
ABSTRACT
Oncogenic KIT or PDGFRA receptor tyrosine kinase (TK) mutations are compelling therapeutic targets in gastrointestinal stromal tumors (GISTs), and the KIT/PDGFRA kinase inhibitor, imatinib, is the standard of care for patients with metastatic GIST. However, approximately 10% of KIT-positive GIST metastases lose KIT expression at the time of clinical progression during imatinib therapy. In the present report, we performed TK-activation screens, using phosphotyrosine-TK double immunoaffinity purification and mass spectrometry, in GIST in vitro models lacking KIT expression. These studies demonstrated tyrosine-phosphorylated EGFR, AXL, and EPHA2 in four of six KIT-negative GIST lines (GIST62, GIST522, GIST54, GIST226, GIST48B, and GIST430B), and tyrosine-phosphorylated focal adhesion kinase (FAK) in each of the six KIT-negative lines. AXL expression was strong in KIT-negative or -weak clinical GIST samples that were obtained from progressing metastases during imatinib therapy. AXL knockdown inhibited viability in three KIT-negative GIST cell lines (GIST62, GIST54, and GIST522), but not in an AXL-negative, KIT-positive GIST control cell line (GIST430). AXL inhibition by R428, a specific AXL kinase inhibitor, reduced viability in AXL-activated GIST54. AXL knockdown in GIST62, GIST522, and GIST54 was accompanied by an increase in p21, p27, and p53 expression. By contrast, gefitinib-mediated EGFR inhibition, PF562271-mediated FAK inactivation, and shRNA-mediated knockdowns of EPHA2 and FAK had no effect on viability or colony formation of the KIT-negative GISTs. These findings highlight the potential relevance of AXL/p53 signaling as a therapeutic target in a subset of GISTs that have lost KIT oncoprotein expression.
KEYWORDS:
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract [Citation1], arising from a transformed progenitor cell committed to differentiation along the interstitial cell of Cajal lineage. GISTs are heterogeneous histologically, including spindle cell (70%), epithelioid cell (20%), and mixed types (10%) [Citation2]. Most GISTs contain oncogenic gain-of-function receptor tyrosine kinase (RTK) mutations involving KIT (~85%) or PDGFRA (~5%), which are accompanied by strong expression of the protein products of these oncogenes [Citation2–Citation4]. Oncogenic events in wild type GISTs lacking KIT/PDGFRA mutations include IGF1R overexpression and loss of succinate dehydrogenase (SDH) function due to SDH mutation or SDHC epimutation [Citation5,Citation6]. KIT oncoproteins remain crucial to the proliferation and survival of GIST cells in patients with metastatic KIT-positive GIST, as evidenced by the clinical successes of KIT kinase inhibition by imatinib and sunitinib [Citation7–Citation9] Indeed, imatinib therapy has become the standard of care in patients with metastatic GIST [Citation10].
Although KIT inhibition by imatinib represents a major therapeutic advance for patients with inoperable GIST, most patients eventually experience clinical progression due to multiple imatinib-resistant mechanisms, which include acquisition of secondary mutations in the KIT kinase domain [Citation11,Citation12], genomic amplification of KIT, and activation of alternate RTKs [Citation2]. Approximately 10% of KIT-positive GISTs lose KIT expression at time of clinical progression during imatinib therapy [Citation13–Citation15]. Previous studies have shown that AXL is activated and overexpressed in GIST cells with loss of KIT, and AXL inhibition effectively suppressed GIST cell growth [Citation16]. However, the mechanism of AXL-mediated cell growth in KIT-negative GIST remains unclear. Recent reports have shown induction of AXL, EGFR, and MET expression with weak activation in GISTs with loss of KIT expression [Citation17], but did not demonstrate crucial oncogenic roles in these GIST cells. In the present study, our strategy was to identify and validate novel RTK candidate oncoproteins in GIST cell lines with loss of KIT oncoprotein expression using the phosphotyrosine (PY)-RTK double immuno-affinity purification approach and tandem mass spectrometry. We followed up several candidate RTK targets with RNAi or specific kinase inhibitors. We demonstrated an essential role of AXL, but not EGFR, EPHA2, or FAK in KIT-negative GIST cell lines.
Results
Double immuno-affinity purification approach for proteomic RTK screen
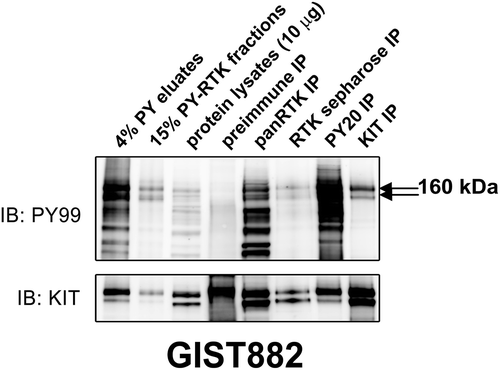
GIST882 expresses strongly phosphorylated KIT due to a mutation at exon 13, and therefore serves as an ideal control for double PY-RTK immunoaffinity purification [Citation7]. As shown in , two strongly phosphorylated bands at 160 (mature KIT) and 145 kDa (immature KIT) were observed in PY-RTK fractions, PY eluates, pan-RTK IP, RTK-sepharose IP, PY20 IP, and KIT IP. Preimmune IP did not demonstrate staining at the same position. The presence of KIT oncoprotein in the 160 kDa band excised from the Coomassie gel was confirmed by mass spectrometry and total KIT immunoblotting (), validating our approach as a proteomic screen for activated RTK in cancer cell lines and frozen tumor tissues.
Figure 1. Validation of the phosphotyrosine (PY) and RTK immunoaffinity double purification approach in GIST882 by immunoblot. Total protein lysate (input) is included as a control. KIT, panRTK, PY, and RTK-sepharose IPs are positive controls. Pre-immune IP is a negative control. PY staining after double purification (PY immunoaffinity column followed by panRTK IP) shows a doublet representing the known KIT oncoprotein. The presence of KIT was corroborated by mass spectrometry analysis.
The EGFR, AXL, EPHA2, and FAK tyrosine kinases were identified in KIT-negative GIST cell lines by double purification and mass spectrometry
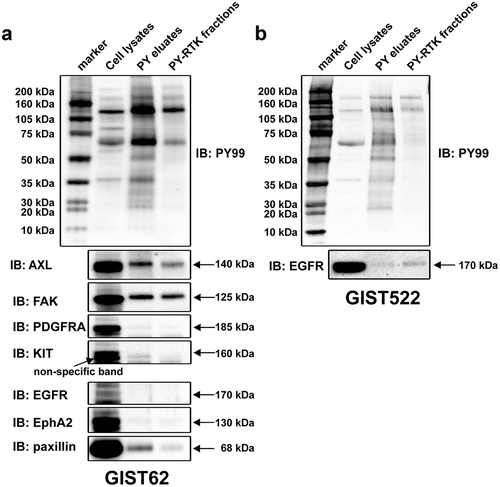
Strongly activated RTKs were identified in GIST62 and GIST522 by PY immunoaffinity purification and mass spectrometry (). Phosphotyrosine staining showed that proteins at ~140 kDa and ~170 kDa were phosphorylated in PY eluates and PY-RTK fractions in GIST62 and GIST522. Six additional phosphorylated receptor tyrosine kinases including EGFR, KIT, AXL, PDGFRA and EPHA2, and non-receptor tyrosine kinases FAK and paxillin were identified by mass spectrometry (). Immunoblotting evaluations of AXL, FAK, EGFR, KIT, PDGFRA, EPHA2, or paxillin in GIST62 (), and EGFR in GIST522 () showed that FAK and AXL in GIST62 demonstrated strong phosphorylation in PY eluates and PY-RTK fractions ().
Table 1. Identification of novel candidate protein tyrosine kinase oncoproteins in KIT-negative cell lines (GIST62 and GIST522) by the phosphotyrosine-RTK immuno-affinity purification and tandem mass spectrometry.
Figure 2. Phosphorylation of purified activated tyrosine kinases in KIT-negative GIST62 and GIST522 were evaluated by staining with PY and specific antibodies (AXL, FAK, EGFR, PDGFRA, KIT, EPHA2, and paxillin) in immunoblots. Total protein lysate (input) is a total protein control. PY and total staining of PY eluates and PY-RTK fractions reveal bands corresponding to AXL and FAK, which were corroborated by mass spectrometry analysis.
Relative expression and phosphorylation levels of candidate tyrosine kinase oncoproteins between KIT-negative GIST cell lines and KIT-positive GIST cell lines
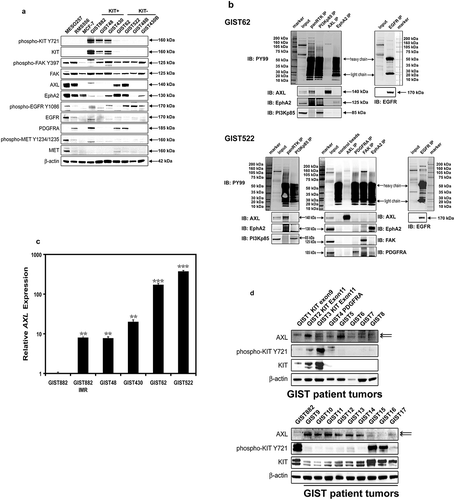
We performed immunoblotting to evaluate the relative expression and/or phosphorylation levels of the candidate tyrosine kinase oncoproteins including FAK, AXL, EPHA2, EGFR, PDGFRA, and MET in KIT-negative GIST cell lines and KIT-positive GIST cell lines (). MESO257, RMS556, and MCF-7 were used as controls. The phosphorylation and expression of FAK were comparable in GIST cell lines (). The expression of AXL and EPHA2 was greater in KIT-negative GIST cell lines than in KIT-positive GIST cell lines, which showed weak-to-absent expression. The expression and phosphorylation of EGFR and MET were nearly undetectable in KIT-positive GIST cell lines, whereas EGFR phosphorylation was moderate in GIST522, GIST48B, and GIST430B (). In addition, although wild-type PDGFRA was expressed in GIST822, GIST62, and GIST522 (), PDGFRA phosphorylation was weak (Figure S1).
Figure 3. (a) Immunoblotting evaluations of the relative expression and phosphorylation levels of candidate tyrosine kinase oncoproteins including FAK, AXL, EPHA2, EGFR, PDGFRA, and MET, between KIT-negative GISTs (GIST62 and GIST522), and KIT-positive GISTs (GIST882, GIST48, and GIST430). MESO257, RMS556, and MCF-7 are included as controls. Actin staining is a loading control. (b) Phosphorylation levels of AXL, EPHA2, FAK, and EGFR were evaluated by immunoprecipitation followed by PY and specific antibody immunoblotting in GIST62 and GIST522. Total protein lysate is a total protein control. (c) Quantitative RT-PCR evaluations of AXL transcript expression in GIST cell lines. AXL expression was greater in KIT-negative GIST62 and GIST522 than in KIT-positive GIST882, GIST48, and GIST430. AXL expression was induced after imatinib selection in GIST882IMR, which has lost KIT oncogene expression, as compared to parental GIST882. (d) Immunoblotting evaluations of the expression of AXL and KIT in KIT-positive or -negative frozen clinical GISTs. Actin staining is a loading control.
AXL, EGFR, and FAK are activated in KIT-negative GIST cell lines
To evaluate phosphotyrosine levels of candidate tyrosine kinase oncoproteins in KIT-negative GIST cell lines, AXL, FAK, EGFR, EPHA2, PDGFRA, panRTK, and PI3-K IPs were performed, followed by phosphotyrosine immunoblotting, which showed that AXL, FAK, and EGFR were phosphorylated (). Total AXL, FAK, EGFR, PDGFRA, EPHA2, and PI3-K immunoblotting validated the corresponding IP findings. As compared to total protein lysates (input), AXL and EGFR IPs efficiently pulled down AXL and EGFR receptors (). Strongly activated RTKs were also identified in GIST48B and GIST430B by PY-RTK immunoaffinity purification (Figure S2(a)). Phosphotyrosine staining showed that a protein at ~140 kDa was phosphorylated in PY eluates and PY-RTK fractions in both cell lines. Immunoblotting evaluation of FAK in GIST48B and GIST430B (Figure S2(a)) demonstrated strong phosphorylation in PY eluates and PY-RTK fractions (Figure S2(a)). However, targeting FAK with PF562271 for 3 and 6 days did not result in anti-proliferative effects in GIST48B as compared to the DMSO control (Figure S2(b)).
AXL expression in GISTs with loss of KIT oncogene
We further investigated AXL mRNA expression level by qRT-PCR in GIST cell lines (). AXL expression was greater in KIT-negative GIST62 and GIST522 than in KIT-positive GIST882, GIST48, and GIST430 (), which was consistent with AXL protein expression (). AXL expression was induced after imatinib selection in GIST882IMR, an isogenic KIT-negative cell line of GIST882 (Figure S3), as compared to parental GIST882. Likewise, AXL expression was strong in KIT-negative or -weak clinical GIST samples that were obtained as clinically progressing metastases during imatinib therapy (). By contrast, AXL expression was weak-to-undetectable in KIT-positive clinical GIST samples ().
Mutation analysis of AXL kinase domain in kit-negative GIST cell lines
We did not identify activating mutations in the kinase region of AXL except for an alternatively spliced product with deletion of exon 10 in GIST62 and GIST522.
EGFR activation is not a key event in GIST cell lines with loss of KIT oncoprotein expression
The phosphorylation of EGFR is stronger in MESO924 than in MESO257, so MESO924 was used as the control cell line to detect phospho-EGFR (). As compared to the phosphorylation of EGFR in MESO924, EGFR activation was present but low in GIST522 and GIST48B ( and ). Due to this observation, we used the EGFR inhibitor gefitinib to determine the inhibition of signaling intermediates and anti-proliferative effects in GIST522 and GIST48B (). EGFR was inactivated after treatment with gefitinib in GIST522, GIST48B, and control positive cell line MESO924 ( and Figure S4). Signaling intermediates which have been shown to be KIT-dependent in GISTs, including AKT, MAPK, and S6 [Citation18,Citation19], remained phosphorylated after EGFR inhibition ().
Figure 4. (a) Immunoblotting evaluations of GIST522 and GIST48B after treatment with gefitinib for 4h in serum-free media. MESO924 is an EGFR-positive control, GIST882 is an EGFR-negative control. (b) Cell viability was evaluated in GIST522 (black bars), GIST48B (light gray bars), and PC-9 (dark gray bars), at day 6 after treatment with gefitinib. Viability was analyzed using Cell-titer Glo® ATP-based luminescence assay. The data were normalized to DMSO and represent the mean values (± s.d.) from quadruplicate cultures. Statistically significant differences between DMSO control and gefitinib treatments are presented as *p < 0.05, ***p < 0.001. (c) Colony growth assays were performed 10 days after treatment with gefitinib (100, 250, 500, 1000, and 2500 nM). EGFR inhibition led to a greater reduction in colony formation and size in PC-9, but not in GIST48B. (d) Quantitations of GIST48B and PC-9 cell colony growth after treatment with gefitinib for 10 days. Statistically significant differences between DMSO control and inhibitor treatments are presented as ***p < 0.001.
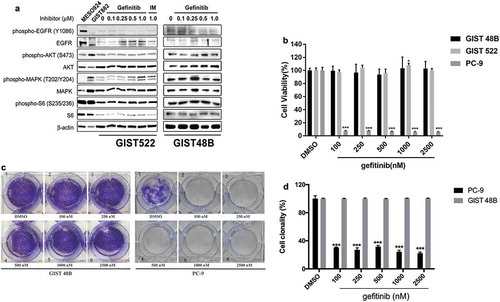
Cellular proliferation was evaluated by an ATP-based cell viability assay in GIST522 and GIST48B after treatment with gefitinib (). Treatment with gefitinib for 6 days did not result in anti-proliferative effects as compared to the DMSO control in GIST522 and GIST48B, but dramatically decreased cell viability in EGFR-activated non-small cell lung cancer PC-9 ().
GIST48B and PC-9 cells were further evaluated by colony formation assays. Colonies of GIST48B cells treated with gefitinib were similar to DMSO-treated cells in the size and number of colonies (), but gefitinib treatment significantly decreased the number of colonies in PC-9, as compared to DMSO controls (). Quantification of GIST48B and PC-9 cell colony growth after treatment with gefitinib for 10 days confirmed that EGFR inhibition resulted in ~70% decrease of the colony number in PC-9, as compared to DMSO control, but not in GIST48B ().
Effects of AXL, EPHA2 and FAK shRNA knockdown on GIST signaling pathways
To evaluate the roles of AXL, EPHA2, and FAK in KIT-negative GIST lines, these genes were stably silenced by lentivirus-mediated shRNAs in GIST62, GIST522, and GIST882. Immunoblotting studies were then performed at 96 h post-infection (). The shRNA knockdowns resulted in greater than 60% inhibition of their intended targets. These studies also evaluated phosphorylation of AKT, MAPK, and other signaling intermediates which have been shown to be KIT-dependent in KIT-positive GISTs [Citation2,Citation18,Citation19]. Immunoblotting showed that AXL, EPHA2 and FAK silencing did not inactivate AKT, MAPK, or S6 in KIT-negative GIST62 and GIST522 cells, or KIT-positive GIST882 (). AXL knockdown resulted in increased STAT1 phosphorylation in both GIST62 and GIST522, and increased STAT3 phosphorylation in GIST62, but not in GIST882 (). AKT, MAPK, and S6 remained activated in GIST62 at 10 days after stable AXL, EPHA2, and FAK silencing (Figure S5).
Figure 5. (a) Immunoblotting evaluations of KIT-negative GIST cell lines (GIST62 and GIST522) and KIT-positive GIST cell line (GIST882) at 96 hours after infection by lentiviral AXL, EPHA2, and FAK shRNA constructs. Immunoblots demonstrate the effects of AXL, EPHA2 and FAK knockdown on signaling intermediates (AKT, MAPK p42/44, S6, STAT1, and STAT3), proliferation markers (Cyclin A and PCNA), and cell cycle checkpoint proteins (p27, p21, and p53). Control lanes for each cell line include uninfected cells (untreated lane) and cells infected with empty lentiviral vector. Actin is a loading control. (b) Immunoblotting evaluations of GIST522 and GIST882 cells at 10 days after infection by lentiviral AXL, EPHA2, and FAK shRNA constructs. AXL shRNA resulted in decreased expression of cyclin A and PCNA, and upregulation of p53, p21, and p27 in GIST522, but not in GIST882. Actin is a loading control. (c) GIST62 and GIST522 cell cultures, evaluated at 6 days after infection by lentiviral AXL shRNA constructs, showing growth inhibition as compared to control cultures infected with empty lentiviral constructs. Scale bars: 100 μm. (d) Cell viability was evaluated in GIST62 (black bars), GIST522 (white bars), and GIST430 (gray bars), at day 8 and day 12 after infection with lentiviral AXL, EPHA2, or FAK shRNA. Viability was analyzed using the Cell-titer Glo® ATP-based luminescence assay. The data were normalized to empty lentiviral infections, and represent the mean values (± s.d.) from quadruplicate cultures. The experiments were performed in triplicate, and statistically significant differences between vector control and shRNAs are defined as *p < 0.05, ** p < 0.01, *** p < 0.001. (e) Cell cycle analyses were performed in GIST62 cells at 4 days after infection by lentiviral AXL shRNA constructs. GIST62 cells showed a G2 block following AXL silencing as compared to empty vector control. Cells treated with AXL shRNA showed increased nuclear fragmentation (pre-G1), the experiments were performed in triplicate.
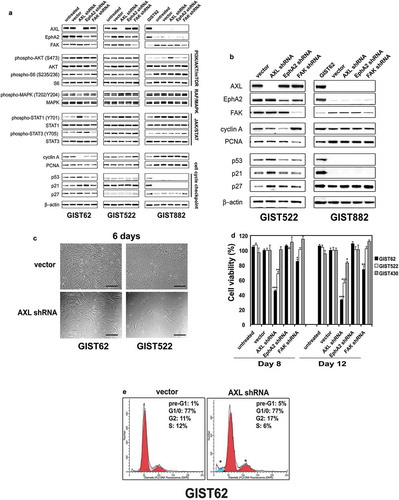
Effects of AXL, EPHA2 and FAK shRNA knockdown on GIST proliferation and cell cycle
AXL and EPHA2 shRNA knockdown decreased expression of the cyclin A proliferation marker in KIT-negative GIST62 and GIST522 lines (,). FAK shRNA knockdown also decreased cyclin A expression in GIST62 at 4 days (), and decreased PCNA expression in GIST522 at 10 days (). Cyclin A and PCNA expression were unaffected by AXL, EPHA2, and FAK shRNA knockdown in GIST882 at 96 hours (), and at 10 days of stable expression of AXL, EPHA2, and FAK shRNAs in the presence of puromycin (). GIST62 and GIST522 cell growth was reduced dramatically after 6 days of AXL shRNA expression ().
Cellular proliferation was evaluated in GIST62, GIST522, and GIST430 after transduction with AXL, EPHA2 or FAK shRNAs. Anti-proliferative effects were greater in GIST62 and GIST522 cells after treatment with AXL shRNA than after treatment with EPHA2 or FAK shRNA. GIST430 cells were unaffected after treatment with AXL, EPHA2 or FAK shRNAs ().
We also evaluated expression of cell cycle-associated proteins p53, p21, and p27 (,). GIST62 and GIST522 cells showed increased p53, p21, and p27 expression after AXL knockdown at day 4 (GIST62) and day 10 (GIST522), but not after EPHA2 or FAK knockdown. p27 expression was not affected in GIST882 cells after AXL or EPHA2 knockdown, and FAK knockdown resulted in an increase of p27 expression in these cells, whereas p21 and p53 were not demonstrably expressed in these cells (,).
Cell cycle analysis in GIST62 showed a block in G2 after AXL silencing with an increase in the G2 peak from 11% after empty vector transduction to 17% after transduction with AXL shRNA, (). This was accompanied by a decrease in the S phase population from 12% with the empty vector control to 6% with AXL shRNA (). In addition, the pre-G1 nuclear fragmentation peak increased from 1% with empty vector control to 5% with AXL shRNA ().
Silencing and inactivation of AXL shows anti-proliferative effects in AXL-activated GIST cells
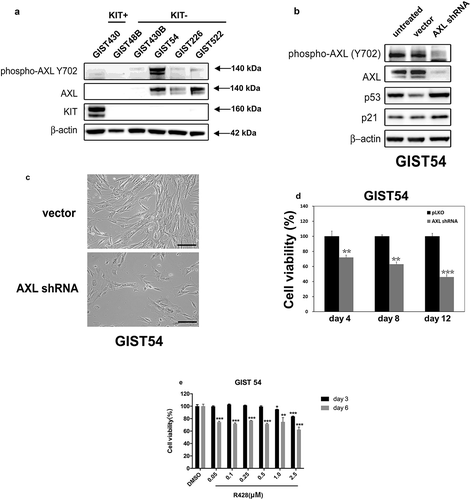
We performed immunoblotting to evaluate AXL activation in GIST cell lines with loss of KIT expression (GIST48B, GIST430B, GIST54, GIST226, and GIST522), with KIT positive cell line GIST430 as the control line (). The expression and phosphorylation of AXL were nearly undetectable in KIT-positive GIST430 and in KIT-negative GIST48B and GIST430B, whereas activation and expression of AXL was seen in GIST54, GIST226, and GIST522 (). ShRNA knockdown resulted in greater than 90% inhibition of AXL in GIST54, and GIST54 cells showed increased p53 and p21 expression after AXL knockdown at 4 days (). Furthermore, GIST54 cell growth was reduced dramatically after 6 days of stable AXL shRNA knockdown (). The shRNA-mediated knockdown resulted in a 30–50% reduction in viability of GIST54 cells (). Anti-proliferative effects were also observed in AXL-activated GIST54 after treatment with R428, a specific AXL kinase inhibitor (). Treatment with R428 in GIST54 at 6 days resulted in a 30–40% reduction in cell viability, as compared to the DMSO control ().
Figure 6. (a) Immunoblotting evaluations of the expression and phosphorylation of AXL between KIT-negative GISTs (GIST48B, GIST430B, GIST62, GIST226, and GIST522), and KIT-positive GISTs (GIST430). Actin staining is a loading control. (b) Immunoblots demonstrate the effects of AXL knockdown on expression of p53 and p21. (c) GIST54 cell cultures, evaluated at 6 days after infection by lentiviral AXL shRNA constructs, showing growth inhibition as compared to control cultures infected with empty lentiviral constructs. Scale bars: 100 μm. (d) Cell viability was evaluated in GIST54, at day 4, day 8, and day 12 after infection with lentiviral AXL. Viability was analyzed using the Cell-titer Glo® ATP-based luminescence assay. The data were normalized to empty lentiviral infections, and represent the mean values (± s.d.) from quadruplicate cultures. The experiments were performed in triplicate, and statistically significant differences between vector control and shRNAs are defined as ** p < 0.01, *** p < 0.001. (e) Cell viability was evaluated in GIST54, at day 3 (black bars) and day 6 (gray bars) after treatment with R428. Viability was analyzed using Cell-titer Glo® ATP-based luminescence assay. The data were normalized to DMSO and represent the mean values (± s.d.) from quadruplicate cultures. Statistically significant differences between DMSO control and R428 treatments are presented as *p < 0.05, ** p < 0.01, *** p < 0.001.
Discussion
The majority of GISTs contain oncogenic gain-of-function mutations in the KIT or PDGFRA receptor tyrosine kinases. These mutations can be early – and even initiating – transforming events [Citation2]. Therefore, KIT/PDGFRA inhibition by small molecule kinase inhibitors such as imatinib, sunitinib or regorafenib has become the mainstay of treatment in patients with inoperable GIST [Citation4,Citation8,Citation18–Citation23]. Although more than 80% of patients with inoperable GIST show marked clinical benefit from imatinib and sunitinib, most of these patients eventually develop clinical resistance to imatinib and other KIT/PDGFRA kinase inhibitors [Citation11]. The drug-resistance mechanisms responsible for a subset of GIST patients involves the loss of KIT expression at the time of clinical progression during imatinib therapy [Citation13–Citation15].
GIST62 and GIST522 are KIT-negative cell lines established from KIT-positive human GISTs, which retain genomic KIT mutations. These cell lines show activation of the same proliferation and survival signaling intermediates that are KIT-dependent in KIT-positive GISTs (), indicating that a novel oncogenic mechanism has supplanted the oncogenic role of KIT in these cells.
In the present study, we use the phosphotyrosine-panRTK double immunoaffinity approach and mass spectrometry to identify novel tyrosine kinase candidates in GISTs with loss of KIT onco-protein expression. GIST882, an imatinib-sensitive cell line expressing strongly activated KIT, was employed to develop the double purification approach (). Phosphotyrosine staining of a double purification shows a doublet representing the known KIT oncoprotein (). This was corroborated by mass spectrometry analysis. Additionally, we identified candidate kinase oncoproteins including AXL, EPHA2, FAK, and EGFR in KIT-negative GIST62 and GIST522 by the double immunoaffinity approach and mass spectrometry ( and ). Tyrosine kinases were also explored by this approach in KIT negative GIST430B and GIST48B cell lines, but no strong activated tyrosine kinases except for FAK were identified (Figure S2(a)).
Immunoblotting evaluation and qRT-PCR demonstrated that expression of AXL and EPHA2 were greater in KIT-negative GIST lines than in corresponding KIT-positive GIST lines (,), and that EGFR was strongly phosphorylated in KIT-negative GIST lines (). AXL mRNA increased after imatinib selection in KIT-positive GIST882 (), which has been shown previously by Mahadevan et al. [Citation16]. In addition, AXL expression is strong in KIT-negative or -weak clinical GISTs that were obtained as clinically progressing metastases during imatinib therapy (). These observations suggest that these receptor tyrosine kinases might play a crucial role in GISTs with loss of KIT expression.
10–15% of non-small cell lung cancers contain oncogenic gain-of-function mutations in EGFR [Citation24,Citation25]. EGFR inhibition by small molecule kinase inhibitors such as gefitinib has become the mainstay of treatment in patients with EGFR-mutant tumors [Citation26]. In the present study, we observed strong EGFR phosphorylation in KIT-negative GIST cells (,). Thus, we hypothesized that EGFR inhibition may provide a relevant salvage option in KIT-negative GISTs resistant to imatinib therapy. However, EGFR inhibition by gefitinib in GIST522 and GIST48B did not affect PI3-K/AKT and RAF/MAPK signaling pathways, cell viability, or colony formation (–). In addition, targeting FAK by shRNA or the inhibitor PF562271 had little effect on cell viability in GIST62, GIST522, and GIST48B ( and S2(b)). These observations suggest that neither EGFR nor FAK is not a targetable tyrosine kinase in GISTs with loss of KIT expression.
AXL is a receptor tyrosine kinase (RTK) with oncogenic potential and transforming activity [Citation27], which is stimulated by its ligand growth arrest specific 6 (Gas6), a vitamin K-dependent growth potentiating factor [Citation28]. AXL is overexpressed in metastatic colon and prostate cancer, mesothelioma, thyroid carcinoma, breast cancer, and melanoma [Citation29–Citation34]. Additionally, Gas6/AXL signaling plays an important role in tumor formation, tumor invasion and metastasis, and tumor cell survival [Citation34–Citation37]. Although AXL has been identified to play a crucial oncogeneic role in GISTs [Citation16] that have converted, during TKI-therapy, to a KIT-independent state, the pathogenic mechanisms of AXL remain unclear. In order to explore the function of AXL, EPHA2, and FAK in KIT-negative GISTs, we stably silenced these genes by lentivirus-mediated shRNAs (). Knockdowns of AXL, EPHA2, and FAK showed little effect on AKT, MAPK, and S6 signaling in GIST62 and GIST522 (). Although GIST62 and GIST522 cell lines preserve activation of the same proliferation and survival signaling intermediates as KIT-positive GISTs (), AKT activation is weaker in KIT-negative GIST cells than in KIT-positive GIST cells (Figure S6). Inhibition of PI3-K showed minimal anti-proliferative effects on GIST62 growth as compared to PI3-K inhibition in KIT-positive GIST882, GIST48 and GIST430 [Citation19]. These observations indicate that PI3-K/AKT may not be a crucial survival pathway in GISTs with loss of KIT expression. AXL knockdown resulted in STAT1 hyperactivation in GIST62 and GIST522, and STAT3 hyperactivation in GIST62, but not in GIST882 (). AXL knockdown dramatically inhibited proliferation in GIST62 and GIST522 (,), accompanied by upregulation of the cell-cycle checkpoint inhibitors p21, p27, and p53 (). Inactivation of AXL by a specific inhibitor R428 or knockdown of AXL by shRNA resulted in a reduction of cell viability in AXL-activated KIT-negative GIST54 through induction of p53/p21 expression (). Recent data have shown that AXL suppresses p53 in melanoma through stabilization of the MDMX-MDM2 complex [Citation38]. Our novel finding also indicates that AXL regulates cell growth in KIT-negative GISTs through a signaling pathway involving AXL/p53/p21, but not PI3K/AKT or RAF/MAPK. Previous studies indicate that STAT1 activation contributes to cell death through upregulation of cell death receptors and ligands such as Fas/FasL or TRAIL/TRAIL-R, induction of nitric oxide synthase expression, and accumulation of p21, leading to cell cycle arrest or apoptosis [Citation39,Citation40]. In addition, STAT1 directly interacts with p53, enhancing DNA damage-induced apoptosis. Different stresses induce different patterns of STAT1 phosphorylation, which determine patterns of gene activation. STAT1 phosphorylation at serine 727 promotes transcriptional activation, while tyrosine 701 phosphorylation is required for dimerization and direct DNA binding [Citation41]. STAT3 plays a pro-apoptotic role by upregulation of Fas, Bax, and p21 [Citation39]. JAK/STAT signaling inhibition by JAK-inhibitor 1 and AG490 results in minimal reduction of cell viability in GIST62 [Citation19], suggesting that JAK/STAT signaling inactivation is not an essential function in these KIT-negative GIST cells. However, our current observations indicate that AXL shRNA inhibits KIT-negative GIST cell growth and cell cycle arrest through STAT1 and STAT3 activation, although the mechanism of this regulation is yet to be established. Our study implicates AXL as a potential therapeutic target in GISTs with loss of KIT oncoprotein expression. Patient-derived orthotopic mouse models have been demonstrated to be good approach for therapeutic target evaluation and anticancer drug discovery, which orthotopic implantation of intact tumour tissue can result in metastasis that mimics that seen in tumor patients [Citation42–Citation44]. . Recently, the effects of regorafenib against an imatinib-resistant recurrent GIST was evaluated in a patient-derived orthotopic xenograft (PDOX) nude mouse model [Citation45] . Thus, further studies in vivo (PDOX) are needed to determine whether AXL inhibition by small molecules can reproduce the effect of AXL expression in GIST.
In the present study, the RTKs EGFR, AXL, and EPHA2 and non-RTK FAK were identified in GISTs with loss of KIT expression. AXL knockdown or inhibition resulted in anti-proliferative effects. By contrast, EGFR, EPHA2 and FAK inhibition had little impact on viability of the KIT-negative GISTs. These findings highlight the potential relevance of AXL/p53 signaling axis as a therapeutic target in a subset of GISTs that have lost KIT oncoprotein expression.
Materials and methods
Antibodies and reagents
An anti-panRTK polyclonal rabbit antibody that recognizes highly conserved epitopes in the tyrosine kinase family was developed by Dr. Fletcher’ lab34. Polyclonal antibodies to AXL and S6 were from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal antibodies to AKT and all phospho-antibodies except phospho-STAT1 (Zymed Laboratories, Invitrogen life Technologies, Carlsbad, CA) were obtained from Cell Signaling Technology (Beverly, MA), Polyclonal antibodies to MAPK were from Zymed Laboratories. Monoclonal mouse antibodies were to PY99, PCNA and p53 (Santa Cruz Biotechnology), cyclin A (Novocastra, Newcastle upon Tyne, UK), p21Cip1/WAF1, STAT1 and STAT3 (Zymed Laboratories), p27Kip1 (BD Transduction Laboratories, San Jose, CA), and β-actin (Sigma-Aldrich, St, Louis, MO). Mouse anti-phosphotyrosine (PY)-sepharose 4B, Protein A- and Protein G-sepharose beads were from Zymed Laboratories. NOVEX Coomassie colloidal blue staining kit, NuPAGE TM 4–12% Bis-Tris Gel, Lipofectamine and plus reagent were from Invitrogen Life Technologies. Phenyl phosphate and polybrene were obtained from Sigma. Gefitinib, R428, and PF562271 were from LC Labs (Woburn, MA) and Selleckchem (Shanghai, China), respectively.
Cell lines
GIST882, GIST48, and GIST430 are KIT-mutant human GIST cell lines established from untreated GIST (GIST882) or from GISTs progressing on imatinib therapy (GIST48 and GIST430) [Citation46]. GIST882IMR, GIST48B, and GIST430B are novel isogenic cell lines of GIST882, GIST48, and GIST430, respectively, which retain the parental oncogenic KIT genomic mutations but have entirely lost KIT protein expression and are therefore KIT-independent. GIST62 was established from an untreated GIST harboring a heterozygous KIT exon 11 in-frame deletion. GIST522 was established from a GIST progressing on imatinib therapy, and has a heterozygous KIT exon 11 in-frame deletion mutation. GIST62, GIST522, GIST48B, GIST430B, GIST54, and GIST226 have undetectable KIT expression and are highly imatinib-resistant. 293T cells were used to prepare lentiviral constructs.
Frozen tumor specimens
All frozen tumor specimens were analyzed histologically, and shown to be composed of >90% neoplastic cells. The Brigham and Women’s Hospital Institutional Review Board approved the experiments, and informed consent was obtained from all subjects.
Development of the double immune-affinity purification approach for proteomic RTK screen
The activated proteins in cancer cell lines or frozen tumor tissues were first purified by a PY affinity column and subsequently purified by panRTK IP. Strongly phosphorylated proteins were identified by tandem mass spectrometry. GIST882 cells expressing strongly activated KIT were used as a positive control. During the double purification, panRTK and KIT IPs were used as a positive control. Whole cell lysates from cell lines were prepared by using lysis buffer (1% NP-40, 50 mM Tris-HCl pH 8.0, 100 mM sodium fluoride, 30 mM sodium pyrophosphate, 2 mM sodium molybdate, 5 mM EDTA, 2 mM sodium orthovanadate) containing protease inhibitors (10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM phenylmethylsulfonyl fluoride). Protein concentrations were determined with the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA, USA). 1 mL of sepharose-conjugated anti-PY was transferred to a disposable 10 mL polypropylene column with a frit and washed with 5X volume of storage buffer (1x PBS containing 0.02% sodium azide) to eliminate 1 mM PY. The column was pre-equilibrated with 5X volume of binding buffer (lysis buffer) before incubating the lysates (10 mg) with beads at 4°C with end-over-end mixing overnight. The flow-through fraction was collected and reloaded on the column 10 times. The column was washed with 1 mL of binding buffer 5 times. The PY proteins were repeatedly eluted with 1.3 mL of elution buffer (100 mM phenyl phosphate in binding buffer) 10 times. The eluates were dialyzed against 1 L of 1x PBS buffer overnight. The sample was concentrated to approximately 100 μL final volume by centricon centrifugal filter devices (Millipore Corporation, Bedford, MA). The majority of PY eluates were used for panRTK IP. Finally, the rest of PY eluates and 15% of panRTK IP fractions are immunoblotted for phosphotyrosine and 85% of panRTK IP fractions were carried out Coomassie gel. Electrophoresis and western blotting were performed as described previously [Citation47]. The hybridization signals were detected by chemiluminescence (ECL, Amersham Pharmacia Biotechnology) and were captured using a FUJI LAS1000-plus chemiluminescence imaging system. Staining and destaining of Coomassie gels were carried out according to the NOVEX Coomassie colloidal blue stain kit (Thermo Fisher Scientific Inc.MA, USA). The strongly phosphorylated protein bands were excised from the Coomassie gel and protein sequences were determined by tandem mass spectrometry.
Identification of novel candidate tyrosine kinase oncoproteins in KIT-negative GIST cell lines by phosphotyrosine-RTK immuno-affinity purification and tandem mass spectrometry
In order to screen activated RTKs in GIST cell lines with loss of KIT oncoprotein expression, strongly phosphorylated tyrosine kinase proteins in GIST62 and GIST522 were firstly purified by PY-RTK immunoprecipitation; and then, as revealed by phosphotyrosine staining in immunoblots, were identified by mass spectrometry. Strongly phosphorylated protein bands at 140 kDa and 170 kDa in GIST62, and 170 kDa in GIST522 were excised from Coomassie gel and protein sequences were determined by mass spectrometry. The blots were then stripped and restained with specific antibodies to validate the candidate tyrosine kinase oncoproteins.
Evaluation of expression and phosphorylation of candidate tyrosine kinase oncoproteins in KIT-negative and KIT-positive GIST cell lines by western blotting
To compare the relative expression and phosphorylation of the candidate tyrosine kinase oncoproteins in KIT-negative and KIT-positive GIST cell lines, 30 μg of protein lysates were separated by SDS-PAGE and immunoblotted with specific phospho- and total antibodies. MESO257, RMS556, and MCF-7 cell lysates were used as controls.
Evaluation of candidate tyrosine kinase oncoprotein activation by Immuno-precipitation and phosphotyrosine immunoblotting
For co-IPs, sepharose-protein G beads with goat polyclonal antibody and sepharose-protein A with rabbit polyclonal antibody were used. 1 mg of protein lysate (500 μL) was preadsorbed for 30 min using 20μl of protein G or protein A beads at 4°C. Then, 2 μg of antibody against rabbit panRTK, AXL, EGFR, EPHA2, FAK, or PI3-Kp85 were added to each supernatant and rocked for 2 h at 4°C. 20 μL of sepharose-protein G or sepharose-protein A beads were added and rocked overnight at 4°C. The lysates were then spun at 10,000 rpm for 2 min at 4°C and beads were washed 3 times with 750 μL of IP buffer for 25 min followed once by 750 μL 10 mM Tris-Cl buffer (pH7.6). 20 μL of loading buffer was added to the beads and boiled for 5 min at 95°C. Tyrosine kinase activation was subsequently evaluated by phosphotyrosine and specific antibody immunoblotting.
RNA preparation and qRT-PCR
AXL RNA expression was evaluated by qRT-PCR in GIST882, GIST882IMR, GIST48, GIST430, GIST62 and GIST522 cells. Total RNA was prepared using the Trizol reagent (Invitrogen, Carlsbad, CA). Reverse transcription PCR was performed with 0.5 μg RNA, using the BioRad iScriptTM cDNA synthesis Kit (BioRad Laboratories, Hercules, CA). qPCR was performed with iQ SYBR green supermix (BioRad) in a reaction volume of 25 μL, using a MyiQ single-color real-time PCR detection system (BioRad). Reactions contained 1 μL cDNA, 400 nM of each primer, and 12.5 μL iQ SYBR green supermix. After 3 min at 95°C, each of the 40 PCR cycles consisted of denaturation for 10 seconds at 95 °C, hybridization of primers and SYBR green, and DNA synthesis for 1 minute at 60 °C. The qRT-PCR assays for AXL (NM_001699) were performed using the following primers: sense: 5ʹ-GACCGGCCAAGTTTTACAGA −3ʹ and anti-sense: 5ʹ-ATAACCTCCACCCTCATCCA-3ʹ [Citation48]; KIT (NM_936229) sense: 5ʹ-ATTGGTATTTTTGTCCAGGAACTGA-3ʹ and anti-sense: 5ʹ-TGGCCCAGATGAGTTTAGTGTCT-3ʹ [Citation49]. As a control, GAPDH (NM_002046) was amplified using the following primers: sense: 5ʹ-GAAGGTGAAGGTCGGAGTCAAC-3ʹ and anti-sense: 5ʹ-TGGAAGATGGTGATGGGATTTC-3ʹ. All primers were obtained from Invitrogen. The comparative Ct (cycle threshold) method was used to determine AXL expression differences in GIST cell lines as compared to the GIST882 cell control. Data (run in triplicate assays) were normalized to GAPDH.
AXL mutation analysis
Total RNA was prepared using Invitrogen Trizol reagent and the manufacturer’s recommended procedure. 2 μg of total RNA was used for each reverse transcription reaction using Sigma Enhanced Avian HS RT-PCR Kit with a 2-step format, as follows: the RT reaction was performed in 20 μL reaction mixture at 42°C, with 1 unit/μL RNAse inhibitor, 1 unit/μL enhanced avian RT, 1.25 μM random nonamer, 250 μM each dATP, dGTP, dCTP and dTTP. 2 μL of the RT reaction was then used for each PCR reaction, at 0.5 μM final concentration of PCR primer pair, 200 μM of dNTP, 0.05 units/μL AccuTaq LA DNA polymerase. PCR reactions were performed as follows: 94 oC for 2 m; 35 cycles of 94 oC for 30 s, 58 oC for 30 s, 68 oC for 1 m 45s; 68 oC for 5 m. The RT-PCR amplified DNA fragments were then separated with an agarose gel and used for AXL cDNA sequencing analysis with appropriate primers. Outer primers used were 1597F (5ʹ-TAATGGACATAGGGCTAAGGCAAGAG-3ʹ) and 2354R (5ʹ-GAGGAAGCTGTGTAGGTCTCCATGTT-3ʹ); and 2217F (5ʹ-GTCTGCATGAAGGAATTTGACCATC-3ʹ) and 2934R (5ʹ-CATCCATGTTGACATAGAGGATTTCG-3ʹ). Inner primers were 1650F (5ʹ-TCTGTGTCCAATCTGACAGTGTGTGT-3ʹ) and 1954R (5ʹ-CGGTAGCTGACTACCAGTTCACCTCT-3ʹ), used for second round PCR and sequencing, and 2537F (5ʹ-CTCCAAGAAGATCTACAATGGGGACT-3ʹ), used as an inner primer for sequencing only.
Lentiviral AXL, EPHA2, and FAK shRNA constructs and virus preparation
shRNA constructs were generated by ligating the following oligomers into the unique AgeI and EcoRI sites of pLKO.1puro (7 kb) lentiviral construct: AXL forward 5ʹ-CCGGCGAAATCCTCTATGTCAACATCTCGAGATGTTGACATAGAGGATTTCGTTTTTG-3ʹ and reverse 5ʹ-AATTCAAAAACGAAATCCTCTATGTCAACATCTCGAGATGTTGACATAGAGGATTTCG-3ʹ. EPHA2 forward 5ʹ-CCGGGCGTATCTTCATTGAGCTCAACTCGAGTTGAGCTCAATGAAGATACGCTTTTTG-3ʹ and reverse 5ʹ-AATTCAAAAGCGTATCTTCATTGAGCTCAACTCGAGTTGAGCTCAATGAAGATACGC-3ʹ. FAK forward 5ʹ-CCGGGCCTTAACAATGCGTCAGTTTCTCGAGAAACTGACGCATTGTTAAGGCTTTTTG-3ʹ and reverse 5ʹ-AATTCAAAAGCCTTAACAATGCGTCAGTTTCTCGAGAAACTGACGCATTGTTAAGGC-3ʹ.
Lentiviruses were produced by cotransfecting pLKO.1puro with AXL, EPHA2, or FAK shRNA, and helper virus packaging plasmids pCMV∆R8.91 and pMD.G (at a 10:10:1 ratio) into 293T cells. Transfections were carried out using lipofectamine and PLUS reagent. Lentiviruses were harvested at 24, 36, 48, and 60 h post-transfection. Viruses were frozen at −80°C in aliquots of appropriate amounts for infection. shRNAs used for AXL, EPHA2, or FAK knockdown were those that accomplished the most efficient knockdowns of their intended targets.
Cell culture and virus infection
GIST62 and GIST882 cell lines were maintained in RPMI 1640 medium plus 15% fetal bovine serum (FBS) containing penicillin/streptomycin and L-glutamine. GIST54/GIST226, and GIST48B/GIST430B were maintained in IMDM medium plus 5% or 15% FBS containing penicillin/streptomycin and L-glutamine, respectively. GIST522, GIST430 and GIST48 were maintained in F10 medium containing 15% FBS, penicillin/streptomycin and L-glutamine. GIST cells were seeded in 6-well plates. Infections were carried out in the presence of 8 μg/mL of polybrene. Cells were lysed for western blot analysis at 96 h post-infection. Following transduction, GIST62, GIST522 and GIST882 were selected with 2.5 μg/mL puromycin for 10 days.
Cell viability analysis
GIST cells were plated at 10,000 cells/well in a 96-well flat-bottomed plate (Falcon, Lincoln, NJ) and cultured in RPMI 1640, IMDM, or F10 medium for 2 days before transduction with lentiviruses containing empty vector, AXL shRNA, EPHA2 shRNA, or FAK shRNA in GIST62, GIST522, and GIST430; or incubated with different concentrations of gefitinib (EGFR inhibitor) in GIST522 and GIST48B, R428 (AXL inhibitor) in GIST54, PF562271 (FAK inhibitor) in GIST48B. Proliferation studies were carried out after 3, 4, 6, 8, and 12 days using the CellTiter-Glo luminescent assay (Promega, Madison, WI), and were quantitated using a Veritas™ Microplate Luminometer from Turner Biosystems (Sunnyvale, CA). Data were normalized to the control group. All assays were performed in quadruplicate wells, and were averaged from two independent transductions in each cell line.
Colony formation assay
Colony formation assays were performed as published previously with minor modifications [Citation12]. In brief, GIST48B and PC-9 cells were plated at 5000 and 3,000 cells/well in 6-well plates, respectively, and cultured in RPMI 1640, for 6 days before treatment with gefitinib. After treatment with inhibitors for 10 days, the medium was removed, the cells were washed with PBS, and the cells were then stained with 0.5% crystal violet in methanol for 5 min. Excess stain was removed by washing with distilled water. Colonies were dissolved by using 900 µL glacial acetic acid and A570 was measured after colonies were photographed. The experiments were performed in duplicate wells and repeated three times.
Cell cycle analysis
After infection with lentivirus for 96 h, GIST62 cells in 6-well plates were trypsinized, spun down, and washed once with Hanks’ Balanced Salt Solution at room temperature. For nuclear staining, a DAPI-containing solution (Nuclear isolation and staining solution, NPE systems, Pembrooke Pines, FL) was added to the cells and the cell suspension was immediately analyzed in a flow cytometer (NPE Quanta, NPE Systems). Data analysis was performed using Modfit LT software 3.1 (Verity Software House, Topsham, ME). The experiments were performed in duplicate wells and repeated three times.
Statistical analysis
Student’s t-tests were performed on data from cells treated with control DMSO or inhibitors, as well as cells treated with empty vector or shRNAs. Statistically significant differences between untreated control and treatment were defined as *P < 0.05, **P < 0.01, and ***P < 0.001.
Author contributions
OW designed research; ZR, TY, NN, WD, PY, NZ, WY, WY, and OW performed research; ZR, TY, NN, EG, WY, and OW analyzed data; EG and OW prepared the manuscript.
Supplemental Material
Download Zip (2 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33:459–465.
- Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813–3825.
- Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580.
- Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710.
- Nannini M, Astolfi A, Paterini P, et al. Expression of IGF-1 receptor in KIT/PDGF receptor-alpha wild-type gastrointestinal stromal tumors with succinate dehydrogenase complex dysfunction. Future Oncol. 2013;9:121–126.
- Killian JK, Miettinen M, Walker RL, et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med. 2014;6:268ra177.
- Tuveson DA, Willis NA, Jacks T, et al. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene. 2001;20:5054–5058.
- Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480.
- Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26:5352–5359.
- Blay JY, Bonvalot S, Casali P, et al. Consensus meeting for the management of gastrointestinal stromal tumors. Report of the GIST Consensus Conference of 20-21 March 2004, under the auspices of ESMO. Ann Oncol. 2005;16:566–578.
- Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–4774.
- Chen W, Kuang Y, Qiu H, et al. Dual Targeting of Insulin Receptor and KIT in Imatinib-Resistant Gastrointestinal Stromal Tumors. Cancer Res. 2017;77:5107–5117.
- Fletcher JA, Corless CL, Dimitrijevic S, et al. Mechanisms of resistance to imatinib mesylate (IM) in advanced gastrointestinal stromal tumor (GIST). Proc Am Soc Clin Oncol. 2003;22:815.
- Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–279.
- Bauer S, Hartmann JT, de Wit M, et al. Resection of residual disease in patients with metastatic gastrointestinal stromal tumors responding to treatment with imatinib. Int J Cancer. 2005;117:316–325.
- Mahadevan D, Cooke L, Riley C, et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene. 2007;26:3909–3919.
- Mahadevan D, Theiss N, Morales C, et al. Novel receptor tyrosine kinase targeted combination therapies for imatinib-resistant gastrointestinal stromal tumors (GIST). Oncotarget. 2015;6:1954–1966.
- Duensing A, Medeiros F, McConarty B, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene. 2004;23:3999–4006.
- Bauer S, Duensing A, Demetri GD, et al. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene. 2007;26:7560–7568.
- Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23:5357–5364.
- Rossi F, Ehlers I, Agosti V, et al. Oncogenic Kit signaling and therapeutic intervention in a mouse model of gastrointestinal stromal tumor. Proc Natl Acad Sci U S A. 2006;103:12843–12848.
- Zhu MJ, Ou WB, Fletcher CD, et al. KIT oncoprotein interactions in gastrointestinal stromal tumors: therapeutic relevance. Oncogene. 2007;26:6386–6395.
- Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295–302.
- Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500.
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139.
- Sequist LV, Bell DW, Lynch TJ, et al. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007;25:587–595.
- Wimmel A, Glitz D, Kraus A, et al. Axl receptor tyrosine kinase expression in human lung cancer cell lines correlates with cellular adhesion. Eur J Cancer. 2001;37:2264–2274.
- Varnum BC, Young C, Elliott G, et al. Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature. 1995;373:623–626.
- Craven RJ, Xu LH, Weiner TM, et al. Receptor tyrosine kinases expressed in metastatic colon cancer. Int J Cancer. 1995;60:791–797.
- Jacob AN, Kalapurakal J, Davidson WR, et al. A receptor tyrosine kinase, UFO/ Axl,and other genes isolated by a modified differential display PCR are overexpressed in metastatic prostatic carcinoma cell line DU145. Cancer Detect Prev. 1999;23:325–332.
- Ito T, Ito M, Naito S, et al. Expression of the Axl receptor tyrosine kinase in human thyroid carcinoma. Thyroid. 1999;9:563–567.
- Meric F, Lee WP, Sahin A, et al. Expression profile of tyrosine kinases in breast cancer. Clin Cancer Res. 2002;8:361–367.
- Quong RY, Bickford ST, Ing YL, et al. Protein kinases in normal and transformed melanocytes. Melanoma Res. 1994;4:313–319.
- Ou WB, Corson JM, Flynn DL, et al. AXL regulates mesothelioma proliferation and invasiveness. Oncogene. 2011;30:1643–1652.
- Kirane A, Ludwig KF, Sorrelle N, et al. Warfarin blocks Gas6-mediated axl activation required for pancreatic cancer epithelial plasticity and metastasis. Cancer Res. 2015;75:3699–3705.
- May CD, Garnett J, Ma X, et al. AXL is a potential therapeutic target in dedifferentiated and pleomorphic liposarcomas. BMC Cancer. 2015;15:901.
- Rea K, Pinciroli P, Sensi M, et al. Novel Axl-driven signaling pathway and molecular signature characterize high-grade ovarian cancer patients with poor clinical outcome. Oncotarget. 2015;6:30859–30875.
- de PA, Luo Z, Gerarduzzi C, et al. AXL receptor signalling suppresses p53 in melanoma through stabilization of the MDMX-MDM2 complex. J Mol Cell Biol. 2017;9:154–165.
- Battle TE, Frank DA. The role of STATs in apoptosis. Curr Mol Med. 2002;2:381–392.
- Kim HS, Lee MS. STAT1 as a key modulator of cell death. Cell Signal. 2007;19:454–465.
- Townsend PA, Scarabelli TM, Davidson SM, et al. STAT-1 interacts with p53 to enhance DNA damage-induced apoptosis. J Biol Chem. 2004;279:5811–5820.
- Hoffman RM. Orthotopic metastatic mouse models for anticancer drug discovery and evaluation: a bridge to the clinic. Invest New Drugs. 1999;17:343–359.
- Hoffman RM. Patient-derived orthotopic xenografts: better mimic of metastasis than subcutaneous xenografts. Nat Rev Cancer. 2015;15:451–452.
- Coleman WB, Tsongalis GJ. Patient-derived mouse models of cancer. Molecular and translational medicine. Series eds. Switzerland: Springer Intl. Publishing AG; 2017. ISSN: 2197-7852.
- Miyake K, Kawaguchi K, Kiyuna T, et al. Regorafenib regresses an imatinib-resistant recurrent gastrointestinal stromal tumor (GIST) with a mutation in exons 11 and 17 of c-kit in a patient-derived orthotopic xenograft (PDOX) nude mouse model. Cell Cycle. 2018;17:722–727.
- Ou WB, Zhu MJ, Demetri GD, et al. Protein kinase C-theta regulates KIT expression and proliferation in gastrointestinal stromal tumors. Oncogene. 2008;27:5624–5634.
- Rubin BP, Singer S, Tsao C, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–8121.
- Malard V, Berenguer F, Prat O, et al. Global gene expression profiling in human lung cells exposed to cobalt. BMC Genomics. 2007;8:147.
- Théou N, Tabone S, Saffroy R, et al. High expression of both mutant and wild-type alleles of c-kit in gastrointestinal stromal tumors. Biochim Biophys Acta. 2004;1688:250–256.