
ABSTRACT
Acute lung injury (ALI) is a critical clinical condition with a high mortality rate, characterized with excessive uncontrolled inflammation and apoptosis. Recently, microRNAs (miRNAs) have been found to play crucial roles in the amelioration of various inflammation-induced diseases, including ALI. However, it remains unknown the biological function and regulatory mechanisms of miRNAs in the regulation of inflammation and apoptosis in ALI. The aim of this study is to identify and evaluate the potential role of miRNAs in ALI and reveal the underlying molecular mechanisms of their effects. Here, we analyzed microRNA expression profiles in lung tissues from LPS-challenged mice using miRNA microarray. Because microRNA-27a (miR-27a) was one of the miRNAs being most significantly downregulated, which has an important role in regulation of inflammation, we investigated its function. Overexpression of miR-27a by agomir-27a improved lung injury, as evidenced by the reduced histopathological changes, lung wet/dry (W/D) ratio, lung microvascular permeability and apoptosis in the lung tissues, as well as ameliorative survival of ALI mice. This was accompanied by the alleviating of inflammation, such as the reduced total BALF cell and neutrophil counts, decreased levels of tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-6) interleukin-1β (IL-1β) and myeloperoxidase (MPO) activity in BAL fluid. Toll-like receptor 4 (TLR4), an important regulator of the nuclear factor kappa-B (NF-κB) signaling pathway, was identified as a novel target of miR-27a in RAW264.7 cells. Furthermore, our results showed that LPS stimulation increased the expression of MyD88 and NF-κB p65 (p-p65), but inhibited the expression of inhibitor of nuclear factor-κB-α (IκB-α), suggesting the activation of NF-κB signaling pathway. Further investigations revealed that agomir-miR-27a reversed the promoting effect of LPS on NF-κB signaling pathway. The results here suggested that miR-27a alleviates LPS-induced ALI in mice via reducing inflammation and apoptosis through blocking TLR4/MyD88/NF-κB activation.
Introduction
Acute lung injury (ALI) is a spectrum of pulmonary condition that results in edema, hypoxemia, and respiratory failure [1]. Recent epidemiologic studies reported that ALI remains an important public health problem globally and a major challenge for clinicians [Citation2]. Despite several treatment strategies have been made to improve the functional outcome, the mortality rate of ALI patients remains high [Citation3,Citation4]. Therefore, it is important and urgent to explore the pathogenesis of ALI and find a new therapeutic strategy for the treatment of ALI.
Accumulating evidence suggests that inflammation play a pivotal role in the pathogenesis of ALI [Citation5]. Importantly, inflammation suppression resulted in reduction of the damage severity of ALI [Citation6,Citation7]. Lipopolysaccharide (LPS), a major biologically active component of the Gramnegative bacterial cell wall, has been widely used to induce an ALI model that is similar with pathological features to ALI in humans by triggering excessive inflammatory mediator [Citation8]. For example, Chi et al. showed that Limonene attenuated the pulmonary inflammatory responses induced by LPS in ALI [Citation9]. Jiang et al. found that Geraniol alleviated lung tissue inflammation induced by LPS in a murine model [Citation10]. Accordingly, focusing on the potential targets of inflammatory processes would provide novel treatment strategies for the prevention and treatment of ALI.
MicroRNAs (miRNAs) are small non-coding RNAs with a length of 18–23 nucleotides that serve key roles in post-transcriptional regulation of gene expression [Citation11]. A large number of studies have pinpointed the essential roles of miRNAs in the onset and development of inflammatory lung diseases, including ALI [Citation12,Citation13]. Extensive studies have been carried out to characterize miRNA expression and function in ALI. For example, Fu et al. showed that downregulation of miR-181a protected mice from LPS-induced acute lung injury by targeting Bcl-2 [Citation14]. Tang et al. recently reported that miR-126-5p played a similar role in the LPS-induced ALI mice by down-regulating vascular endothelial growth factor-A (VEGFA) [Citation15]. However, the importance of miRNAs in the pathogenesis of ALI is largely unknown, especially their effect on inflammation. Therefore, an understanding of the role of miRNAs in regulating inflammatory response presents an attractive approach to control ALI disease.
In the present study, we adopted microarray analysis to investigate the expression profiles of miRNAs in lung tissue of ALI mice and picked up a miRNA, miR-27a, as a potential candidate in the regulation of inflammatory progress in ALI. Moreover, we demonstrated that overexpression of miR-27a in vivo attenuated LPS induced ALI through suppressing inflammatory response via regulating TLR4/MyD88/NF-κB pathway. Our findings provide novel research strategies for the molecular therapeutic target of ALI to some extent.
Materials and methods
Animals
BALB/c mice, weighing 18–20 g each, were purchased from Shanghai Slack Laboratory Animal Co, Ltd (Shanghai, China). We confirmed that all animal experiments were carried out in accordance with the guidelines of the Guide for the Care and Use of Laboratory Animals of the Institutional Animal Care and Use Committee (IACUC) set by the Zhongshan Hospital, Fudan University. The protocols were approved by the Animal Care and Use Committee of the Zhongshan Hospital, Fudan University (Shanghai, China). Pentobarbital sodium was used as an anesthetic to minimize pain during all procedures. All animals were housed in plastic cages at 22 ± 2°C with free access to food and water on a 12 h light/dark cycle.
Establishment of ALI model
The mice model of LPS-induced ALI was established as previous reported [Citation13]. Mice were randomly divided into different groups: a control group with intra-tracheal instillation of 1.5 mg/kg normal saline (NS) (n = 6) and an ALI group (n = 6) (LPS group) with intra-tracheal instillation of 3 mg/kg LPS (Escherichia coli 055:B5; Sigma), LPS +agomir-27a (3 mg/kg LPS plus 8 mg/kg agomir-27a) (n = 3) and LPS + agomir-NC (3 mg/kg LPS plus 2 mg/kg agomir-NC) (n = 3). MiR-27a agomirs and miR-27a agomirs negative control were synthesized by RiboBiotech (Ribo-Bio Co., Ltd., China). Agomirs of miR-27 (2 mg/kg) or control agomirs were injected intravenously (tail vein) every third day until the ALI was induced as described above. Twenty-four hours after injection of LPS, mice were humanely killed by overdose of anesthesia. Chest was opened and left lung was clamped. Right lung was lavaged using 0.9% saline. Bronchoalveolar lavage fluid (BALF) was collected and centrifuged for 15 min at 4°C using a cooling centrifuge (4000 rpm). Cell pellet was collected and used for the determination of cell counts. The BALF supernatants were stored at −80°C until further analysis. Small piece of the left lung was weighed and homogenized in 0.1 mol/L phosphate buffer (pH 7.4) in an ice bath. The remaining of left lung was dissected and washed with ice-cold saline then then fixed in 10% neutral buffered formalin for 24 h and submitted for histopathological assessments, western blot assay and terminal deoxynucleotidyl transferase mediated dUTP nick end labeling (TUNEL) staining.
In addition, four mice in control and ALI group were selected to subject to miRNA microarray analysis. The survival experiments were performed in four group mice (n = 10/group) (LPS, Control, LPS + agomir-27a, LPS + agomir-NC). Survival of mice was monitored for 96 h.
Microarray analysis
Total RNA isolation was performed with miRNeasy mini kit (QIAGEN) following the manufacturer’s protocol. RNA concentration and qualification were determined by Nanodrop spectrophotometer (ThermoFisher Scientific, USA). Total RNA was labeled using the miRCURY™ Hy3™/Hy5™ power labeling kit (Exiqon Inc., Woburn, MA, USA) and hybridized on the miRCURY™ LNA Array (v18.0) (Exiqon, Copenhagen, Denmark). After washing, the slides were scanned using an Axon GenePix 4000 B microarray scanner (Axon Instruments, Foster City, CA, USA). After washing and staining, the arrays were scanned in an Agilent G2565BA Microarray Scanner System (Agilent Technologies, Santa Clara, CA, USA). Scanned images were then imported into GenePix Pro 6.0 software (Axon) for grid alignment and data extraction. Replicated miRNAs were averaged, and miRNAs with intensities ≥50 in all samples were used to calculate a normalization factor. Expressed data were normalized by median normalization. After normalization, the miRNAs that were significantly differentially expressed were identified by Volcano Plot filtering. Finally, Tthe expression data were subjected to hierarchical clustering and subsequently depicted in a heat map format using GeneSpring GX, version 7.3 (Agilent Technologies, California, United Stages).
Quantitative real-time PCR analysis
For miRNAs analysis, total RNA was isolated from lung tissues and splenocytes using miRNeasy mini kit (QIAGEN) following the manufacturer’s protocol. Reverse transcription was performed using miRNA specific stem-loop primers (Applied Biosystems, Foster City, CA, USA). The qRT-PCR assays were carried out using SYBR Green Master Mixture (Roche) reagent on a 7500 Fast Real-Time PCR System (Applied Biosystems, USA). U6 was used as an internal control. The primer sequences were as follows: miR-27a, RT: 5ʹ-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGCGGAA-3ʹ Forward: 5ʹ-CGGCGGTTTCACAGTGGCTAAG-3ʹ, Reverse: 5ʹ-CCAGTGCAGGGTCCGAGGTAT-3ʹ; U6 forward: 5ʹ-GCTTCGGCAGCACATATACTAAAAT-3ʹ, reverse: 5ʹCGCTTCACGAATTTGCGTGTCAT-3ʹ. The relative expression of miR-27a was shown as fold difference relative to U6.
BALF collection and analysis
Following euthanasia, mice lungs were lavaged three times with 0.5 mL of ice-cold PBS as previously described [Citation16]. The pellet was re-suspended in PBS and total cells were counted with a hemacytometer. The BALF was centrifuged 1500 rpm for 10 min at 4°C and the supernatant was collected and stored in −80°C freezer until use. The remaining cell pellet was re-suspended in 200 μL of PBS and centrifuged 700 rpm for 5 min. For differential cell counting, cell deposits were then stained using the Diff Quick staining system (International Reagents Corp., Japan). A microscopic cell count was then conducted using an Olympus BX61WI microscope at 400x magnification in which 300 cells were counted on the slide (100 in three separate frames of view) and averaged to 100 cells to deduce the percentage of neutrophils.
Measurement of inflammatory cytokines by enzyme-linked immunosorbent assay (ELISA)
ELISA kits for the detection of IL-1β (Cat no. E-EL-H0149c), IL-6 (Cat no. E-EL-H0102c) and TNF-α (Cat no. E-EL-H0109c) protein levels were obtained from Elabscience Biotechnology Co., Ltd (Wuhan, China). The levels of IL-6, IL-1β, and TNF-α in BALF and cells were analyzed with corresponding ELISA kits according to the manufacturer’s instructions. Each experiment was independently performed three times.
Measurement of wet-to-dry ratio of the lungs
At 24 h after challenge with LPS, mice were euthanized, the right lung was then removed and the wet weight was determined. Subsequently, the lungs were incubated at 60°C for 3 to 4 days to remove all moisture, then the dry weight was measured and the ratio of wet-to-dry weight calculated.
Oxygenation index (PaO2/FiO2) analysis
At 24 h after ALI (or control), mice were anesthetized and given endotracheal intubation with a 18-gauge catheter. The mice were mechanically ventilated with pure oxygen at 7 mL/kg (120 breaths/min). After 20 min-ventilation, the arterial blood was obtained from carotid artery and measured using a commercial blood gas analyzer (model ABL8000; Radiometer Copenhagen, Westlake, Ohio).
Evaluation of lung permeability
Lung permeability was assessed using the Evans blue dye extravasation method, as described previously [Citation17]. In brief, Evans blue dye (Sigma-Aldrich; 30 g/L) was injected (45 mg/kg) into the external jugular vein 30 min before the animals were euthanized. After adequate perfusion with PBS, Evans blue dye was extracted from the lung using formamide for 18 h at 60°C and measured as the absorbance of the supernatant at 620 nm on a microplate reader (BioTek, Winooski, Vermont, United States) and is reported as the amount of EB per wet tissue weight (μg/g).
Histopathological analysis
For assessment of lung injury, the left lungs of mice were excised at 24 h after the LPS challenge, and fixed in 4% (v/v) paraformaldehyde, embedded in paraffin, sectioned at 4-μm thickness, stained with H&E solution (Sigma-Aldrich) and then examined under a microscope. The histopathological changes were scored following a classic lung injury score standard as described by Eveillard, Soltner [Citation18] and Parsey MV [Citation19].
Myeloperoxidase activity assay
Lung tissues were homogenized and the supernatant was subjected to myeloperoxidase activity assay using a commercially available kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Absorbance was determined at 460 nm.
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL) staining
After being deparaffinized with xylene, the sections of lung tissues were rehydrated with ethanol at graded concentrations of 100%–70% (v/v), followed by washing with water. Then, the lung tissues segment was subjected to 100 μL proteinase K (20 μg/ml, Roche) for 15 min at RT, and then washed three times with PBS. TUNEL solution preparation and staining were performed using TUNEL Apoptosis Detection Kit (Alexa Fluor 488) (Roche, Basel, Switzerland). Cell quantification was obtained using an inverted fluorescence microscope (DP73; Olympus) at 400 × magnification. TUNEL-positive cells were counted in three fields of view per section.
Immunohistochemistry
4-μm-thick formalin-fixed, paraffin-embedded serial sections of lung tissues were used for immunohistochemistry analysis of TLR4 expression. The sections were immersed in xylene for 15 minutes before being rehydrated in water by using an ethanol gradient. Then the sections were immersed in citric acid (pH 6.0; DAKO) for 10 minutes, after the samples were cooled to room temperature, the sections were washed with water and PBS buffer for 15 minutes before incubated with 3% H2O2 for 10 minutes. Then the sections were blocked with 5% BSA for 30 minutes before incubated with the primary antibody overnight at 4°C. Color development was performed by using a DAB color development kit (DAKO). Sections were scanned by Pannoramic Scanning Electron Microscope to view the images.
Cell culture
RAW 264.7 cells were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and grown in DMEM/F12 (Abcam, Cambridge, MA) media supplemented with 10% fetal bovine serum (FBS, Gibco, Rockville, MD) and 1% pen-strep at 37°C in a humidified atmosphere with 5% CO2.
Cell transfection
The miR-27a mimics, mimics negative control (mimics NC), miR-27a inhibitor, and inhibitor NC were bought from GenePharma (Shanghai, China). TLR4 expression vector was constructed by inserting overall sequence of TLR4 into the pcDNA 3.1 vector (Invitrogen). Specific small interfering RNAs (siRNAs) for TLR4 were purchased from GenePharma. Cellular transfection was performed by using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the protocol.
Cell apoptosis
After 48 h transfection, RAW 264.7 cells were harvested by ice-cold PBS and stained with FITC-Annexin V and propidium iodide (PI) in binding buffer for 15 min at room temperature in the dark. Then, cell apoptosis was analyzed on a FACScan flow cytometer (FCM; Bechman Coulter, CA).
Luciferase assay
The 3´-UTR of TLR4, with wild-type or mutant (Mut) binding sites for miR-27a, was amplified and cloned into the pGL3 vector (Promega, Madison, WI, USA) to generate the plasmid pGL3-WT-TLR4-3´-UTR or pGL3-Mut-TLR4-3´-UTR, respectively. For the luciferase reporter assay, RAW 264.7 cells were co-transfected with the luciferase reporter vectors and miR-27a mimics, miR-27a inhibitor or corresponding negative control (10 pmol/ml, GenePharma) using Lipofectamine 2000 reagent. The pRL-TK plasmid (Promega, Madison, USA) was used as a normalizing control. After 24 h of incubation, luciferase activity was analyzed using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol.
Western blot
Protein samples from tissues and cells were prepared using addition of radio immunoprecipitation assay buffer containing 1 × protease inhibitory cocktail and phosphatase inhibitors (RIPA buffer, Cell Signaling Technology, Danvers, MA, USA). We extracted nuclear proteins using a nuclear protein extraction kit (Pierce). Briefly, the prepared samples were incubated on ice for 15 min, after which 12.5 µl of 10% Nonidet P-40 was added and the contents were mixed on a vortex and then centrifuged for 1 min at 4°C at 14,000 g. The nuclear pellet was resuspended in 25 µl of ice-cold nuclear extraction buffer (20 mM Hepes, pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF, 2.0 µg/ml leupeptin, 2.0 µg/ml aprotinin, 0.5 mg/ml benzamidine) and incubated on ice for 30 min with intermittent mixing. The tube was centrifuged for 5 min (14,000g) at 4°C, and the supernatant (nuclear extract) was stored at −80°C. Protein quantification was directly measured using a BCA protein assay kit (Thermo Fisher Scientific, Inc.) according to the instructions provided by the manufacturer. Next, proteins (25 µg each sample) were resolved on 10% SDS–PAGE gels, transferred onto PVDF membranes (Millipore, Billerica, MA, USA), as descripted previously. After blocking with Tris-buffered saline and Tween (TBST) containing 5% skim milk, the blots were incubated overnight at 4°C with a primary antibody against TLR-4 (Cat no. 14,358, 1:1,000), p-p65 (Cat no. 3033, 1:1,000), MyD88 (Cat no. 4283, 1:1,000), IκB-α (Cat no. 4814, 1:1,000) or β-actin (Cat no. 4970, 1:2,000), and then incubated with a HRP-conjugated secondary antibody (Cat no. 7074, 1:5,000) at room temperature for 1 h. All antibodies were supplied by Cell Signaling Technology, Inc. (Danvers, MA, USA). A chemiluminescence detection system (Millipore, Billerica, MA, USA) was used for visualization of the results and quantification of the bands was performed using Quantity One software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Data are presented as the means ± standard deviation (SD) of results derived from three independent experiments performed in triplicate. Statistical differences were analyzed using the Student’s t test or one-way analysis of variance (ANOVA) with the Tukey’s test. Kaplan-Meier method was performed to calculate the survival rates, and log-rank test was conducted to compare the survival distributions between four groups. P values <0.05 were considered significant.
Results
miR-27a was downregulated in LPS-induced acute lung injury in mice
As we known, intratracheal administration of LPS has gained wide acceptance as a clinically relevant model of ALI [Citation20,Citation21]. In this study, we first established the animal model as described previously [Citation13]. Compared with the control group, the lung of mice in LPS group showed marked inflammatory alterations characterized by edema, necrosis, and neutrophil infiltration ()). Subsequently, the histopathological changes were scored and the result showed that LPS significantly induced the histological damage ()), which indicated the success of the model establishment. To determine the potential involvement of miRNAs in the pathogenesis of ALI, we used microarray analysis to determine miRNA levels in lung tissues between ALI and control groups. Our data revealed that compared with normal group, 39 miRNAs were downregulated and 20 miRNAs were upregulated in ALI group ()). Among them, we observed that miR-27a was one of the most dysregulated miRNAs, which has been shown to have a protective effect in many injury models [Citation22–Citation24]. Furthermore, miR-27a has been demonstrated to play an important role in regulating inflammatory response [Citation24–Citation26]. However, whether miR-27a plays a similar role in ALI remains unknown.
Figure 1. miR-27a was downregulated in LPS-induced acute lung injury in mice. Groups of mice (n = 10/group) were challenged with 1 mg/kg LPS or PBS for 24 h. Lung tissue samples, splenocytes and the BALF were collected and analyzed. (a) Lung tissues from each experimental group were processed for histological evaluation. Data represent the mean ± SD of three independent experiments. **p < 0.01 vs. control group. (b) Heat map of miRNA profiles represented the significantly regulated miRNAs (n = 3/group). (C-E) MiR-27a expression was validated by qRT-PCR in lung tissues, BALF and splenocytes of mice challenged with LPS (n = 3/group). Data represent the mean ± SD of three independent experiments. **p < 0.01 vs. control group.
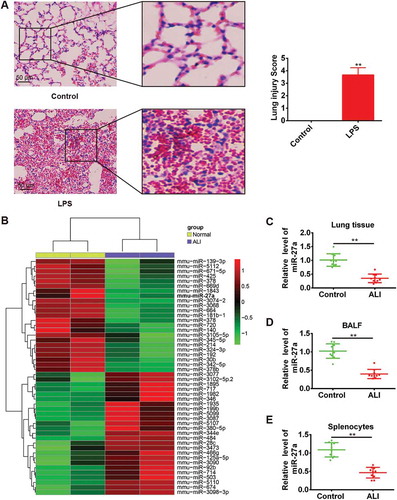
To validate the expression trend of miR-27a in lung tissues obtained from miRNA microarray assay, quantitative real-time-PCR (qRT-PCR) was performed to detect miR-27a in lung tissues from all ALI mice. As shown in ), the expression level of miR-27a in lung tissues was significantly decreased in mice challenged with LPS compared with the control groups. Consistent with our findings from the analysis of miR-27a expression in lung tissues, the expression of miR-27a in BAL fluid ()) and splenocytes ()) was also down-regulated. Given the importance of splenocytes in inflammatory response, we speculate that miR-27a might be involved in the development of ALI.
Increased miR-27a ameliorated LPS induced ALI in mice
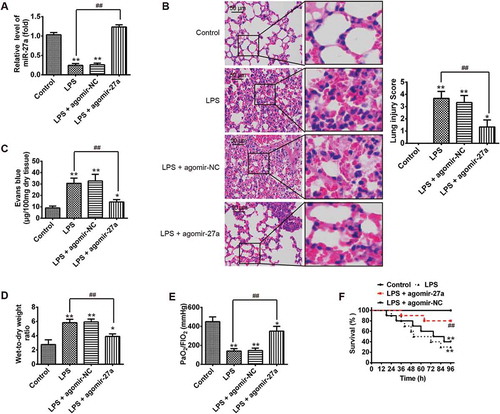
Having verified the expression of miR-27a was down-regulated in LPS-induced ALI mice, then we explored whether raising the expression of miR-27a in vivo alleviates the process of ALI. To confirm our assumption, the mice were given agomiR-27a (2 mg/kg) by tail intravenous injection at 24 h prior to LPS treatment. After LPS administration for 24 h, all animals were euthanized by CO2 asphyxiation. Subsequently, lung tissue samples were collected for analysis. We first detected the miR-27a level in lung tissue samples by qRT-PCR. It was found that the level of miR-27a was significantly increased in lung tissue samples from ALI mice after agomiR-27a injection ()). Pathological analysis indicated that agomir-27a really relieved the severity and distribution of lung lesions compared with the LPS plus agomir-NC group ()). Lung microvascular permeability was assessed by EB extravasation. As shown in ), compared with the control group, LPS caused a marked increase in EB extravasation, whereas injected with agomir-27a effectively inhibited LPS induced the increase in EB extravasation. The lung wet-to-dry (W/D) ratio and oxygenation index (PaO2/FiO2) were evaluated to indicate the pulmonary edema. 24 hours after LPS challenge, the lung W/D ratio increased significantly, while the PaO2/FiO2 decreased in mice with ALI. However, administration of agomir-27a dramatically decreased W/D ratio and increased PaO2/FiO2 (). Furthermore, results in ) demonstrated that mice survival rate in LPS + agomir-27a group was significantly longer than LPS group. Collectively, all data suggest that miR-27a is able to alleviate the development and pathological process of ALI in vivo.
Figure 2. Increased miR-27a ameliorated LPS induced ALI in mice. Groups of mice were given agomir-27a or agomir NC (2 mg/kg) by tail intravenous injection 24 h prior to 1 mg/kg LPS treatment. The mice were sacrificed after LPS administration for 24 h and then lung tissues were collected for analysis. (a) The miR-27a level in lung tissue samples were measured by qRT-PCR (n = 3/group). (b) Lung tissues from each experimental group were processed for histological evaluation (n = 3/group). (c, d) The Evans blue content and lung wet/dry assay (n = 3/group). (e) Oxygenation index (PaO2/FIO2) were determined (n = 3/group). Data represent the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 vs. control group. ##p < 0.01 vs. LPS alone group. (f) The survival rates were observed during 96 h after exposure to LPS (n = 10/group). **p < 0.01 vs. control group. ##p < 0.01 vs. LPS alone group.
Enforced expression of miR-27a attenuated the LPS-induced inflammation response
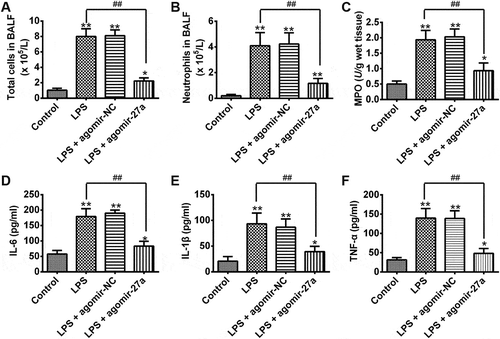
Given the importance of miR-27a in inflammatory response, we sought to determine the effects of enforced expression of miR-27a on LPS-induced inflammatory response. First, we detected the total cell and neutrophil counts in BALF from ALI mice injected with agomir-27a. As shown in ), B, LPS significantly increased the total inflammatory cell and neutrophil counts in the BALF compared with that in control group. However, upregulation of miR-27a significantly suppressed the increased total inflammatory cell and neutrophil counts induced by LPS. We also measured the myeloperoxidase (MPO) activity which is commonly used to assess the quantification of neutrophil accumulation in tissues [Citation17]. The results showed that LPS significantly increased the MPO activity in the lung tissues compared with that in the control group. Agomir-27a treatment obviously inhibited LPS induced MPO activity ()). To further assess the anti-inflammatory effects of miR-27a, we detected the levels of pro-inflammatory cytokines in BAL fluid. We found that LPS significantly increased the levels of pro-inflammatory cytokines including IL-6, IL-1β and TNF-α in the BAL fluid compared with that in the control group, whereas agomir-27a treatment obviously inhibited the levels of pro-inflammatory cytokines induced by LPS (). These results indicate that enforced expression of miR-27a has a protective effect on ALI through suppressing inflammatory response.
Figure 3. Overexpression of miR-27a attenuated LPS-induced inflammatory response. Groups of mice were given agomir-27a or agomir NC (2 mg/kg) by tail intravenous injection 24 h prior to 1 mg/kg LPS treatment. The mice were sacrificed after LPS administration for 24 h, and then the BALF were collected for analysis. (a,b) The total counts of cells and neutrophils from the BALF were counted using a hemocytometer (n = 3/group). (c) MPO activity in lung tissues was measured at 24 h after LPS challenge (n = 3/group). (d-f) IL-6, IL-1β and TNF-α levels in BALF were measured at 24 h after LPS challenge (n = 3/group). Data represent the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 vs. control group. ##p < 0.01 vs. LPS alone group.
Overexpression of miR-27a inhibited the apoptosis in lps-induced ALI mice
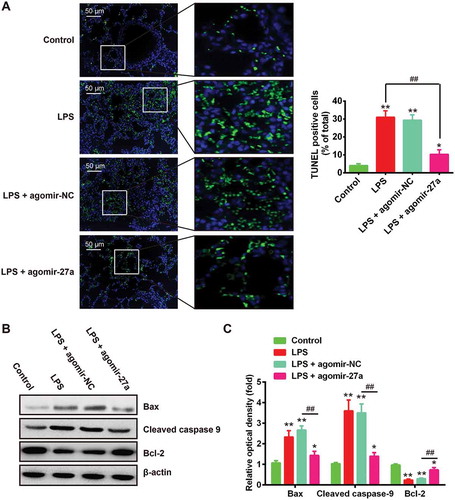
Apoptosis is another key pathologic feature of ALI [Citation27]. To determine whether miR-27a inhibited the apoptosis in LPS-induced ALI mice, we quantified the number of apoptotic events in the lungs of ALI mice after agomir-27a treatment by TUNEL staining assay. As expected, LPS significantly increased the numbers of TUNEL-positive cells compared with that in the control group, whereas the TUNEL-positive cells induced by LPS were obviously inhibited after agomir-27a injection ()). Previous studies have shown the critical role of apoptosis associated proteins Bcl-2 and Bax during the development of ALI [Citation28]. Thus, we detected the expressions of Bcl-2 and Bax, as well as cleaved-capase-9. The results on this study demonstrated that the expression of Bax and cleaved-capase-9 was significantly increased in LPS alone group compared with control group, while the levels of Bcl-2 protein was markedly reduced compared with the control group. However, treatment with agomir-27a significantly inhibited the expression of Bax and cleaved-capase-9 induced by LPS and reversed the inhibitory effect of LPS on the expression of Bcl-2 (). Taken together, these data suggest that enhanced expression of miR-27a attenuates LPS-induced apoptosis in ALI mice.
Figure 4. Overexpression of miR-27a inhibited LPS-induced apoptosis. Groups of mice were given agomir-27a or agomir NC (2 mg/kg) by tail intravenous injection 24 h prior to 1 mg/kg LPS treatment. The mice were sacrificed after LPS administration for 24 h, and then the BALF were collected for analysis. (a) Apoptotic cells in the tissues were observed and the number of TUNEL positive cells was counted in 5 to 10 fields for each slide (n = 3/group). (b) The protein expression levels of Bcl-2, Bax and cleaved-caspase 9 were detected by Western Blot (n = 3/group). Data represent the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 vs. control group. ##p < 0.01 vs. LPS alone group. (C) The bands were semi-quantitatively analyzed by using Image J software, normalized to β-actin density. Data represent the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 vs. control group. ##p < 0.01 vs. LPS + agomir-NC group.
TLR4 was a direct target of miR-27a
To uncover the molecular mechanism by which miR-27a performs a suppressive role in ALI progression, miRanda and Targetscan were used to predicate the potential targets of the miR-27a. Toll-like receptor 4 (TLR4) was considered as one of the candidates after analysis. Previous studies showed that TLR4 mediates microbial infection of immune and inflammatory responses and is involved in the pathogenesis of ALI [Citation29]. Furthermore, it has been reported that miR-27a attenuated the inflammatory response during microglial activation by targeting TLR4 [Citation26]. Thus, TLR4 was selected for further analysis. As suggested in , the complementary sequence of miR-27a was found in the 3´-UTR of TLR4 mRNA. To experimentally validate whether TLR4 was a direct target of miR-27a, we constructed the dual-luciferase reporter system containing the 3´-UTR of TLR4 along with the putative miR-27a binding sites. Luciferase reporter gene assay showed that the activity of luciferase was remarkably decreased after co-transfection with WT constructive luciferase reporter plasmid harboring the TLR4 3´UTR and miR-27a mimics. By contrast, the activity of luciferase was notably increased following co-transfection with WT constructive luciferase reporter plasmid and miR-27a inhibitor. However, these effects were abrogated when the TLR4 sequence was mutated ()). To further identify the correlation between the miR-27a levels and the TLR4 protein, the protein levels in RAW 264.7 cells were examined by Western blot. The expression of TLR4 at protein level was significantly downregulated after overexpression of miR-27a, whereas upregulated after knockdown of miR-27a in RAW 264.7 cells ()). We also assessed the effect of agomir-27a on the expression of TLR4 protein in vivo by Western Blot. As shown in ), we found that LPS significantly increased the expression level of TLR4 compared with control group, whereas agomir-27a treatment attenuated the promoting effect of LPS on the expression of TLR4 ()). In addition, the expression of TLR4 in lung tissues was tested by immunohistochemistry. Consistently, LPS significantly increased the expression level of TLR4 compared with control group, whereas injectiond with agomir-27a effectively inhibited LPS induced the expression of TLR4 ()). All above findings indicated that miR-27a may exert anti-inflammatory effect through suppressing the expression of TLR4.
Figure 5. TLR4 was a direct target of miR-27a. (a, b) The putative binding site of miR-27a and TLR4 is shown. (c) Luciferase assay of HEK293 cells co-transfected with firefly luciferase constructs containing the TLR4 wild-type or mutated 3´-UTRs and miR-27a mimics, mimics NC, miR-27a inhibitor or inhibitor NC, as indicated (n = 3). Data represent the mean ± SD of three independent experiments. **p < 0.01 vs mimics NC, ## p < 0.01 vs inhibitor NC. (d) The expression of TLR4 protein after transfection with miR-27a mimic or miR-27a inhibitor was measured by Western Blot. Data represent the mean ± SD of three independent experiments. **p < 0.01 vs. inhibitor NC. ##p < 0.01 vs. mimics NC. (e) Expression of TLR4 was measured using Western Blot in lung tissues from ALI mice injection with agomir-27a or agomir NC (n = 3/group). Data represent the mean ± SD of three independent experiments. **p < 0.01 vs. control group. ##p < 0.01 vs. LPS + agomir-NC group. (f) Expression of TLR4 was measured using IHC in lung tissues from ALI mice injection with agomir-27a or agomir NC (n = 3).
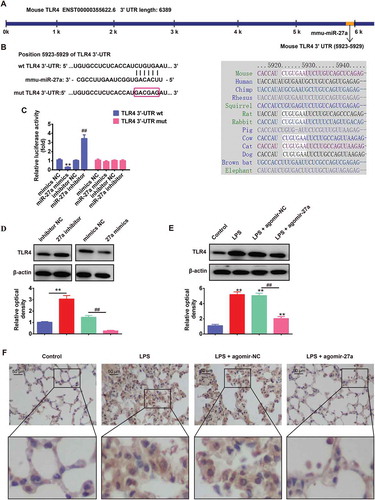
miR-27a regulates the inflammatory response and apoptosis in LPS treated RAW 264.7 cells through targeting TLR4
To confirm the role of TLR4 in the observed protective effects of miR-27a overexpression on ALI, we overexpressed the expression of TLR4 in miR-27a mimics transfected RAW 264.7 cells, and then treated with LPS for 6 h, followed by the assessment of cell apoptosis and pro-inflammatory cytokines (IL-1β, TNF-α and IL-6). As shown in ), overexpression of miR-27a markedly suppressed the expression of IL-1β, TNF-α and IL-6 induced by LPS, whereas these inhibitory effects was attenuated when TLR4 was overexpressed. Moreover, overexpression of miR-27a reduced LPS induced cell apoptosis, while the inhibitory effect was attenuated when TLR4 was overexpressed ()). In contrast, silencing TLR4 could mimic the effect of miR-27a on LPS-stimulated RAW 264.7 cells and resulted in decreased secretion of IL-1β, TNF-α and IL-6 and cell apoptosis (–h)). Collectively, these results revealed that miR-27a overexpression attenuated LPS-induced cell apoptosis and inflammation through downregulating of TLR4.
Figure 6. miR-27a regulates the inflammatory response and apoptosis in LPS treated RAW 264.7 cells through targeting TLR4. RAW 264.7 cells were co-transfected with pcDNA-TLR4 and miR-27a mimics or treated with si-TLR4 alone, and then treated with LPS for 6 h, followed by the assessment of cell apoptosis and inflammatory response. (a-c, e-g) IL-6, IL-1β and TNF-α levels were measured using ELISA assay (n = 3). (d, h) Cell apoptosis was determined by flow cytometry. Data represent the mean ± SD of three independent experiments. *p < 0.05, **p 0. 0.01 vs. control group. ##p * 0.01 vs. LPS alone group.
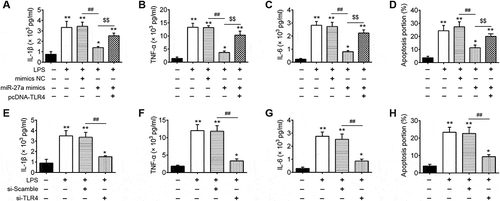
Overexpression of miR-27a blocked TLR4/MyD88/NF-κB pathway in LPS-induced ALI mice
As the TLR4/MyD88/NF-κB pathway is a key regulator involved in inflammatory process [Citation30,Citation31], we further explored the regulation of miR-27a on the activation of the TLR4/MyD88/NF-κB pathway in lung tissue. We extracted nuclear proteins from lung tissues of different groups and subjected them to western blot for detection of the NF-κB pathway transcription factor nuclear p-p65 (one important NF-κB subunit for its activation) [Citation32], myeloid differentiation factor 88 (MyD88) and IκB-α. As shown in , the protein expression of MyD88 and nuclear p-p65 was significantly increased, while the expression of IκB-α was decreased in the LPS group compared with that in the control group. In contrast, when agomir-27a was applied (agomir-27a + LPS group), it was observed that the expression of MyD88 and nuclear p-p65 became lower, while the expression of IκB-α was significantly increased. Consistent with the results in Western Blot, immunohistochemistry staining exhibited that LPS promoted the expression of p-p65, whereas the increased expression of p-p65 was significantly decreased after agomir-27a treatment ()). These results highlighted that miR-27a suppressed LPS-induced inflammatory response by attenuating the release of pro-inflammatory cytokines through the inhibition of TLR4/MyD88/NF-κB activation ().
Figure 7. Overexpression of miR-27a blocked TLR4/NF-κB pathway in LPS-induced ALI mice. Groups of mice were given agomir-27a or agomir NC (2 mg/kg) by tail intravenous injection 24 h prior to 1 mg/kg LPS treatment. The mice were sacrificed after LPS administration for 24 h, and then the BALF were collected for analysis. (a) The levels of MyD88, nuclear p-p65 and p-IκB-α were measured by Western Blot (n = 3). (b) The bands were semi-quantitatively analyzed by using Image J software, normalized to β-actin density. Data represent the mean ± SD of three independent experiments. **p < 0.01 vs. control group. ##p < 0.01 vs. LPS + agomir-NC group. (c) Expression of nuclear p-p65 was measured using IHC in lung tissues from ALI mice injection with agomir-27a or agomir NC.
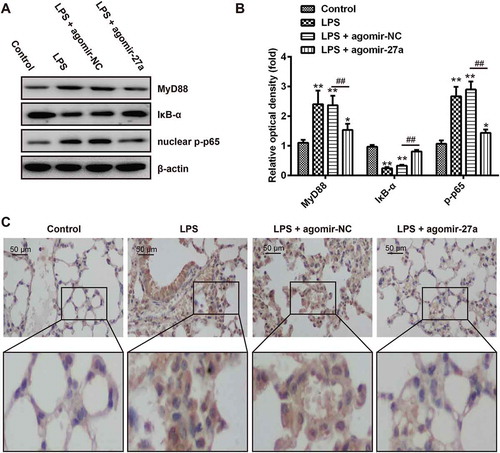
Figure 8. Scheme summarizing the protective effects of miR-27a on LPS-induced acute lung injury via the inhibiting TLR4/MyD88/NF-κB activation. LPS can induce NF-κB activation via TLR4-MyD88 signaling, IκBα acts as an inhibitor of NF-κB, Once the pathway is activated and IκBα is degraded, the NF-κB subunit p65 translocates from the cytoplasm to nucleus, which triggers the transcription of target genes, including TNF-α, IL-1β, and IL-6, and thus regulates inflammatory responses. However, miR-27a attenuates the release of pro-inflammatory cytokines by inhibiting TLR4/MyD88/NF-κB activation.
Discussion
In the present study, we reported the beneficial effects of miR-27a overexpression against LPS-induced ALI in mice, which could be explained in part by its properties of anti-inflammation and anti-apoptosis. Furthermore, we demonstrated that miR-27a suppressed the inflammatory response through the inhibition of TLR4/MyD88/NF-κB activation. Thus, miR-27a may be a potential therapeutic target for ALI.
Recent studies demonstrated that miRNAs function as gene expression switches in key processes of the ALI [Citation33,Citation34]. For example, Wu et al. showed that miR-326 targeting the BCL2A1 gene activated the NF-κB signaling pathway, resulting in aggravated inflammatory response and lung injury of septic shock with ALI in mice [Citation35]. Shi et al. found that miR-21 inhibitor could aggravate the injury of lung tissue in hyperoxia-induced ALI rats [Citation36]. Zhao et al. found that the expression of miR-7 increased significantly in lung tissue in murine LPS-induced ALI model and miR-7 deficiency could significantly attenuate the pathologies of ALI [Citation37]. Several studies have reported the role of miRNAs as biomarkers of ALI, and their potential use as therapeutic targets for this condition [Citation38–Citation41]. Microarray data from various experimental models reveal that ALI induces aberrant expression of miRNAs and the potential targets for these miRNAs that are associated with many pathophysiological processes such as inflammation and apoptosis [Citation42,Citation43]. In the present, by using miRNA microarray analyses, we found a large set of miRNAs were significantly deregulated in injured lung tissues compared with normal group, and the expression of miR-27a was further confirmed by qRT-PCR analysis.
It should be noted that several other studies have demonstrated aberrant expression of miR-27a in many injury models. For example, Boris et al. found that miR-27a was decreased in the brain tissue suffered from traumatic brain injury (TBI), and administration of miR-27a mimics attenuated neuronal apoptosis via inhibition of pro-apoptotic Bcl-2 protein [Citation44]. Similarly, Sun et al. showed that overexpression of miR-27a protected against brain injury via suppressing FoxO3a-mediated neuronal autophagy following TBI [Citation45]. Cai et al. reported that miR-27a protected hippocampal neurons against oxygen-glucose deprivation-induced injury [Citation22]. However, the role of miR-27a in ALI is unclear. In this study, we found that overexpression of miR-27a alleviated lung injury after ALI, as evidenced by the reduced histopathological changes, lung wet/dry (W/D) ratio, lung microvascular permeability and apoptosis in the lung tissues, and increased oxygenation index and survival of ALI mice. However, the molecular mechanism of miR-27a mediated improvement in ALI is unclear.
Recent studies reported that inflammatory response played important roles in initiation and maintenance of ALI [Citation46,Citation47]. Pro-inflammatory cytokines such as TNF-α and IL-1β increase the permeability of pulmonary epithelium, furthermore induced lung tissue damage and accumulation of neutrophils which lead to lung edema [Citation31]. IL-6 is increased in the BALF of patients and higher levels increase mortality, which is identified as one of the biomarkers in monitoring ALI [Citation48]. In addition, MPO activity is a sensitive and specific marker for ALI that is used to assess the quantification of neutrophil accumulation in tissues [Citation49]. Notably, miR-27a has been reported to be associated with inflammatory mediator production [Citation26], which promoted us to verify whether miR-27a also attenuates LPS-induced ALI through suppressing the inflammatory response. As expected, the present study showed that overexpression of miR-27a significantly reduced the numbers of total cells in BALF and the MPO activity in lung tissues compared with the LPS group. In addition, we also found that overexpression of miR-27a effectively reduced TNF-α, IL-6 and IL-1β secretion in BALF. These results suggest that the improvement of ALI by miR-27a in the lung is associated with its inhibition of inflammatory response.
TLR4 is a sensory receptor for LPS and has a very important role in the up-regulation of inflammation and the release of inflammatory cytokines in animal models and also in in vitro cellular models of ALI [Citation50]. For example, disruption of the TLR4 inhibits lung inflammatory responses associated with ALI, confirming the role of TLR receptor signaling in inflammation associated lung damage [Citation30,Citation50–Citation53]. In addition, neutrophil accumulation in the lung, which is a characteristic feature of ALI, is attenuated in TLR4 mutant mice, indicating the involvement of TLR4 signaling in lung inflammatory responses [Citation54,Citation55]. Interestingly, we noticed that the expression of TLR4 had also been reported influencing by miR-27a. For example, Lv et al. demonstrated that miR-27a is associated with microglial activation and the inflammatory response by targeting TLR4 in lipopolysaccharide (LPS)-induced cell model [Citation26]. Another study showed that increasing expression of miR-27a ameliorates inflammatory damage to the blood-spinal cord barrier after spinal cord ischemia: reperfusion injury in rats by downregulating TLR4/NF-κB/IL-1βsignaling pathway [Citation24,Citation56,Citation57]. Therefore, we determined whether miR-27a exerts effective protection in modulating LPS-induced ALI through targeting TLR4. Our results showed that TLR4 was a potential target of miR-27a and miR-27a negatively regulated the protein level of TLR4 by targeting its 3´-UTR in vitro and in vivo. Moreover, overexpression of TLR4 reversed the inhibitory effects of miR-27a overexpression on inflammatory response and apoptosis. In addition, we performed western blot analysis to evaluate the expression of pivotal protein involved in the TLR4/NF-κB signaling pathway. The results showed that agomiR-27a downregulated the levels of MyD88 and nuclear p-p65 and upregulated the levels of IκB-α induced by LPS. These indicate that miR-27a exerts its effects on inflammatory response in ALI by modulating TLR4/MyD88/NF-κB pathway.
A variety of studies have shown that, in the development and manifestations of ALI, apoptosis is thought to be closely related to the severity of ALI [Citation58,Citation59]. For example, extensive apoptosis of pulmonary alveolar type II epithelial cells has been shown to be responsible for the impairment of the epithelial barrier function and the remodeling of certain mesenchymal cells in ALI [Citation60]. In addition, uncontrolled activation of apoptosis pathway result in inflammation and causes destruction of lung tissues, suggesting that inhibition of apoptosis maybe a promising therapeutic strategy for ALI [Citation61]. In this study, we found that overexpression of miR-27a significantly decreased the apoptotic index (number of TUNEL-positive cells) in ALI mice. It has been found that intrinsic and extrinsic pathways are involved in the apoptosis [Citation62,Citation63]. In our study, we found that overexpression of miR-27a significantly upregulated expression of anti-apoptotic protein Bcl-2 and downregulated expressions of pro-apoptotic proteins Bax and cleaved-caspase 9. These results implied that miR-27a has a protection effect in ALI by inhibiting apoptosis.
In conclusion, the present study proved the level of miR-27a was significantly down-regulated in lung tissues of ALI mice and overexpression of miR-27a protected against ALI through a mechanism involving suppressing inflammation via modulating TLR4/MyD88/NF-κB pathway (). Our findings provide a previously unknown role of miR-27a in the pathology of ALI, which could aid the development of new therapeutic strategies against ALI.
Disclosure statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this Article.
Additional information
Funding
Notes on contributors
Zhe Luo
Zhe Luo and Jing Cang was responsiblefor the main conceive of the study and the draft of the manuscript. MinJie Ju, BoFei Liu, HongYu He, ZhunYong Gu, YiMei Liu, Ying Su and DuMing Zhu helped to design the study and performed the statistical analysis. MinJie Ju, BoFei Liu and HongYu He helped to revise the manuscript and participated in its design. All authors have read and approved the final manuscript
References
- Mendez JL, Hubmayr RD. New insights into the pathology of acute respiratory failure. Curr Opin Crit Care. 2005;11:29–36.
- Bellani G, Laffey JG, Pham T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. Jama. 2016;315:788–800.
- Stapleton RD, Wang BM, Hudson LD, et al. Causes and timing of death in patients with ARDS. Chest. 2005;128:525–532.
- Singh N. Acute lung injury and acute respiratory distress syndrome. Lancet. 2007;370: 383–384. author reply 4–5.
- Su ZQ, Mo ZZ, Liao JB, et al. Usnic acid protects LPS-induced acute lung injury in mice through attenuating inflammatory responses and oxidative stress. Int Immunopharmacol. 2014;22:371–378.
- Herold S, Gabrielli NM, Vadasz I. Novel concepts of acute lung injury and alveolar-capillary barrier dysfunction. Am J Physiol Lung Cellular Mol Physiol. 2013;305:L665–81.
- Dengler V, Downey GP, Tuder RM, et al. Neutrophil intercellular communication in acute lung injury. Emerging Roles of Microparticles and Gap Junctions. Am J Respir Cell Mol Biol 2013;49:1–5.
- Lee WL, Downey GP. Neutrophil activation and acute lung injury. Curr Opin Crit Care. 2001;7:1–7.
- Chi G, Wei M, Xie X, et al. Suppression of MAPK and NF-kappaB pathways by limonene contributes to attenuation of lipopolysaccharide-induced inflammatory responses in acute lung injury. Inflammation. 2013;36:501–511.
- Jiang K, Zhang T, Yin N, et al. Geraniol alleviates LPS-induced acute lung injury in mice via inhibiting inflammation and apoptosis. Oncotarget. 2017;8:71038–71053.
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297.
- Song L, Zhou F, Cheng L, et al. MicroRNA-34a suppresses autophagy in alveolar type II epithelial cells in acute lung injury by inhibiting FoxO3 expression. Inflammation. 2017;40:927–936.
- Fang Y, Gao F, Hao J, et al. microRNA-1246 mediates lipopolysaccharide-induced pulmonary endothelial cell apoptosis and acute lung injury by targeting angiotensin-converting enzyme 2. Am J Transl Res. 2017;9:1287–1296.
- Fu X, Zeng L, Liu Z, et al. MicroRNA-206 regulates the secretion of inflammatory cytokines and MMP9 expression by targeting TIMP3 in mycobacterium tuberculosis-infected THP-1 human macrophages. Biochem Biophys Res Commun. 2016;477:167–173.
- Tang R, Pei L, Bai T, et al. Down-regulation of microRNA-126-5p contributes to overexpression of VEGFA in lipopolysaccharide-induced acute lung injury. Biotechnol Lett. 2016;38:1277–1284.
- Fang L, Gao Y, Liu F, et al. Shuang-huang-lian attenuates lipopolysaccharide-induced acute lung injury in mice involving anti-inflammatory and antioxidative activities. Evidence-Based Complementary and Alternative Medicine: eCAM. 2015;2015:283939.
- Duan Y, Learoyd J, Meliton AY, et al. Inhibition of Pyk2 blocks lung inflammation and injury in a mouse model of acute lung injury. Respir Res. 2012;13:4.
- Eveillard M, Soltner C, Kempf M, et al. The virulence variability of different acinetobacter baumannii strains in experimental pneumonia. J Infect. 2010;60:154–161.
- Parsey MV, Tuder RM, Abraham E. Neutrophils are major contributors to intraparenchymal lung IL-1 beta expression after hemorrhage and endotoxemia. J Immunology. 1998;160:1007–1013.
- Tianzhu Z, Shumin W. Esculin Inhibits the Inflammation of LPS-induced acute lung injury in mice via regulation of TLR/NF-kappaB pathways. Inflammation. 2015;38:1529–1536.
- Mei SH, McCarter SD, Deng Y, et al. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Medicine. 2007;4:e269.
- Cai Q, Wang T, Yang WJ, et al. Protective mechanisms of microRNA-27a against oxygen-glucose deprivation-induced injuries in hippocampal neurons. Neural Regener Res. 2016;11:1285–1292.
- Xue WL, Bai X, Zhang L. rhTNFR:Fc increases Nrf2 expression via miR-27a mediation to protect myocardium against sepsis injury. Biochem Biophys Res Commun. 2015;464:855–861.
- Sun C, Liu Z, Li S, et al. Down-regulation of c-Met and Bcl2 by microRNA-206, activates apoptosis, and inhibits tumor cell proliferation, migration and colony formation. Oncotarget. 2015;6:25533–25574.
- Cheng Y, Du L, Jiao H, et al. Mmu-miR-27a-5p-dependent upregulation of MCPIP1 Inhibits the inflammatory response in LPS-Induced RAW264.7 macrophage cells. BioMed Res Int. 2015;2015:607692.
- Lv YN, Ou-Yang AJ, Fu LS. MicroRNA-27a negatively modulates the inflammatory response in lipopolysaccharide-stimulated microglia by targeting TLR4 and IRAK4. Cell Mol Neurobiol. 2017;37:195–210.
- Sweeney RM, Griffiths M, McAuley D. Treatment of acute lung injury: current and emerging pharmacological therapies. Semin Respir Crit Care Med. 2013;34:487–498.
- Bao YP, Yi Y, Peng LL, et al. Roles of microRNA-206 in osteosarcoma pathogenesis and progression. Asian Pacific J Cancer Prev. 2013;14:3751–3755.
- Fan HY, Qi D, Yu C, et al. Paeonol protects endotoxin-induced acute kidney injury: potential mechanism of inhibiting TLR4-NF-kappaB signal pathway. Oncotarget. 2016;7:39497–39510.
- Yao H, Sun Y, Song S, et al. Protective effects of dioscin against lipopolysaccharide-induced acute lung injury through inhibition of oxidative stress and inflammation. Front Pharmacol. 2017;8:120.
- Zhao G, Zhang T, Ma X, et al. Oridonin attenuates the release of pro-inflammatory cytokines in lipopolysaccharide-induced RAW264.7 cells and acute lung injury. Oncotarget. 2017;8:68153–68164.
- Schmitz ML, Baeuerle PA. The p65 subunit is responsible for the strong transcription activating potential of NF-kappa B. EMBO J. 1991;10:3805–3817.
- Li W, Qiu X, Jiang H, et al. Downregulation of miR-181a protects mice from LPS-induced acute lung injury by targeting Bcl-2. Biomed Pharmacother. 2016;84:1375–1382.
- Guo Z, Gu Y, Wang C, et al. Enforced expression of miR-125b attenuates LPS-induced acute lung injury. Immunol Lett. 2014;162:18–26.
- Wu CT, Huang Y, Pei ZY, et al. MicroRNA-326 aggravates acute lung injury in septic shock by mediating the NF-kappaB signaling pathway. Int J Biochem Cell Biol. 2018;101:1–11.
- Shi L, He Y, Bai B, et al. Effects of microRNA-21 inhibitor on apoptosis of type II alveolar epithelial cells in rats with hyperoxia-induced acute lung injury. Zhonghua wei zhong bing ji jiu yi xue. 2017;29:244–248.
- Zhao J, Chen C, Guo M, et al. MicroRNA-7 deficiency ameliorates the pathologies of acute lung injury through elevating KLF4. Front Immunol. 2016;7:389.
- Zhou T, Garcia JG, Zhang W. Integrating microRNAs into a system biology approach to acute lung injury. Transl Res. 2011;157:180–190.
- Rajasekaran S, Pattarayan D, Rajaguru P, et al. MicroRNA regulation of acute lung injury and acute respiratory distress syndrome. J Cell Physiol. 2016;231:2097–2106.
- Bhargava M, Wendt CH. Biomarkers in acute lung injury. Transl Res. 2012;159:205–217.
- Ferruelo A, Penuelas O, Lorente JA. MicroRNAs as biomarkers of acute lung injury. Ann Transl Med. 2018;6:34.
- Neudecker V, Brodsky KS, Clambey ET, et al. Neutrophil transfer of miR-223 to lung epithelial cells dampens acute lung injury in mice. Sci Transl Med. 2017;9:eaah5360.
- Zhao X, Wang X, Wang F, et al. Poly r(C) binding protein 1-mediated regulation of microRNA expression underlies post-sevoflurane amelioration of acute lung injury in rats. J Cell Physiol. 2017;233:3048–305.
- Sabirzhanov B, Zhao Z, Stoica BA, et al. Downregulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. J Neuroscience. 2014;34:10055–10071.
- Sun L, Zhao M, Wang Y, et al. Neuroprotective effects of miR-27a against traumatic brain injury via suppressing FoxO3a-mediated neuronal autophagy. Biochem Biophys Res Commun. 2017;482:1141–1147.
- Brandenberger C, Kling KM, Vital M, et al. The role of pulmonary and systemic immunosenescence in acute lung injury. Aging Dis. 2018;9:553.
- Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–163.
- Chen C, Shi L, Li Y, et al. Disease-specific dynamic biomarkers selected by integrating inflammatory mediators with clinical informatics in ARDS patients with severe pneumonia. Cell Biol Toxicol. 2016;32:169–184.
- Chen JJ, Huang CC, Chang HY, et al. Scutellaria baicalensis ameliorates acute lung injury by suppressing inflammation in vitro and in vivo. Am J Chin Med. 2017;45:137–157.
- Sun H, Cai S, Zhang M, et al. MicroRNA-206 regulates vascular smooth muscle cell phenotypic switch and vascular neointimal formation. Cell Biol Int. 2017;41:739–748.
- Deng G, He H, Chen Z, et al. Lianqinjiedu decoction attenuates LPS-induced inflammation and acute lung injury in rats via TLR4/NF-kappaB pathway. Biomed Pharmacother. 2017;96:148–152.
- Zhang T, Wang J, Wang S, et al. Timosaponin B-II inhibits lipopolysaccharide-induced acute lung toxicity via TLR/NF-kappaB pathway. Toxicol Mech Methods. 2015;25:665–671.
- Kanzler H, Barrat FJ, Hessel EM, et al. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13:552–559.
- Ben DF, Yu XY, Ji GY, et al. TLR4 mediates lung injury and inflammation in intestinal ischemia-reperfusion. J Surg Res. 2012;174:326–333.
- Chong AJ, Shimamoto A, Hampton CR, et al. Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg. 2004;128:170–179.
- Shi DL, Shi GR, Xie J, et al. MicroRNA-27a inhibits cell migration and invasion of fibroblast-like synoviocytes by targeting follistatin-like protein 1 in rheumatoid arthritis. Mol Cells. 2016;39:611–618.
- Li XQ, Lv HW, Wang ZL, et al. MiR-27a ameliorates inflammatory damage to the blood-spinal cord barrier after spinal cord ischemia: reperfusion injury in rats by downregulating TICAM-2 of the TLR4 signaling pathway. J Neuroinflammation. 2015;12:25.
- Tian WF, Weng P, Sheng Q, et al. Biliverdin protects the isolated rat lungs from ischemia-reperfusion injury via antioxidative, anti-inflammatory and anti-apoptotic effects. Chin Med J. 2017;130:859–865.
- Ferrari RS, Andrade CF. Oxidative stress and lung ischemia-reperfusion injury. Oxid Med Cell Longev. 2015;2015:590987.
- Bardales RH, Xie SS, Schaefer RF, et al. Apoptosis is a major pathway responsible for the resolution of type II pneumocytes in acute lung injury. Am J Pathol. 1996;149:845–852.
- Dosreis GA, Borges VM, Zin WA. The central role of Fas-ligand cell signaling in inflammatory lung diseases. J Cell Mol Med. 2004;8:285–293.
- Borner C. The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol Immunol. 2003;39:615–647.
- Fan TJ, Han LH, Cong RS, et al. Caspase family proteases and apoptosis. Acta Biochim Biophys Sin. 2005;37:719–727.