cycle arrest in epithelium. In summary, in spite of the adverse effect of TGF-β signaling on cell cycle and growth in different cell types, the ligand TGF-β and the receptor TGFBR2 exhibit the similar function. This may attribute to TGF-β and TGFBR2 share the common downstream molecular complex of TGF-β signaling. In addition, the reasons for the opposite function of TGF-β signaling in fibroblasts and epithelium need to be further explored.
To date, the powerful gene regulators microRNAs have been implicated in many pathophysiological processes, including cell development, proliferation, apoptosis, and carcinogenesis [Citation20,Citation38,Citation39]. Accumulating evidence has shown that many miRNAs are also critically involved in the pathogenesis of human tissue fibrosis of in several organs [Citation40]. In the development of fibrosis in SSc, the existence of a pro-fibrosis or anti-fibrosis role has been recognized for selected microRNAs that regulate the well-known pro-fibrotic factor, TGF-β, or directly target the predicted fibrotic genes [Citation41].
To explain the epigenetic alternation of TGFBR2 in SSc, we performed an in silico prediction of microRNA targets and identified that miR-3606-3p could potentially bind to three target sites of the TGFBR2 3′-UTR. We first detected the expression level of miR-3606-3p in SSc primary fibroblasts and dermal tissues. Our findings revealed a decrease in miR-3606-3p among SSc patients compared to normal donors, indicating the contrary role of TGFBR2 and miR-3606-3p in SSc pathogenesis. To further explore the association between TGFBR2 and miR-3606-3p, we performed a luciferase reporter assay to determine whether or not TGFBR2 is the directly target of miR-3606-3p. The result showed that miR-3606-3p could selectively target one putative site of the TGFBR2 3′-UTR. Furthermore, our data revealed an important result that overexpression of miR-3606-3p in SSc fibroblasts significantly inhibited TGFBR2 mRNA and protein expression. Collectively, these findings provide strong evidence that, in SSc, miR-3606-3p may function as a novel anti-fibrotic miRNA by directly targeting TGFBR2. Given the essential role of miR-3606-3p in the expression of TGFBR2, a key component of the TGF-β signaling pathway and thus of SSc pathogenesis, we went on to show that transfection of SSc primary fibroblasts with miR-3606-3p mimics or si-TGFBR2 yielded significant cell cycle arrest and proliferation repression. As expected, this is the first evidence supporting the identification of a cytostatic role of miR-3606-3p in SSc fibroblasts, further improving our understanding of the pro-fibrotic function of TGFBR2 [Citation18]. In addition, we cannot exclude the possibility that miR-3606-3p’s activity as a fibrosis suppressor could be through other targets. To better establish the relationship between miR-3606-3p and TGFBR2, the experiment where both miR-3606-3p and TGFBR2 are overexpressed was added. Results showed that no significant differences in the expression levels of TGFBR2, Collagen I and p-SMAD2/3 were observed in the cells co-transfected miRNA-3606-3p mimics and pcDNA-3.1-TGFBR2 plasmid, compare with the negative control (NC and pcDNA-3.1-vector group). These results further indicated that miR-3606-3p inhibited collagen expression and p-SMAD2/3 activity largely depending on regulating TGFBR2 level (). More importantly, owing to the common alteration of TGFBR2 and the consistent pro-fibrotic role of TGFBR2 in other fibrotic diseases, we speculated the possibility that miR-3606-3p might also inhibit fibrosis of other organs.
In conclusion, this is the first report in SSc revealing a downregulation of miR-3606-3p together with an increase in TGFBR2 expression. Mechanistically, miR-3606-3p inhibits TGFBR2 expression via direct targeting of the TGFBR2 3′-UTR, thereby repressing proliferation of SSc fibroblasts. Our study sheds light on the mechanistic interaction of miR-3606-3p and TGFBR2 in SSc progression. The mediation of TGFBR2 downregulation by miR-3606-3p provides new insight into potential therapeutic strategies for SSc.
Declaration of interest
Author declares no conflict of interest.
Supplemental Material
Download Zip (1.4 MB)Supplementary material
Supplementary material can be accessed here.
Additional information
Funding
Related Research Data
References
- LeRoy EC, Black C, Fleischmajer R, et al. Systemic sclerosis: classification, subsets and pathogenesis. J Rheumatol. 1988;15:202–205.
- Kawakami T, Ihn H, Xu W, et al. Increased expression of TGF-beta receptors by scleroderma fibroblasts: evidence for contribution of autocrine TGF-beta signaling to scleroderma phenotype. J Invest Dermatol. 1998;110:47–51.
- Garret SM, Frost DB, Feghali-Bostwick C. The mighty fibroblast and its utility in scleroderma research. J Scleroderma Related Disord. 2017;2:69–134.
- Shreiner AB, Murray C, Denton C, et al. Gastrointestinal manifestations of systemic sclerosis. J Scleroderma Related Disord. 2016;1:247–256.
- Baraut J, Farge D, Jean-Louis F, et al. Transforming growth factor-beta increases interleukin-13 synthesis via GATA-3 transcription factor in T-lymphocytes from patients with systemic sclerosis. Arthritis Res Ther. 2015;17:196.
- Derk CT. Transforming growth factor-beta (TGF-beta) and its role in the pathogenesis of systemic sclerosis: a novel target for therapy? Recent Pat Inflamm Allergy Drug Discov. 2007;1:142–145.
- Wu T, Chu H, Tu W, et al. Dissection of the mechanism of traditional Chinese medical prescription-Yiqihuoxue formula as an effective anti-fibrotic treatment for systemic sclerosis. BMC Complement Altern Med. 2014;14:224.
- La J, Reed E, Chan L, et al. Downregulation of TGF-beta receptor-2 expression and signaling through inhibition of Na/K-ATPase. PloS one. 2016;11:e0168363.
- Phan TT, Lim IJ, Aalami O, et al. Smad3 signalling plays an important role in keloid pathogenesis via epithelial-mesenchymal interactions. J Pathol. 2005;207:232–242.
- Zhang Q, Ye H, Xiang F, et al. miR-18a-5p inhibits sub-pleural pulmonary fibrosis by targeting TGF-beta receptor II. Mol Therapy. 2017;25:728–738.
- Fierro-Fernandez M, Busnadiego O, Sandoval P, et al. miR-9-5p suppresses pro-fibrogenic transformation of fibroblasts and prevents organ fibrosis by targeting NOX4 and TGFBR2. EMBO Rep. 2015;16:1358–1377.
- Li M, Krishnaveni MS, Li C, et al. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest. 2011;121:277–287.
- Miguel V, Busnadiego O, Fierro-Fernandez M, et al. Protective role for miR-9-5p in the fibrogenic transformation of human dermal fibroblasts. Fibrogenesis Tissue Repair. 2016;9:7.
- Meng J, Li L, Zhao Y, et al. MicroRNA-196a/b mitigate renal fibrosis by targeting TGF-beta receptor 2. J Am Soc Nephrology. 2016;27:3006–3021.
- Yu F, Chen B, Fan X, et al. Epigenetically-regulated MicroRNA-9-5p Suppresses the activation of hepatic stellate cells via TGFBR1 and TGFBR2. Cel Physiol Biochem. 2017;43:2242–2252.
- De Oliveira FL, Araujo-Jorge TC, De Souza EM, et al. Oral administration of GW788388, an inhibitor of transforming growth factor beta signaling, prevents heart fibrosis in Chagas disease. PLoS Negl Trop Dis. 2012;6:e1696.
- Asano Y, Ihn H, Yamane K, et al. Impaired Smad7-Smurf-mediated negative regulation of TGF-beta signaling in scleroderma fibroblasts. J Clin Invest. 2004;113:253–264.
- Pannu J, Gore-Hyer E, Yamanaka M, et al. An increased transforming growth factor beta receptor type I: typeII ratio contributes to elevated collagen protein synthesis that is resistant to inhibition via a kinase-deficient transforming growth factor beta receptor type II in scleroderma. Arthritis Rheum. 2004;50:1566–1577.
- Dong C, Zhu S, Wang T, et al. Deficient Smad7 expression: a putative molecular defect in scleroderma. Proceedings of the National Academy of Sciences of the United States of America 2002; 99:3908–3913.
- Shi X, Zhan L, Xiao C, et al. miR-1238 inhibits cell proliferation by targeting LHX2 in non-small cell lung cancer. Oncotarget. 2015;6:19043–19054.
- Makino K, Jinnin M, Kajihara I, et al. Circulating miR-142-3p levels in patients with systemic sclerosis. Clin Exp Dermatol. 2012;37:34–39.
- Makino K, Jinnin M, Hirano A, et al. The downregulation of microRNA let-7a contributes to the excessive expression of type I collagen in systemic and localized scleroderma. J Immunology. 2013;190:3905–3915.
- Honda N, Jinnin M, Kajihara I, et al. TGF-beta-mediated downregulation of microRNA-196a contributes to the constitutive upregulated type I collagen expression in scleroderma dermal fibroblasts. J Immunology. 2012;188:3323–3331.
- Li H, Yang R, Fan X, et al. MicroRNA array analysis of microRNAs related to systemic scleroderma. Rheumatol Int. 2012;32:307–313.
- Honda N, Jinnin M, Kira-Etoh T, et al. miR-150 down-regulation contributes to the constitutive type I collagen overexpression in scleroderma dermal fibroblasts via the induction of integrin beta3. Am J Pathol. 2013;182:206–216.
- Makino K, Jinnin M, Aoi J, et al. Discoidin domain receptor 2-microRNA 196a-mediated negative feedback against excess type I collagen expression is impaired in scleroderma dermal fibroblasts. J Invest Dermatol. 2013;133:110–119.
- Chu H, Wu T, Wu W, et al. Involvement of collagen-binding heat shock protein 47 in scleroderma-associated fibrosis. Protein Cell. 2015;6:589–598.
- Lu J, Liu Q, Wang L, et al. Increased expression of latent TGF-beta-binding protein 4 affects the fibrotic process in scleroderma by TGF-beta/SMAD signaling. Lab Invest. 2017;97:591–601.
- Shirdel EA, Xie W, Mak TW, et al. NAViGaTing the micronome–using multiple microRNA prediction databases to identify signalling pathway-associated microRNAs. PloS one. 2011;6:e17429.
- Peoples C, Medsger TA Jr., Lucas M, et al. Gender differences in systemic sclerosis: relationship to clinical features, serologic status and outcomes. J Scleroderma Related Disord. 2016;1:177–240.
- Pattanaik D, Brown M, Postlethwaite BC, et al. Pathogenesis of systemic sclerosis. Front Immunol. 2015;6:272.
- Chu H, Shi Y, Jiang S, et al. Treatment effects of the traditional Chinese medicine Shenks in bleomycin-induced lung fibrosis through regulation of TGF-beta/Smad3 signaling and oxidative stress. Sci Rep. 2017;7:2252.
- Liu Q, Chu H, Ma Y, et al. Salvianolic acid B attenuates experimental pulmonary fibrosis through inhibition of the TGF-beta signaling pathway. Sci Rep. 2016;6:27610.
- Chu H, Jiang S, Liu Q, et al. Sirtuin1 protects against systemic sclerosis-related pulmonary fibrosis by decreasing proinflammatory and profibrotic processes. Am J Respir Cell Mol Biol. 2018;58:28–39.
- Wang JC, Lai S, Guo X, et al. Attenuation of fibrosis in vitro and in vivo with SPARC siRNA. Arthritis Res Ther. 2010;12:R60.
- Zhang Y, Alexander PB, Wang XF. TGF-beta family signaling in the control of cell proliferation and survival. In: Cold spring harbor perspectives in biology. 2017. p. 9.
- Lei Z, Xu G, Wang L, et al. MiR-142-3p represses TGF-beta-induced growth inhibition through repression of TGFbetaR1 in non-small cell lung cancer. FASEB J. 2014;28:2696–2704.
- Artlett CM, Sassi-Gaha S, Hope JL, et al. Mir-155 is overexpressed in systemic sclerosis fibroblasts and is required for NLRP3 inflammasome-mediated collagen synthesis during fibrosis. Arthritis Res Ther. 2017;19:144.
- Jafarinejad-Farsangi S, Farazmand A, Gharibdoost F, et al. Inhibition of MicroRNA-21 induces apoptosis in dermal fibroblasts of patients with systemic sclerosis. Int J Dermatol. 2016;55:1259–1267.
- Jiang X, Tsitsiou E, Herrick SE, et al. MicroRNAs and the regulation of fibrosis. FEBS J. 2010;277:2015–2021.
- Aslani S, Sobhani S, Gharibdoost F, et al. Epigenetics and pathogenesis of systemic sclerosis; the ins and outs. Hum Immunol. 2018;79:178–187.