
ABSTRACT
A different expression signature of miRNA in oral squamous cell carcinoma (OSCC) has been validated. MicroRNA-16 (miR-16) as one of the distinctly dysregulated miRNAs in OSCC, its functional role in progression of OSCC remains not fully clear. Herein, miR-16 expression was significantly lower in OSCC tissues compared to that in adjacent normal tissues (n = 131). A lower level of miR-16 was found to be associated with poor prognosis on a cohort of 131 patients with OSCC, and on an extensive public data (457) from TCGA database. Additionally, expression of TLK1 was significantly higher in OSCC tissues compared to that in adjacent normal tissues, which is negatively correlated with miR-16 expression in OSCC. Bioinformatics analyses exhibited that TLK1 is a potential downstream effector of miR-16 by directly targeting the 3′-untranslated regions (3′-UTR) of mRNA. Forced expression of miR-16 in OSCC cell lines inhibits cell proliferation in vitro, and tumor growth in vivo by inhibition of TLK1. Mechanistically, downregulation of TLK1 by miR-16 enhances higher level of DNA damage leading to a significant increase of G2/M arrest in SCC9 cells. And, overexpression of TLK1 substantially reduces DNA damage and G2/M arrest by activation of TLK1-dependent cell cycle checkpoint response. To conclude, miR-16 is downregulated in OSCC and serves as tumor suppressor in OSCC progression by targeting TLK1, which has potential to be the novel therapeutic targets and diagnostic biomarkers for OSCC.
Introduction
Oral squamous cell carcinoma (OSCC) is the most common malignancy of the oral cavity with significant local invasiveness, regional and distant metastases[Citation1].Unpredictable metastasis is the major causative event for cancer deaths like OSCC[Citation2].The development of OSCC metastasis is attributed to host factors, as well as intrinsic characteristics of cancer cells and tissue microenvironment[Citation3]. A process of metastasis consists of a multistep, including proliferation, angiogenesis, detachment from the primary tumor and invasion into surrounding tissue, extravasation into other organs, highly correlated with poor prognosis [Citation4]. Therefore, it is necessary to understand the etiology and progression of OSCC for exploring novel therapeutic strategies.
MicroRNAs (miRNAs) are a class of small, non-coding RNA transcripts with the length of 18–22 nucleotides. MiRNAs could modulate gene expression through interacting with the 3′-untranslated region (3′-UTR) of target mRNAs, thereby maintaining post-transcriptional gene repression of targets [Citation5]. An increasing numbers of studies confirmed that miRNAs played important roles in the development, invasion and metastasis of diverse tumor types, including digestive carcinoma, head and neck cancers, nervous system cancers and reproductive system cancers[Citation6]. As the diversity of the targets of miRNAs in tumor context, miRNAs have been proved to involve in multiple oncogenic cellular processes of tumor cells, such as growth, invasion, metastasis, antigenic regulation[Citation7]. Among these miRNAs, miR-16 has been well documented as a prominent tumor-suppressor in glioma, non-small cell lung cancer, prostate cancer and chronic lymphocytic leukemia [Citation8–Citation11]. Most importantly, miR-16 was found to significantly downregulate in OSCC samples compared to control adjacent normal tissues [Citation12]. This study therefore focused specifically on the role of miR-16 in OSCC progression.
Tousled-like kinase 1 (TLK1), belonging to the family of serine-threonine kinases, was suggested to be implicated in chromatin assembly, DNA repair of double strand breaks (DSBs), and chromosome segregation [Citation13]. TLK1 also has been shown to have multiple functions in a broad range of physiological and pathological processes in a cell-cycle dependent manner [Citation14].As the target of cell cycle checkpoint kinases, TLK1 plays important role in DNA damage-induced checkpoint arrest [Citation15]. TLK1 expression was intimately linked to the radio tolerance of prostate cancer cells [Citation16].However, whether TLK1 was involved in OSCC has not yet been reported. Based on bioinformatics analysis, we hypothesized that aberrant miR-16 expression was associated with OSCC progression via regulating TLK1.The aim of this study was to further clarify how miR-16 regulate OSCC progression. This study profiled the expression of miR-16 and TLK1 in 131 OSCC tissues and five OSCC cell lines, and validated the interaction between miR-16 and TLK1 in OSCC cells. Moreover, we determined the role of miR-16 in OSCC growth both in vitro and in vivo.
Material and methods
Clinical specimen collection
The OSCC tissues (n = 131) were obtained from the diagnosed patients underwent surgery at the Affiliated Cancer Hospital of Central South University. All the patients have complete clinical and pathological data including age, classification of tumor, and lymph node metastasis (TNM) staging. The present study was approved by the Ethics Committees of the First Affiliated Hospital of Zhengzhou University and the Affiliated Cancer Hospital of Central South University, and written informed consent was obtained from all subjects. All of the patients had not received any treatment before the surgery.
Quantitative real-time PCR (qRT-PCR)
Total RNAs were extracted from clinical tissues using TRIzol reagent (Invitrogen, CA, USA) according to the manufacturer’s introductions, and RNAiso plus Reagent (Takara, Dalian, China) was used to obtain the total RNAs of cell lines. The purity and concentration of extracted RNA were determined by spectrophotometry (PerkinElmer Inc, Boston, MA) at optical density of 260 nm and 280 nm. For detecting the relative expression of TLK1 mRNA, total RNAs (2 μg) were reverse transcribed into cDNA using Revert Aid First Strand cDNA Synthesis kit (Thermo Fisher). For miR-16 expression, the reverse transcription was performed using miRNA First-Strand Synthesis (Creative Biogene, NY, US) according to the manufacturer’s instructions. Then, qRT-PCR was performed in 20 μL reaction systems with GeneAmp RNA PCR Kit (Applied Biosystems, CA, USA) on an ABI 7500 Real-Time PCR System (Applied Biosystems, CA, USA). The expression of TLK1 mRNA was normalized to that of GAPDH, and miR-16 expression was normalized to that of U6. The specific primers used in qRT-PCR were as follows:
miR-16: (forward) 5′-TAGCAGCAGCTAAATA TTGGCG-3′ and (reverse)
5′-CCAGTATACTATGTGCTCGTGA-3′;
TLK1: (forward) 5′-ACTCTCAGGCGTCCAGC-3′ and (reverse)
5′-AGCATCTACCATGATGCTTG-3′;
GAPDH: (forward) 5′-TGCACCACCAACTGCTT AG-3′ and (reverse)
5′-AGAGGCAGGGATGATGTT-3′;
U6: (forward) 5′-CTCGCTTCGGCAGCACA-3′ and (reverse)
5′-AACGCTTCACGAATTTGCGT-3′.
Cell culture and transfection
The human OSCC cell lines Tca83, HN4, Cal27, SCC9, SCC25, and human oral keratinocytes (HOK) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). HN4, SCC9, SCC25 and HOK were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM)-F12 (Gibco, Grand Island, NY, USA). Cal27 cells were cultured in DMEM medium (Gibco), and Tca83 cells were cultured in RPMI 1640 medium (Gibco). All medium was supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin. Cells were maintained in a 5% CO2 humidified incubator at 37°C. For overexpression, SCC9 cells were infected with lentivirus vectors-expressing miR-16 or TLK1 or lentivirus empty vector for 24 hours with 6 μg/mL polybrene (Sigma-Aldrich, St. Louis, MO). MiR-16-stable expressed cells were selected using puromycin (1 μg/mL) for 7 days.
Immunohistochemistry (IHC)
After frozen in 10% neutral buffered formalin, the tissue slides (4 μm) from tumors were prepared with xylene dewaxing, gradient ethanol dehydration and rehydration. The tissue slides were then blocked with methanol containing 3% hydrogen peroxide for 10 min followed by blocking with 5% bovine serum albumin (BSA) for 30 min. Subsequently, tumor sections were incubated with primary antibodies against TLK1 (1:2000, Cell Signaling Technology) for 2 hours at room temperature and incubated with the secondary antibody (Beyotime, Shanghai, China) for 1 hour at room temperature. The reaction was performed with 3,3′-Diaminobenzidine (DAB) for 10 min at room temperature in accordance with the manufacturer’s instructions. The stained sections were observed by two independent oncologists using an Axiovert 200M inverted microscope (Zeiss, Oberkochen, Germany). The score of each section was evaluated by the indicators containing the numbers of staining-positive cell and the intensity of cell staining and repeated on three random selected sections. The results of each sample were determined by the scores of sections by the two oncologists.
Western blot analysis
Total protein was extracted from clinical tissues and cell lines using RIPA buffer (Beyotime, Shanghai, China) according to the manufacturer’s introductions. The concentration of extracted protein sample was determined using BCA Protein Assay Kit (Beyotime, Shanghai, China). And the proteins were separated using SDS-PAGE and transferred to polyvinyl difluoride membranes (PVDF, Millipore, Bedford, MA). After being blocked with 5% BSA for 1 h at room temperature, the membranes were incubated with antibodies specific for TLK1 (1:1000, Cell Signaling Technology) or β-actin (1:500, Cell Signaling Technology) at 4°C overnight, and incubated with the horseradish peroxidase-conjugated secondary antibody (Beyotime, Shanghai, China) as appropriate for 1 hour at room temperature. The blot signals were visualized enhanced chemiluminescence (ECL) substrate (Thermo Fisher Scientific Inc.).
Luciferase reporter assay
To validate the interaction between miR-16 and TLK1, the putative miR-16 binding sites in the 3′-UTR of TLK1 were mutagenized using QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). For luciferase reporter assay, the luciferase plasmids containing the 3′-UTR of TLK1 (pMirTarget-TLK1-Wild type) or the mutated 3′-UTR of TLK1 (pMirTarget-TLK1-Mutant) were employed. Briefly, SCC9 cells (5 × 105 cells/well) were cultured in 96-well plates overnight and were then transfected with pMirTarget-TLK1-Wild type or pMirTarget-TLK1-Mutant together with lentivirus vectors expressing miR-16 or lentivirus empty vector using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. After 48 hours of transfection, the luciferase activities were measured by dual-luciferase reporter system (Promega, Madison, WI, USA), and then calculated the relative activity of each group according to the manufacturer’s instructions.
Immunofluorescence
After 24 hours of transfection, the cells were fixed in 4% paraformaldehyde for 15 min at room temperature. The fixed cells were permeabilized with 0.2% TritonX-100 for 20 min and subsequently blocked with 5% BSA in PBS for 1 hour at room temperature. The cells were incubated with anti-γ-H2AX antibody (1:500, Cell Signaling Technology, Beverly, MA) or anti-TLK1 antibody (1:1000, Cell Signaling Technology, Beverly, MA) at 4°C overnight. After being washed with PBST, the cells were incubated with the fluorescently-labeled secondary antibodies for 1 hour at room temperature. Cell nuclei were stained with DAPI to visualize DNA. The results were detected by Axiovert 200M inverted microscope (Zeiss, Oberkochen, Germany).
Cell proliferation assay
SCC9 cells were cultured in 96-well plates (3 × 104 cells/well). CCK-8 reagent (10 μL) (Dojindo Laboratories, Kumamoto, Japan) was added into each well and incubated with the cells for 2 hours at 37°C. Optical density (OD) value of each well was measured at 450 nm using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA). In colony formation assay, SCC9 cells were cultured in 6-well plates (1 × 104 cells/well) with 1mL medium in a 5% CO2 humidified incubator at 37°C. After 2 weeks, colonies were washed with PBS and then stained with 0.1% crystal violet in 4% paraformaldehyde for 10 min.
Flow cytometry for cell cycle analysis
After 48 hours of transfection, the cells were collected and fixed with 70% ice-cold ethanol at −20°C for 4 hours. The fixed cells were washed once with PBS and subsequently resuspended in PI staining buffer (0.5 mL) (BD Biosciences, San Jose, CA, USA) containing RNase A and Triton X-100 for 30 min at room temperature in the dark. The analysis of cell cycle was performed using a BD AccuriC6 flow cytometer (BD Biosciences), and data were analyzed with FlowJo software (TreeStar Inc., Ashland, OR).
Establishment of xenograft mouse model
Twelve male BALB/c nude mice (5 weeks old) were purchased from Animal Research Center of the First Affiliated Hospital of Zhengzhou University and maintained in a standard condition. Animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Ethics Committees of the First Affiliated Hospital of Zhengzhou University and the Affiliated Cancer Hospital of Central South University. miR-16- or vector-transfected SCC9 cells (3 × 106 cells in 100 μL normal saline) were obtained as described above and injected into mice by tail vein. Tumor diameter was measured with caliper every 3 days starting at the third day after tumor cell inoculation. Tumor volume was calculated with the formula: Volume (mm3) = length× width2/2. After the last measurement, all mice were sacrificed to collect the lung tissues for H&E staining and analysis of TLK1 expression.
TCGA data analysis
The TCGA Data of the patients with oral squamous cell carcinomas (OSCC) were downloaded from The Cancer Genome Atlas (TCGA, https://cancergenome.nih.gov/), which is a public database providing the RNA level for various cancers. The numbers of the patients with complete RNA sequencing data for miR-16 and TLK1 were 525 and 500, respectively. And the patients without being followed up were excluded for the overall survival analysis.
Statistical analysis
Each experiment was repeated three times (n = 3). Data were expressed as the mean ± SD. The significance between two groups was analyzed using Student’s t test. The correlation between miR-16 expression in tumor tissues and the overall survival of OSCC patients was analyzed by Kaplan–Meier method with the log-rank test. Correlation between the levels of miR-16 and the mRNA expression of TLK1 in 131 patients with OSCC was analyzed using Chi-square test. Fisher’s exact test was used to analyze the correlation between clinical pathologic features and the miR-16 expressions in patients with OSCC. Cox proportional hazards regression analysis with calculation of the hazard ratio (HR) and 95% confidence interval (95% CI) was performed to analyze the factors associated with the overall survival of patients with OSCC. A p value < 0.05 was considered as statistically significant.
Results
miR-16 is downregulated in OSCC, and correlated with poor prognosis
To investigate the clinicopathological and prognostic significance of miR-16 in patients with OSCC, the differential expressions of miR-16 in OSCC tissues and the adjacent normal tissues were determined by qRT-PCR. It showed that miR-16 expression levels in OSCC tissues were markedly lower than that in the corresponding adjacent normal tissues (n = 131) (Figure 1(a)). We also found lower levels of miR-16 in five OSCC cell lines (Tca83, HN4, Cal27, SCC9, SCC25) were than that in human oral keratinocytes (HOK) cells ()). Moreover, compared to non-metastatic OSCC patients (n = 76), miR-16 expression was significantly lower in patients with metastatic OSCC (n = 55) ()).
Figure 1. Down-regulated miR-16 expression in OSCC tissues correlated with OSCC aggressiveness. (a) The relative expression of miR-16 in OSCC tissues (n = 131) and adjacent normal tissues (n = 131) as determined by qRT-PCR. Tumor: OSCC tissues; Non-tumor: adjacent normal tissues. (b) The expression levels of miR-16 in five OSCC cell lines and HOK were showed. HOK: human oral keratinocytes; (c) Comparison of miR-16 expressions between non-metastatic (n = 76) and metastatic (n = 55) OSCC tissues. (d) Kaplan–Meier analysis of the overall survival of OSCC patients with different miR-16 expression in tumor tissues. (e) Kaplan-Meier analysis for overall survival of 521 OSCC patients by log-rank test according to different miR-16 levels based on TCGA database. *p < 0.05, **p < 0.01, ***p < 0.001 vs. Control group.
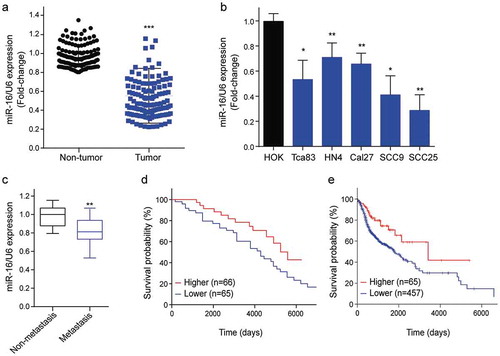
Next, we analyzed the correlation of the miR-16 expression and tumor phenotype. The median value of all 131 OSCC samples was chosen as the cut-off point for separating tumors with lower expression of miR-16 from higher expression miR-16. Correlation analysis showed that lower expression of miR-16 in OSCC was significantly associated with positive lymph metastasis and higher TNM subtype (p<0.001, ). Univariate analysis revealed that miR-16 expression was significantly associated with the overall survival of patients with OSCC (p<0.001, ). Multivariate Cox regression analysis indicated that lower expression of miR-16 is an independent prognostic factor for poor survival of patients with OSCC (p = 0.003, ). Kaplan–Meier survival analysis revealed that OSCC patients with lower miR-16 levels (n = 65) have shorter overall survival time than those with higher miR-16 levels (n = 66) (p = 0.001, )). Next, we took advantage of the extensive cohorts available as TCGA datasets to further investigate the correlation between miR-16 expression and prognosis in OSCC. Likewise, Kaplan–Meier survival analysis showed that patients with lower miR-16 levels (n = 457) have shorter overall survival time than those with higher miR-16 levels (n = 65) (p = 0.003, )). These results suggested an important role of miR-16 in aggressiveness and prognosis of OSCC.
Table 1. Correlation between clinical pathologic features and the miR-16 expressions in patients with oral squamous cell carcinoma.
Table 2. Univariable and multivariable analyses of factors and overall survival of patients with OSCC.
TLK1 is aberrant in OSCC, and negatively correlated with miR-16 expression
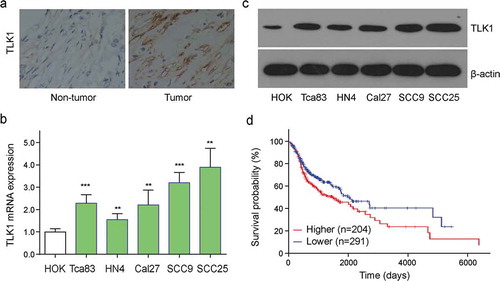
It reported that tousled-like kinase 1 (TLK1) was identified to be involved in cancer progression [Citation17]. Next, we investigated whether the expression of TLK1 was dysregulated in OSCC. Here, we found that OSCC tissue had a higher level of TLK1 expression compared to the adjacent normal tissues (Figure 2(a)). To confirm whether this change occurred in OSCC cells, the transcriptional and protein expressions of TLK1 were detected in five OSCC cell lines (Tca83, HN4, Cal27, SCC9, and SCC25) as well as HOK cells. The results showed that the levels of TLK1 were obviously higher in OSCC cell lines (Tca83, HN4, Cal27, SCC9, and SCC25) than that in HOK cells ()). TCGA datasets-based Kaplan–Meier survival analysis showed that patients with higher TLK1 levels (n = 204) have shorter overall survival time than those with lower TLK1 levels (n = 291) (p = 0.016, )). Next, we further analyzed whether miR-16 level was correlated with TLK1 expression in OSCC. Notably, Chi-square test showed that the expressions of TLK1 and miR-16 in 131 OSCC specimens were negatively correlated significantly (p<0.001, ).
Table 3. Correlations between the levels of miR-16 and the mRNA expression of TLK1 in 131 patients with OSCC.
Figure 2. TLK1 was upregulated in OSCC tissues and cells. (a) Representative images of TLK1 expression in OSCC tissues and adjacent normal tissues by immunohistochemistry (IHC) staining. (b) Relative TLK1 mRNA expressions in five OSCC cell lines and HOK cells. (c) The protein levels of TLK1 in five OSCC cell lines were analyzed by Western blot. (d) Kaplan-Meier analysis for overall survival of 495 OSCC patients by log-rank test according to different TLK1 levels based on TCGA database. **p < 0.01, ***p < 0.001 vs. HOK.
TLK1 is a potential target of miR-16 in OSCC cell
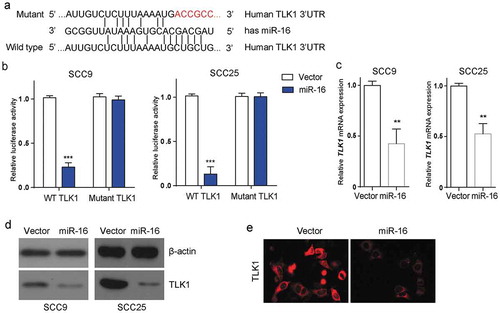
Next, we investigated whether miR-16 could regulate TLK1 expression at post-transcriptional level. Bioinformatics analysis exhibited that TLK1 was a putative target of miR-16 by targeting the 3`-UTR of TLK1 mRNA (Figure 3(a)). Then, we cloned the 3`-UTR of TLK1 mRNA (TLK1-WT) and the mutant (TLK1-Mut) in SCC9 and SCC25 cells. The mutant was randomly designed following the principles that it could not produce another miR-16 binding site. Luciferase reporter assay showed that miR-16 overexpression significantly reduced the luciferase activity in the luciferase plasmids containing TLK1-WT, but not in the TLK1-Mut ()). Moreover, miR-16 overexpression significantly decreased the transcriptional level of TLK1 in SCC9 and SCC25 cells ()). Concomitantly, TLK1 protein expression was also markedly decreased in miR-16-overexpressed SSC9 cells compared with that in control by western blot and immunofluorescence staining ()). The data indicated that miR-16 inhibited TLK1 expression in SCC9 cells via directly targeting the 3′-UTR of TLK1.
Figure 3. TLK1 is a potential target of miR-16 in OSCC cells. (a) miR-16 was identified to potentially interact with the 3-UTR of TLK1 (Wild type). The putative miR-16 binding sites were mutated to generate mutated TLK1 3-UTR (Mutant). (b) Luciferase reporter assay was performed to determine the interaction between miR-16 and the 3`-UTR of TLK1 mRNA in SCC9 and SCC25 cells. SCC9 cells were transfected with lentivirus vector-expressing miR-16 (miR-16) or its negative control (Vector). (c) Analysis of TLK1 mRNA expression by qRT-PCR. (d) After transfection, SCC9 and SCC25cells were subjected to western blot for the level of TLK1 protein. (e) Immunofluorescence staining of TLK1 in SCC9 cells with miR-16 overexpression. **p < 0.01, ***p < 0.001 vs. Vector. The experiments were performed in triplicate.
Forced expression of miR-16 suppresses OSCC progression in vitro and in vivo
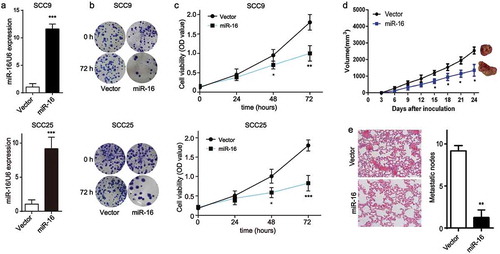
Next, we determined whether miR-16 played a role in OSCC proliferation. qRT-PCR quantification showed that miR-16 expression was efficiently elevated by lentivirus transfection of miR-16 in SCC9 and SCC25 cells (Figure 4(a)). Meanwhile, compared with that in control cells, the colony-forming activity was dramatically reduced in miR-16-overexpressed SCC9 and SCC25 cells ()). Moreover, CCK-8 assay showed that miR-16 overexpression significantly decreased the cell viability in SCC9 and SCC25 cells ()). Our findings suggested that miR-16 overexpression obviously inhibits SCC9 proliferation in vitro. Next, xenograft mouse model was established to investigate the role of miR-16 in tumor growth in vivo. miR-16 overexpression in SCC9 cells significantly inhibited the in vivo tumor growth activity compared with the vector cells, as shown in ). Specifically, fewer metastatic nodes in lung tissue were observed in the miR-16-overexpressed xenograft models compared with that in the vector models ()). These data demonstrated that miR-16 had a vital role in OSCC progression.
Figure 4. Forced expression of miR-16 suppresses OSCC progression in vitro and in vivo. SCC9 and SCC25 cells were transfected with miR-16-contained lentivirus vector (miR-16) or its negative control (Vector). (a) The expression levels of miR-16 were detected after transfection in SCC9 and SCC25 cells. (b) Colony formation ability and (c) cell viability were examined in miR-16- and vector- SCC9 and SCC25 cells. (d, e) BALB/c nude mice were randomly divided into 2 groups with 6 mice in each group. MiR-16-overexpressed SCC9 cells (miR-16) or vector cells (Vector) were injected into the nude mice, respectively. (d) Tumor volume was measured every 3 days starting at the third day after tumor cell injection. (e) Representative images and quantification of H&E staining of metastatic nodes in lung tissue. *p < 0.05, **p < 0.01, vs. Vector.
Overexpression of TLK1 reverses miR-16-mediated DNA damage and G2/M arrest
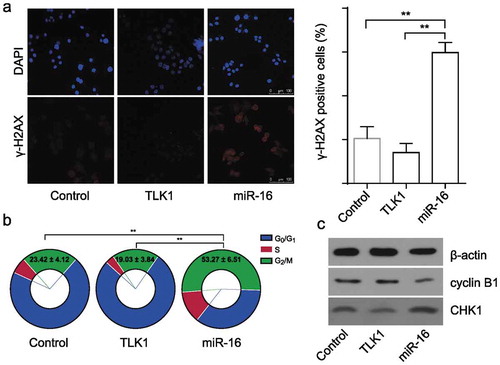
Previous studies demonstrated that TLK1 was vital to initiate and maintain cell cycle checkpoint response avoid of cellular DNA damage [Citation18]. And, TLK1 activity peaks in S-phase and lacking TLK1 display a prolonged G2/M arrest [Citation19]. To gain further insight into the potential role of TLK1 in OSCC, the effects of miR-16 on DNA damage (γ-H2AX foci) and cell cycle in the SCC9 cells were investigated. It showed that miR-16 overexpression in SCC9 cells resulted in a significantly increased number of γ-H2AX foci and G2/M arrest. And, the increased levels of DNA damage and G2/M arrest were effectively reduced by TLK1 overexpression (Figure 5(a,b)). Cell cycle associated protein analysis showed that miR-16 overexpression led to a significantly increased level of CHK1, but decreased level of Cyclin B1 in SCC9 cells, which were effectively reduced by TLK1 overexpression ()) It indicates that forced expression of miR-16 enhances DNA damage level leading to a significant increase of G2/M arrest in SCC9 cells by inactivation of TLK1-dependent checkpoint response.
Figure 5. Overexpression of TLK1 reverses miR-16-mediated DNA damage and G2/M arrest. (a) Representative images of γ-H2AX foci in miR-16-overexpressed (miR-16), TLK1-overexpressed or vector SCC9 cells by immunofluorescence staining. Quantification of the number of γ-H2AX foci in miR-16-overexpressed (miR-16), TLK1-overexpressed or vector SCC9 cells. (b) Flow cytometry analysis of cell cycle in miR-16-overexpressed (miR-16), TLK1-overexpressed or vector SCC9 cells. (c) Western blot analysis of cell cycle associated proteins in miR-16-overexpressed (miR-16), TLK1-overexpressed or vector SCC9 cells. **p < 0.01 vs. Vector. The experiments were performed in triplicate.
Discussion
The aberrant expression of miR-16 has been demonstrated to commonly contribute to the pathogenesis of various human cancers. For example, miR-16 was significantly reduced in human brain glioma tissues and negatively correlated with malignancy; moreover, the forced miR-16 significantly inhibited glioma cell proliferation, migration and invasion [Citation8,Citation20]. Additionally, in hepatocellular carcinoma and chronic lymphocytic leukemia, up-regulated miR-16 could inhibit cell proliferation and induce cell cycle arrest through negative regulating B cell lymphoma 2 (Bcl2) [Citation21,Citation22]. These findings suggested that miR-16 may serve as a tumor suppressor. Low expression of miR-16 was initially found in cancer tissues from DMBA-induced oral carcinogenesis Syrian hamsters, a classic animal model of OSCC [Citation23]. Subsequently, miR-16 was identified to down-regulate in 61 primary OSCC tissue specimens [Citation12]. Although most of these works showed miR-16 as having a critical role in OSCC context, the functional involvement of miR-16 in OSCC is still unknown.
We performed the expression analysis of miR-16 in 131 paired OSCC tumor samples and adjacent normal tissues. Results showed that miR-16 was significantly downregulated in OSCC tumor samples, which was basically consistent with the previous results. Similar results were observed in the OSCC cell lines. In addition, a lower expression of miR-16 was confirmed in metastatic OSCC tissues compared to non-metastatic OSCC tissues, implying the importance of miR-16 in OSCC metastasis. It was reported that downregulation of serum miR-16 correlated with the metastasis and lymph node involvement in prostate cancer [Citation24]. In the current study, Kaplan–Meier survival analysis revealed a strong correlation between lower miR-16 level and the shorter overall survival of OSCC patients. Moreover, lower miR-16 level significantly correlated with positive lymph metastasis and higher TNM subtype. Mayakannan et al. found the low expression of miR-16 in OSCC samples, however, their data failed to demonstrate an association between miR-16 and clinical characteristics [Citation12]. It should be noted that the sample size of this study is about 2 times of that of the aforementioned study. A large number of samples are conducive to the study of subtypes and to draw a reliable conclusion. Combined with the previous results, our data indicated an overall significant predictive value of miR-16 in OSCC.
Another finding of the present study was that miR-16 could inhibit growth of OSCC cells SCC9. Results showed that miR-16 overexpression significantly suppressed OSCC cell proliferation in vitro, as well as SCC9 cell tumor growth and lung metastasis in the xenograft models. Indeed, an innovative mesothelioma treatment based on miR-16 mimic was recently reported to be applied to the treatment for 26 recurrent malignant pleural mesothelioma patients, showing that it is a new safe therapeutic strategy [Citation25]. Although further studies are needed to clarify whether miR-16 modulates the other tumor properties of OSCC cell, including migration and invasion, this study demonstrated a link between miR-16 and OSCC growth and metastasis, suggesting that miR-16 promises to be one of the intervention targets for OSCC treatment.
Our data revealed that TLK1 expression was substantially increased in OSCC tissues and cell lines, and negatively correlated with miR-16 level in OSCC specimens. TLK1 plays important role in DNA damage-induced checkpoint arrest, thus involves in cell cycle regulation. Cell cycle checkpoint precisely maintained by multiple signaling pathways and molecules is critical for cellular biological process [Citation26]. It has been demonstrated that siRNA-mediated depletion of TLK1 induced a G2/M arrest by prolonging G2/M checkpoint activation, which is an important cell cycle checkpoint [Citation19]. In the present study, overexpression of TLK1 also resulted in a significant decrease of G2/M arrest in SCC9 cells through promoting the repair of damaged DNA. Most importantly, miR-16 was identified to inhibit TLK1 expression in SCC9 cells via interacting with the 3′-UTR of TLK1, reinforcing TLK1 as a target of miR-16. A recent experimental study confirmed that miR-16 feedback loop with p53 and Wip1 dominated the fate determination between apoptosis and senescence bistable upon DNA damage[Citation27]. We also observed that miR-16 overexpression reduced xenograft growth and lung metastasis accompanied by a decrease in the lung expression of TLK1. It appeared that miR-16 inhibited SCC9 cells proliferation; the mechanism may be related with the decreased TLK1.
In summary, miR-16 expression was found to be significantly lower in OSCC, and was correlated with the poor prognosis. TLK1, as a target of miR-16, was highly expressed in OSCC tissues and could modulate cell cycle in vitro, as well as cell growth and lung metastasis in vivo in SCC9 cells. Thus, our findings highlight a functional role and clinical significance of miR-16/TLK1 axis in OSCC progression.
Disclosure statement
The authors declare no conflicts of interest.
Additional information
Funding
References
- Delaine-Smith RM, Reilly GC. The effects of mechanical loading on mesenchymal stem cell differentiation and matrix production. In: Litwack G, editor. Vitamins and hormones: stem cell regulators. San Diego: Elsevier Academic Press Inc; 2011. p. 417–480.
- Dutton JM, Graham SM, Hoffman HT. Metastatic cancer to the floor of mouth: the lingual lymph nodes. Head Neck J Sci Spec Head Neck. 2002;24(4):401–405.
- Malik UU, Zarina S, Pennington SR. Oral squamous cell carcinoma: key clinical questions, biomarker discovery, and the role of proteomics. Arch Oral Biol. 2016;63:53–65.
- Liang L, Zhang T, Kong Q, et al. A meta-analysis on selective versus comprehensive neck dissection in oral squamous cell carcinoma patients with clinically node-positive neck. Oral Oncol. 2015;51(12):1076–1081.
- Farazi, T.A., Hoell JI, Morozov P, et al. MicroRNAs in human cancer. Adv Exp Med Biol. 2013;774:1–20.
- Benati M, Montagnana M, Danese E, et al. Evaluation of mir-203 expression levels and DNA promoter methylation status in serum of patients with endometrial cancer. Clin Lab. 2017;63(10):1675–1681.
- Cai Y, He J, Zhang D. Long noncoding RNA CCAT2 promotes breast tumor growth by regulating the Wnt signaling pathway. Onco Targets Ther. 2015;8:2657–2664.
- Chen X, Li D, Gao Y, et al. Long intergenic noncoding RNA 00152 promotes glioma cell proliferation and invasion by interacting with miR-16. Cell Physiol Biochem. 2018;46(3):1055–1064.
- Bandi N, Diks SH, Den Dunnen WFA, et al. miR-15a and miR-16 are implicated in cell cycle regulation in a Rb-dependent manner and are frequently deleted or down-regulated in non-small cell lung cancer. Cancer Res. 2009;69(13):5553–5559.
- Musumeci M, Coppola V, Addario A, et al. Control of tumor and microenvironment cross-talk by miR-15a and miR-16 in prostate cancer. Oncogene. 2011;30(41):4231–4242.
- Cutrona G, Matis S, Colombo M, et al. Effects of miRNA-15 and miRNA-16 expression replacement in chronic lymphocytic leukemia: implication for therapy. Leukemia. 2017;31(9):1894–1904.
- Manikandan M, Deva Magendhra Rao AK, Arunkumar G, et al. Oral squamous cell carcinoma: microRNA expression profiling and integrative analyses for elucidation of tumourigenesis mechanism. Mol Cancer. 2016;15:28.
- Sunavala-Dossabhoy G, De Benedetti A. Tousled homolog, TLK1, binds and phosphorylates Rad9; TLK1 acts as a molecular chaperone in DNA repair. DNA Repair (Amst). 2009;8(1):87–102.
- Takayama Y, Kokuryo T, Yokoyama Y, et al. Silencing of Tousled-like kinase 1 sensitizes cholangiocarcinoma cells to cisplatin-induced apoptosis. Cancer Lett. 2010;296(1):27–34.
- Timiri Shanmugam PS, Nair RP, De Benedetti A, et al. Tousled kinase activator, gallic acid, promotes homologous recombinational repair and suppresses radiation cytotoxicity in salivary gland cells. Free Radic Biol Med. 2016;93:217–226.
- Ronald S, Sunavala-Dossabhoy G, Adams L, et al. The expression of Tousled kinases in CaP cell lines and its relation to radiation response and DSB repair. Prostate. 2011;71(13):1367–1373.
- Norton KS, McClusky D, Sen S, et al. TLK1B is elevated with eIF4E overexpression in breast cancer. J Surg Res. 2004;116(1):98–103.
- Roe JL, Durfee T, Zupan JR, et al. TOUSLED is a nuclear serine/threonine protein kinase that requires a coiled-coil region for oligomerization and catalytic activity. J Biol Chem. 1997;272(9):5838–5845.
- Kelly R, Davey SK. Tousled-like kinase-dependent phosphorylation of Rad9 plays a role in cell cycle progression and G2/M checkpoint exit. PLoS One. 2013;8(12):e85859.
- Yang TQ, Lu X-J, Wu T-F, et al. MicroRNA-16 inhibits glioma cell growth and invasion through suppression of BCL2 and the nuclear factor-kappaB1/MMP9 signaling pathway. Cancer Sci. 2014;105(3):265–271.
- Tsang WP, Kwok TT. Epigallocatechin gallate up-regulation of miR-16 and induction of apoptosis in human cancer cells. J Nutr Biochem. 2010;21(2):140–146.
- Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102(39):13944–13949.
- Yu T, Demichelis S, Crespo M, et al. The expression profile of microRNAs in a model of 7,12-dimethyl-benz[a]anthrance-induced oral carcinogenesis in Syrian hamster. J Exp Clin Cancer Res. 2009;28:64.
- Zidan HE, Abdul-Maksoud RS. Diagnostic and prognostic value of serum miR-15a and miR-16-1 expression among egyptian patients with prostate cancer. IUBMB Life. 2018;70(5):437–444.
- Viteri S, Rosell R. An innovative mesothelioma treatment based on miR-16 mimic loaded EGFR targeted minicells (TargomiRs). Transl Lung Cancer Res. 2018;7(Suppl 1):S1–s4.
- Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246(4930):629–634.
- Issler MVC, Mombach JCM. MicroRNA-16 feedback loop with p53 and Wip1 can regulate cell fate determination between apoptosis and senescence in DNA damage response. PLoS One. 2017;12(10):e0185794.