
ABSTRACT
Rapamycin inhibits cell proliferation, yet preserves (re)-proliferative potential (RPP). RPP is a potential of quiescent cells that is lost in senescent cells. mTOR drives conversion from quiescence to senescence (geroconversion). By suppressing geroconversion, rapamycin preserves RPP. Geroconversion is characterized by proliferation-like levels of phospho-S6K/S6/4E-BP1 in nonproliferating cells arrested by p16 and/or p21. mTOR-driven geroconversion is associated with cellular hyperfunction, which in turn leads to organismal aging manifested by age-related diseases.
Introduction
In brief: proliferative potential is not actual proliferation
Rapamycin and other inhibitors of mTOR (mammalian Target of Rapamycin) maintain proliferative potential in non-proliferating cells [Citation1,Citation2]. This should not be misunderstood to mean stimulation of proliferation. In fact, rapamycin slows proliferation. The potential to proliferate is not actual proliferation; rather it is a hidden feature of quiescent cells that renders quiescence reversible. Rapamycin maintains the potential to proliferate in non-proliferating cells, enabling these cells can re-start proliferation when needed. We can use the term Re-Proliferative Potential (RPP) instead of proliferative potential to avoid confusion with actual proliferation.
In brief: is irreversible arrest reversible?
In senescence, cell cycle arrest cannot be reversed through growth stimulation using methods such as serum stimulation. However, this seemingly irreversible arrest can be reversed by switching off p16, p53/p21 and Rb [Citation3–Citation9]. Still, in practical terms, this arrest is irreversible because after re-entering the cell cycle from senescence, cells cannot proliferate (lost RPP) and eventually die in the attempt [Citation9,Citation10]. In Alzheimer’s disease, for example, senescent neurons die after re-entering the cell cycle [Citation11–Citation13].
In brief: cell cycle arrest is not growth arrest
Growth of cellular mass and the cell cycle can be dissociated [Citation14–Citation20]. Normally, when a cell is arrested, it does not grow; it exists in a state known as quiescence, or G0 arrest [Citation21–Citation23]. During conversion from quiescence to senescence, or geroconversion, cellular size continues to increase exponentially until the cells acquire the senescent phenotype [Citation17,Citation24]. Geroconversion is thus a form of growth rather than of growth arrest [Citation1,Citation25], and it is driven by mTOR [Citation22,Citation23,Citation26]. mTOR stimulates, rather than inhibits, cellular growth.
In brief: senescence is not just cell cycle arrest, and cell cycle arrest is not yet senescence
To become senescent, an arrested cell must undergo geroconversion [Citation22–Citation29]. As we will discuss, geroconversion is associated not only with loss of RPP, but also with a cellular hypertrophic (a large cell morphology), hypersecretory and hyperinflammatory phenotype (or senescence-associated secretory phenotype [SASP]) as well as lysosomal hyperactivation (β-Gal staining) and, most importantly, cell type-specific hyperfunction, which contributes to age-related diseases.
Two-step geroconversion model of cellular senescence
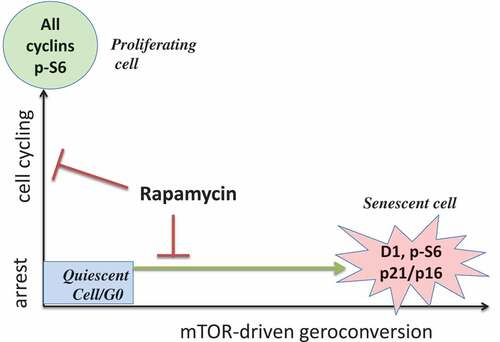
In vitro, senescence involves two conflicting events: (i) cell cycle arrest and (ii) growth stimulation [Citation15]. Growth stimulation drives conversion from arrest to senescence (geroconversion) [Citation22,Citation30]. Thus, a senescence program consists of arrest followed by geroconversion (), which is driven in part by the growth-promoting mTOR pathway [Citation1,Citation22,Citation25,Citation30]. When the cell cycle is arrested but mTOR is still active, senescence develops [Citation1].
Figure 1. mTOR-driven geroconversion from quiescence to senescence.

Senescence cannot be completely understood in the realm of cell cycle arrest alone; mTOR-driven geroconversion must also be considered (). In a two-dimensional model, markers of cell cycle arrest (p16 and p21) are accompanied by growth markers (phospho-S6 and cyclin D1). This two-dimensional view of senescence (cell cycle arrest plus geroconversion, ) enables us to not only to reconcile seemingly contradictory published findings, but also to manipulate and suppress cellular senescence. Rapalogs such as rapamycin and everolimus, as well as the pan-mTOR inhibitors, all suppress geroconversion and maintain quiescence [Citation1,Citation2,Citation31–Citation34].
Figure 2. Two dimensional model: arrest vs mTOR activity.
Geroconversion and markers of senescence
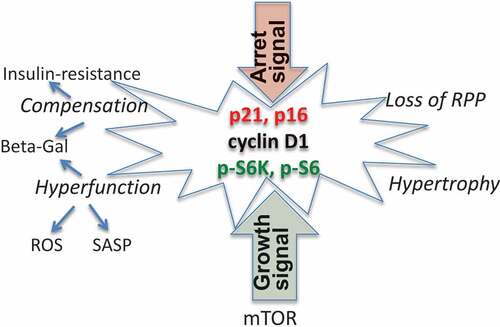
During geroconversion, the mTOR and ERK/MAPK pathways are active, while cell cycling is blocked by p16/p21 (). In a futile attempt to overcome the p16/p21-induced block, cyclin D1 is hyper-induced. At such high levels, cyclin D1 is a marker of senescence rather than of proliferation [Citation10,Citation35–Citation38]. mTOR-driven geroconversion is associated with cellular hypertrophy [Citation17,Citation24,Citation39] and hyperfunctions such as hypersecretion (or SASP) [Citation40–Citation44], ROS production [Citation45], and lysosomal activation (β-Gal staining) [Citation46–Citation50]. These hyperfunctions in turn provoke compensatory reactions such as growth factor and insulin resistance [Citation51–Citation57], further lysosomal hyperactivation [Citation49], and loss of RPP [Citation10,Citation38].
Figure 3. Senescent cells.
So-called golden marker of senescence and mTOR inhibitors
When the conflicting model of cellular senescence outlined above was published in 2003 [Citation15], the golden marker of senescence was permanent arrest. We therefore tested whether rapamycin could prevent this “golden marker.” Certainly, rapamycin decreases other markers of cellular senescence such as cellular hypertrophy [Citation2,Citation8,Citation17,Citation33,Citation58,Citation59] and the hypersecretory and hyperinflammatory phenotypes [Citation8,Citation40,Citation60–Citation63]. But these effects were anticipated, as rapamycin is a known antihypertrophic [Citation25] and anti-inflammatory agent [Citation64]. In contrast, the prediction that rapamycin would preserve proliferative potential (RPP) is counterintuitive. After all, rapamycin inhibits proliferation, which makes it crucial to confirm this prediction.
HT-p21 and HT-p16 cells respectively express IPTG-inducible p21 and p16 [Citation1,Citation9,Citation65,Citation66]. Cell cycle arrest can be switched off and on in these cells through the addition and removal of IPTG. If arrest was induced for only 1–2 d, proliferation re-started in most cells after removal of the IPTG. When arrest lasted longer than 3–4 d, however, cell proliferation did not re-start after IPTG removal [Citation9]. When cells were treated with IPTG in the presence of rapamycin, cell proliferation re-started after the IPTG and rapamycin were washed out. Thus, rapamycin preserved RPP in p21/p16-arrested cells. Rapamycin similarly preserved RPP during cell cycle arrest caused by pharmacologic inhibitors of CDK4/6, DNA damaging drugs, HDACi and phorbol ester [Citation29,Citation58,Citation65–Citation67]. Suppression of senescence by rapamycin was further confirmed in vitro and in vivo [Citation26,Citation40,Citation62–Citation81]. In addition to rapamycin, everolimus and ridaforolimus (two rapalogs), pan-mTOR inhibitors, nutlin-3a (a p53-inducer), hypoxia and contact inhibition all inhibit mTOR and thus maintain RPP in arrested cells [Citation2,Citation32–Citation34,Citation58,Citation82–Citation86]. Suppression of senescence by pan-mTOR inhibitors is closely associated with dephosphorylation of 4E-BP1 at both rapamycin-sensitive and -insensitive sites [Citation2,Citation31–Citation34].
Irreversible proliferative arrest due to loss of RPP
Although senescent cell cycle arrest is often said to be irreversible, it is technically reversible, if the correct method is used. It cannot be reversed using serum, nutrients, growth factors or other stimuli. Serum reverses quiescence caused by serum withdrawal, but serum stimulation causes senescence when the cell cycle is blocked by p21 or p16 [Citation1,Citation58]. Similarly, quiescence caused by contact inhibition can be reversed by splitting cell cultures, but splitting senescent cultures only deepens senescence because mTOR is activated in sparse cell cultures [Citation84,Citation87,Citation88]. It has therefore been suggested that the term “irreversible” be narrowed to “irreversible by mitogenic or oncogenic stimuli” [Citation7].
Consider the mTOR-driven model of senescence. In quiescent cells, mTOR is deactivated (by serum/nutrient withdrawal, contact inhibition, hypoxia, etc.) and cyclin D1 is low; cells do not cycle and do not grow. Growth stimuli activate mTOR and induce cyclin D1, causing proliferation. However, strong growth stimuli can cause proliferation that is followed by arrest and geroconversion. For example, oncogenic Ras and Akt activate mTOR and induce cyclinD1, causing proliferation. But they can simultaneously induce p53, p21 and p16, thereby blocking the cell cycle [Citation8,Citation34]. This block cannot be reversed by growth stimulation, which only deepens the block and enhances mTOR-dependent geroconversion, but it can be reversed by inactivating p53, p21 and p16, for instance [Citation3,Citation15,Citation89]. Once the cell cycle is unblocked, senescent cells re-enter the cell cycle but cannot undergo mitosis [Citation9,Citation10]). Moreover, these cells are hypermotile and literally tear themselves apart and eventually die (see micro-video in ref [Citation10].). Thus, while cell cycle arrest is formally reversible, the loss of RPP renders it irreversible in practical terms. However, because rapamycin maintains RPP, cells in culture can regenerate once the cell cycle is unblocked.
Molecular definition of senescence
Although senescence can be defined as arrest that is irreversible by mitogenic or oncogenic (mTOR-activating) stimuli, this definition cannot be easily used in practice. Furthermore, RPP is a “potential” and is therefore difficult to test, especially in vivo. Defining senescence based on β-Gal staining is also problematic. β-Gal-staining is a marker of lysosomal hyperfunction [Citation46–Citation50]. Consequently, serum-starved and contact-inhibited cells are β-Gal-positive too [Citation46,Citation84], although these cells are not senescent (). So can we distinguish β-Gal-positive quiescent and senescent cells? In β-Gal-positive quiescent cells, levels of phosphorylated S6, S6K and 4E-BP1 are low or undetectable (). In contrast, these proteins are highly phosphorylated in senescent cells (). In β-Gal-positive quiescent cells, insulin and other growth factors induce phospho-S6, whereas in senescent and proliferating cells, phospho-S6 is not further induced upon stimulation.
Figure 4. Characteristics of the main nonproliferative conditions.
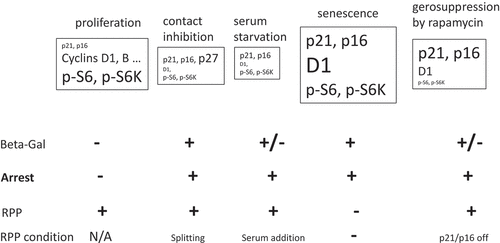
We can define senescence as practically irreversible arrest, a non-proliferative state, associated with proliferation-like mTOR activity (high levels of phospo-S6/S6K/4E-BP1). In addition, high levels of phospho-ERK and cyclin D1 coexist with p21 and/or p16 (), and are associated with hypertrophy and hyperfunctions, including SASP, lysosomal hyperfunction (β-Gal staining), lipid synthesis (oil red O staining), ROS and lactate production. We suggest such cells can be identified using double-staining for phospho-S6 plus p16/p21, phospho-S6 plus β-Gal, or p16/p21 plus cyclin D1. A combination of all these markers may be the most valuable ().
Cell culture and the organism
Rapamycin inhibits growth and slows geroconversion, which is a continuation of growth. In analogous fashion, organismal aging is a continuation of developmental growth [Citation90–Citation98]. Rapamycin (at high doses) slows cell proliferation within the organism, causing leucopenia, thrombocytopenia and mucositis and also decelerates organismal aging and its manifestations: age-related diseases [Citation92].
In cultured cells, the senescence program consists of two steps: arrest plus geroconversion. Because most cells within organisms are quiescent, senescence consists of slow geroconversion. Why is it so slow? Contact inhibition and high cell density [Citation84], hypoxia [Citation83,Citation99], and serum/nutrient starvation each deactivate mTOR. Within the organism, most cells are confluent or contact inhibited, and oxygen tension (1–3% O2) as well as levels of nutrients/growth factors are low. These growth-limiting conditions may maintain quiescence for decades during a human lifespan.
In vitro, senescence is induced in sparse cell cultures in the presence of 21% oxygen and high levels of growth factors and nutrients. For example, glucose levels in DMEM are 5-fold higher than in normal blood, corresponding to levels associated with diabetic coma and causing complete insulin-resistance [Citation55]. As a result, geroconversion is a fast event in vitro, especially in cancer cells. In fact, mTOR activity is much higher in cultured cells than in the organism and is inversely related to Akt activity [Citation87].
Geroconversion and disease
mTOR-driven geroconversion is associated with enhanced tissue-specific functions (hyperfunctions), which drive age-related diseases. For example, vascular smooth muscle cell contraction, hypertrophy and hyperplasia all contribute to hypertension and atherosclerosis. Hyperfunction of adipocytes and hepatocytes increase blood cholesterol levels, contributing to atherosclerosis. Atherosclerosis, hypertension and thrombosis (due to platelet hyperfunction) can culminate in stroke and infarction and subsequent loss of organ function. Therefore, initial hyperfunction eventually leads to dysfunction and functional decline [Citation90,Citation100].
It was known by 2006 that rapamycin delays most age-related diseases [Citation90]. As predicted [Citation90], rapamycin prolongs the lifespan of mice [Citation60,Citation75,Citation101–Citation122]. Rapamycin and everolimus have been tested in healthy volunteers [Citation123,Citation124] and in the elderly [Citation125–Citation127]. A combination of rapamycin with several conventional life-extending drugs, known as the Koschei formula, is already being used for the elderly in the Alan Green Clinic in Little Neck, New York (https://rapamycintherapy.com).
“Repetitio est mater studiorum”
Rapamycin and other mTOR inhibitors suppress geroconversion. In the presence of rapamycin, time moves slower for cell growth, cycling and geroconversion [Citation128]. Rapamycin does not reverse cell cycle arrest and does not stimulate proliferation. Like all gerosuppressants, rapamycin slows cell growth and geroconversion in arrested cells. Rapamycin also maintains RPP in quiescent/arrested cells. To observe this effect, the cell cycle block must be relieved – for example, by decreasing p16 and p21.
Two paradoxical effects of rapamycin
Notably, a two-dimensional model of aging predicts contradictory events.
1. Paradoxical prevention of cell cycle arrest by rapamycin
In replicative senescence, geroconversion starts before cell cycle arrest [Citation30]. Therefore, to prevent geroconversion, rapamycin should be added to proliferating cells. Although rapamycin may itself cause cell cycle arrest, transient treatment or low doses of rapamycin can trick cells, thereby preventing replicative senescence [Citation34,Citation59,Citation129]. However, this phenomenon is not as it seems. Cells exhibit clonal proliferation after rapamycin as well as a change in the karyotype of chromosome 3 in the region containing the nucleolar organizer [Citation129]. This proliferation is thus linked to chromosomal rearrangement, but a selective advantage is detected only in cells in which mTOR activity was inhibited. Similarly, rapamycin increases the efficiency of the cellular reprogramming of induced pluripotent stem cells (iPSCs) [Citation130–Citation132]. For example, brief treatment with nanomolar concentrations of rapamycin enhances cellular reprogramming, though more sustained treatment decreases reprogramming [Citation132].
2. Paradoxical senescence-like state
In cell culture, high concentrations of rapamycin cause arrest in some cell types [Citation133–Citation136]. In the arrested cells, rapamycin slows geroconversion but does not block it completely [Citation2,Citation65]. This initially-reversible arrest may, in theory, slowly lead to geroconversion in the cultured cells. Thus, rapamycin does not cause senescence per se: it merely cannot suppress geroconversion completely. Rapamycin-induced senescence has been observed in vitro [Citation135]. As an illuminating example, consider p53. Nutlin-3a, a p53-inducing drug, causes senescence in some cell types [Citation137,Citation138]. More precisely, nutlin-3a-induced p53 arrests the cell cycle, after which mTOR drives geroconversion in the arrested cells [Citation82,Citation138]. On the other hand, p53 (depending on its level and cell type) may slow geroconversion by inhibiting mTOR [Citation42,Citation138–Citation140]. For example, a 3-day exposure to nutlin-3a causes reversible quiescence [Citation141,Citation142], whereas p21 and p16 induce senescence in the same cells [Citation82]. By inhibiting geroconversion, nutlin-3a slows p21- and p16-induced senescence [Citation82]. Suppression of senescence by p53 was observed in several models [Citation60,Citation143–Citation145].
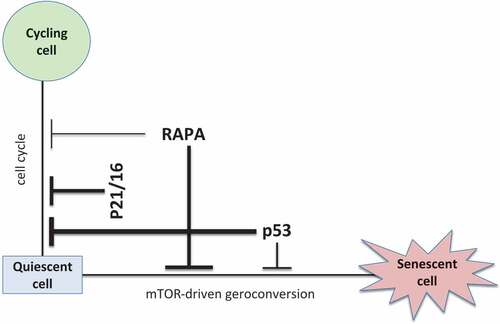
What is the difference between rapamycin, p53, and p21/p16? As illustrated in , Rapamycin causes cell type-specific arrest but slows geroconversion universally. P53 readily and universally causes arrest but inhibits geroconversion under certain conditions [Citation139]. P21 and p16 firmly and universally block the cell cycle without affecting geroconversion.
Figure 5. Paradoxical effects of p53 and rapamycin.
Disclosure statement
No potential conflict of interest was reported by the author.
References
- Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008;7:3355–3361.
- Leontieva OV, Blagosklonny MV. Gerosuppression by pan-mTOR inhibitors. Aging (Albany NY). 2016;8(12):3535–3551.
- Shay JW, Pereira-Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res. 1991;196(1):33–39.
- Beausejour CM, Krtolica A, Galimi F, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. Embo J. 2003;22(16):4212–4222.
- Sage J, Miller AL, Perez-Mancera PA, et al. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424(6945):223–228.
- Dirac AM, Bernards R. Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J Biol Chem. 2003;278(14):11731–11734.
- Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15(7):397–408.
- Astle MV, Hannan KM, Ng PY, et al. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene. 2012;31(15):1949–1962.
- Chang BD, Broude EV, Fang J, et al. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene. 2000;19(17):2165–2170.
- Leontieva OV, Lenzo F, Demidenko ZN, et al. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle. 2012;11(24):4642–4649.
- Bonda DJ, Evans TA, Santocanale C, et al. Evidence for the progression through S-phase in the ectopic cell cycle re-entry of neurons in Alzheimer disease. Aging (Albany NY). 2009;1(4):382–388.
- Khurana V, Lu Y, Steinhilb ML, et al. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol. 2006;16(3):230–241.
- Song B, Davis K, Liu XS, et al. Inhibition of Polo-like kinase 1 reduces beta-amyloid-induced neuronal cell death in Alzheimer’s disease. Aging (Albany NY). 2011;3(9):846–851.
- Kerkhoff E, Rapp UR. High-intensity Raf signals convert mitotic cell cycling into cellular growth. Cancer Res. 1998;58(8):1636–1640.
- Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003;4:358–362.
- Sumikawa E, Matsumoto Y, Sakemura R, et al. Prolonged unbalanced growth induces cellular senescence markers linked with mechano transduction in normal and tumor cells. Biochem Biophys Res Commun. 2005;335(2):558–565.
- Demidenko ZN, Blagosklonny MV. Quantifying pharmacologic suppression of cellular senescence: prevention of cellular hypertrophy versus preservation of proliferative potential. Aging (Albany NY). 2009;1(12):1008–1016.
- Kobayashi Y, Lee SS, Arai R, et al. ERK1/2 mediates unbalanced growth leading to senescence induced by excess thymidine in human cells. Biochem Biophys Res Commun. 2012;425(4):897–901.
- Takauji Y, En A, Miki K, et al. Combinatorial effects of continuous protein synthesis, ERK-signaling, and reactive oxygen species on induction of cellular senescence. Exp Cell Res. 2016;345(2):239–246.
- Polymenis M, Kennedy BK. Unbalanced Growth, Senescence and Aging. Adv Exp Med Biol. 2017;1002:189–208.
- Blagosklonny MV, Pardee AB. The restriction point of the cell cycle. Cell Cycle. 2002;1(2):103–110.
- Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY). 2012;4(3):159–165.
- Terzi MY, Izmirli M, Gogebakan B. The cell fate: senescence or quiescence. Mol Biol Rep. 2016;43(11):1213–1220.
- Wright J, Dungrawala H, Bright RK, et al. A growing role for hypertrophy in senescence. FEMS Yeast Res. 2013;13(1):2–6.
- Blagosklonny MV, Hall MN. Growth and aging: a common molecular mechanism. Aging (Albany NY). 2009;1(4):357–362.
- Cho S, Hwang ES. Status of mTOR activity may phenotypically differentiate senescence and quiescence. Mol Cells. 2012;33(6):597–604.
- Sousa-Victor P, Gutarra S, Garcia-Prat L, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506(7488):316–321.
- Sousa-Victor P, Perdiguero E, Munoz-Canoves P. Geroconversion of aged muscle stem cells under regenerative pressure. Cell Cycle. 2014;13(20):3183–3190.
- Leontieva OV, Blagosklonny MV. Tumor promoter-induced cellular senescence: cell cycle arrest followed by geroconversion. Oncotarget. 2015;5(24):12715–12727.
- Blagosklonny MV. Geroconversion: irreversible step to cellular senescence. Cell Cycle. 2014;13(23):3628–3635.
- Sousa-Victor P, Garcia-Prat L, Munoz-Canoves P. Dual mTORC1/C2 inhibitors: gerosuppressors with potential anti-aging effect. Oncotarget. 2015;6(27):23052–23054.
- Walters HE, Deneka-Hannemann S, Cox LS. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging (Albany NY). 2016;8(2):231–244.
- Leontieva OV, Demidenko ZN, Blagosklonny MV. Dual mTORC1/C2 inhibitors suppress cellular geroconversion (a senescence program). Oncotarget. 2015;6(27):23238–23248.
- Kolesnichenko M, Hong L, Liao R, et al. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012;11(12):2391–2401.
- Berardi P, Meyyappan M, Riabowol KT. A novel transcriptional inhibitory element differentially regulates the cyclin D1 gene in senescent cells. J Biol Chem. 2003;278(9):7510–7519.
- Burton DG, Sheerin AN, Ostler EL, et al. Cyclin D1 overexpression permits the reproducible detection of senescent human vascular smooth muscle cells. Ann N Y Acad Sci. 2007;1119:20–31.
- Averous J, Fonseca BD, Proud CG. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene. 2008;27(8):1106–1113.
- Leontieva OV, Demidenk ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Deth Diff. 2013;20(9):1241–1249.
- Bilinski T, Paszkiewicz T, Zadrag-Tecza R. Energy excess is the main cause of accelerated aging of mammals. Oncotarget. 2015;6(15):12909–12919.
- Laberge RM, Sun Y, Orjalo AV, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17(8):1049–1061.
- Hasty P, Sharp ZD, Curiel TJ, et al. mTORC1 and p53: clash of the gods? Cell Cycle. 2013;12(1):20–25.
- Wiley CD, Schaum N, Alimirah F, et al. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci Rep. 2018;8(1):2410.
- Serrano M. The inflammTORy powers of senescence. Trends Cell Biol. 2015;25(11):634–636.
- Garbers C, Kuck F, Aparicio-Siegmund S, et al. Cellular senescence or EGFR signaling induces Interleukin 6 (IL-6) receptor expression controlled by mammalian target of rapamycin (mTOR). Cell Cycle. 2013;12(21):3421–3432.
- Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008;7:3344–3354.
- Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92(20):9363–9367.
- Severino J, Allen RG, Balin S, et al. Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp Cell Res. 2000;257(1):162–171.
- Lee BY, Han JA, Im JS, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5(2):187–195.
- Young AR, Narita M. Spatio-temporal association between mTOR and autophagy during cellular senescence. Autophagy. 2011;7(11):1387–1388.
- Itahana K, Campisi J, Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007;371:21–31.
- Zhang H, Bajraszewski N, Wu E, et al. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730–738.
- Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–1656.
- Tremblay F, Krebs M, Dombrowski L, et al. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes. 2005;54:2674–2684.
- Selman C, Tullet JM, Wieser D, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–144.
- Leontieva OV, Demidenko ZN, Blagosklonny MV. Rapamycin reverses insulin resistance (IR) in high-glucose medium without causing IR in normoglycemic medium. Cell Death Dis. 2014;5:e1214.
- Demidenko ZN, Blagosklonny MV. The purpose of the HIF-1/PHD feedback loop: to limit mTOR-induced HIF-1alpha. Cell Cycle. 2011;10(10):1557–1562.
- Blagosklonny MV. TOR-centric view on insulin resistance and diabetic complications: perspective for endocrinologists and gerontologists. Cell Death Dis. 2013;4(12):e964.
- Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY). 2010;2(12):924–935.
- Pospelova TV, Leontieva OV, Bykova TV, et al. Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle. 2012;11(12):2402–2407.
- Christy B, Demaria M, Campisi J, et al. p53 and rapamycin are additive. Oncotarget. 2015;6(18):15802–15813.
- Singh M, Jensen MD, Lerman A, et al. Effect of low-dose rapamycin on senescence markers and physical functioning in older adults with coronary artery disease: results of a pilot study. J Frailty Aging. 2016;5(4):204–207.
- Wang R, Yu Z, Sunchu B, et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell. 2017;16(3):564–574.
- Herranz N, Gallage S, Mellone M, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015;17(9):1205–1217.
- Attur MG, Patel R, Thakker G, et al. Differential anti-inflammatory effects of immunosuppressive drugs: cyclosporin, rapamycin and FK-506 on inducible nitric oxide synthase, nitric oxide, cyclooxygenase-2 and PGE2 production. Inflamm Res. 2000;49(1):20–26.
- Demidenko ZN, Zubova SG, Bukreeva EI, et al. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–1895.
- Leontieva OV, Blagosklonny MV. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle. 2013;12(18):3063–3069.
- Romanov VS, Abramova MV, Svetlikova SB, et al. p21(Waf1) is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle. 2010;9(19):3945–3955.
- Hinojosa CA, Mgbemena V, Van Roekel S, et al. Enteric-delivered rapamycin enhances resistance of aged mice to pneumococcal pneumonia through reduced cellular senescence. Exp Gerontol. 2012;47(12):958–965.
- Luo Y, Li L, Zou P, et al. Rapamycin enhances long-term hematopoietic reconstitution of ex vivo expanded mouse hematopoietic stem cells by inhibiting senescence. Transplantation. 2014;97(1):20–29.
- Gu Z, Tan W, Ji J, et al. Rapamycin reverses the senescent phenotype and improves immunoregulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling pathway. Aging (Albany NY). 2016;8(5):1102–1114.
- Singh M, Jensen MD, Lerman AK, et al. Effect of low-dose rapamycin on senescence markers and physical functioning in older adults with coronary artery disease: results of a pilot study. J Frailty Aging. 2016;5(4):204–207.
- Houssaini A, Breau M, Kebe K, et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight. 2018;3(3). pii: 93203. DOI:10.1172/jci.insight.93203
- Gao C, Ning B, Sang C, et al. Rapamycin prevents the intervertebral disc degeneration via inhibiting differentiation and senescence of annulus fibrosus cells. Aging (Albany NY). 2018;10(1):131–143.
- Carroll B, Nelson G, Rabanal-Ruiz Y, et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J Cell Biol. 2017;216(7):1949–1957.
- Chen C, Liu Y, Liu Y, et al. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009;2(98):ra75.
- Coudre C, Alani J, Ritchie W, et al. HIF-1alpha and rapamycin act as gerosuppressant in multiple myeloma cells upon genotoxic stress. Cell Cycle. 2015;15(16):2174–2182.
- Barilari M, Bonfils G, Treins C, et al. ZRF1 is a novel S6 kinase substrate that drives the senescence programme. Embo J. 2017;36(6):736–750.
- Selman C, Sinclair A, Pedroni SM, et al. Evidence that hematopoietic stem cell function is preserved during aging in long-lived S6K1 mutant mice. Oncotarget. 2016;7(21):29937–29943.
- Dou X, Sun Y, Li J, et al. Short-term rapamycin treatment increases ovarian lifespan in young and middle-aged female mice. Aging Cell. 2017;16(4):825–836.
- Chiao YA, Kolwicz SC, Basisty N, et al. Rapamycin transiently induces mitochondrial remodeling to reprogram energy metabolism in old hearts. Aging (Albany NY). 2016;8(2):314–327.
- Sung J, Lee K, Kim J, et al. Interaction between mTOR pathway inhibition and autophagy induction attenuates adriamycin-induced vascular smooth muscle cell senescence through decreased expressions of p53/p21/p16. Exp Gerontol. 2017;16(16):30440–30443. pii: S0531–5565.
- Demidenko ZN, Korotchkina LG, Gudkov AV, et al. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010;107(21):9660–9664.
- Leontieva OV, Natarajan V, Demidenko ZN, et al. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci U S A. 2012;109(33):13314–13318.
- Leontieva OV, Demidenko ZN, Blagosklonny MV. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc Natl Acad Sci U S A. 2014;111(24):8832–8837.
- Leontieva OV, Demidenko ZN, Blagosklonny MV. S6K in geroconversion. Cell Cycle. 2013;12(20):3249–3252.
- Pospelova TV, Demidenko ZN, Bukreeva EI, et al. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009;8:4112–4118.
- Leontieva OV, Blagosklonny MV. Gerosuppression in confluent cells. Aging (Albany NY). 2014;6(12):1010–1018.
- Sun P. Contact inhibition against senescence. Oncotarget. 2014;5(17):7212–7213.
- Ferbeyre G, de Stanchina E, Lin AW, et al. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol. 2002;22(10):3497–3508.
- Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006;5(18):2087–2102.
- Blagosklonny MV. TOR-driven aging: speeding car without brakes. Cell Cycle. 2009;8(24):4055–4059.
- Blagosklonny MV. Answering the ultimate question “what is the proximal cause of aging?”. Aging (Albany NY). 2012;4(12):861–877.
- Blagosklonny MV. Aging is not programmed: genetic pseudo-program is a shadow of developmental growth. Cell Cycle. 2013;12(24):3736–3742.
- Gems D, de la Guardia Y. Alternative perspectives on aging in caenorhabditis elegans: reactive oxygen species or hyperfunction? Antioxid Redox Signal. 2013;19(3):321–329.
- Gems D, Partridge L. Genetics of longevity in model organisms: debates and paradigm shifts. Annu Rev Physiol. 2013;75:621–644.
- de la Guardia Y, Gilliat AF, Hellberg J, et al. Run-on of germline apoptosis promotes gonad senescence in C. elegans. Oncotarget. 2016;7(26):39082–39096.
- Wang H, Zhang Z, Gems D. Monsters in the uterus: teratoma-like tumors in senescent C. elegans result from a parthenogenetic quasi-program. Aging (Albany NY). 2018;10(6):1188–1189.
- Wang H, Zhao Y, Ezcurra M, et al. A parthenogenetic quasi-program causes teratoma-like tumors during aging in wild-type C. elegans. NP J Aging Mech Dis. 2018;4:6.
- Brugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18(23):2893–2904.
- Blagosklonny MV. Prospective treatment of age-related diseases by slowing down aging. Am J Pathol. 2012;181(4):1142–1146.
- Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–395.
- Anisimov VN, Zabezhinski MA, Popovich IG, et al. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010;176(5):2092–2097.
- Anisimov VN, Zabezhinski MA, Popovich IG, et al. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle. 2011;10(24):4230–4236.
- Miller RA, Harrison DE, Astle CM, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011;66(2):191–201.
- Wilkinson JE, Burmeister L, Brooks SV, et al. Rapamycin slows aging in mice. Aging Cell. 2012;11(4):675–682.
- Comas M, Toshkov I, Kuropatwinski KK, et al. New nanoformulation of rapamycin Rapatar extends lifespan in homozygous p53-/- mice by delaying carcinogenesis. Aging (Albany NY). 2012;4(10):715–722.
- Komarova EA, Antoch MP, Novototskaya LR, et al. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53± mice. Aging (Albany NY). 2012;4(10):709–714.
- Livi CB, Hardman RL, Christy BA, et al. Rapamycin extends life span of Rb1± mice by inhibiting neuroendocrine tumors. Aging (Albany NY). 2013;5(2):100–110.
- Neff F, Flores-Dominguez D, Ryan DP, et al. Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest. 2013;123(8):3272–3291.
- Fok WC, Chen Y, Bokov A, et al. Mice fed rapamycin have an increase in lifespan associated with major changes in the liver transcriptome. PLoS One. 2014;9(1):e83988.
- Leontieva OV, Gm P, Blagosklonny MV. Weekly administration of rapamycin improves survival and biomarkers in obese male mice on high-fat diet. Aging Cell. 2014;13(4):616–622.
- Miller RA, Harrison DE, Astle CM, et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell. 2014;13(3):468–477.
- Popovich IG, Anisimov VN, Zabezhinski MA, et al. Lifespan extension and cancer prevention in HER-2/neu transgenic mice treated with low intermittent doses of rapamycin. Cancer Biol Ther. 2014;15(5):586–592.
- Siegmund SE, Yang H, Sharma R, et al. Low-dose rapamycin extends lifespan in a mouse model of mtDNA depletion syndrome. Hum Mol Genet. 2017;26(23):4588–4605.
- Khapre RV, Kondratova AA, Patel S, et al. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging (Albany NY). 2014;6(1):48–57.
- Johnson SC, Yanos ME, Kayser EB, et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science. 2013;342(6165):1524–1528.
- Johnson SC, Yanos ME, Bitto A, et al. Dose-dependent effects of mTOR inhibition on weight and mitochondrial disease in mice. Front Genet. 2015;6:247.
- Bitto A, Ito TK, Pineda VV, et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife. 2016;5. pii: e16351
- Johnson SC, Kaeberlein M. Rapamycin in aging and disease: maximizing efficacy while minimizing side effects. Oncotarget. 2016;7(29):44876–44878.
- Flynn JM, O’Leary MN, Zambataro CA, et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. 2013;12(5):851–862.
- Ye L, Widlund AL, Sims CA, et al. Rapamycin doses sufficient to extend lifespan do not compromise muscle mitochondrial content or endurance. Aging (Albany NY). 2013;5(7):539–550.
- Dai DF, Karunadharma PP, Chiao YA, et al. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell. 2014;13(3):529–539.
- Tortorici MA, Parks V, Matschke K, et al. The evaluation of potential pharmacokinetic interaction between sirolimus and tacrolimus in healthy volunteers. Eur J Clin Pharmacol. 2013;69(4):835–842.
- Brattstrom C, Sawe J, Jansson B, et al. Pharmacokinetics and safety of single oral doses of sirolimus (rapamycin) in healthy male volunteers. Ther Drug Monit. 2000;22(5):537–544.
- Mannick JB, Morris M, Hockey HP, et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci Transl Med. 2018;10:449.
- Mannick JB, Del Giudice G, Lattanzi M, et al. mTOR inhibition improves immune function in the elderly. Sci Transl Med. 2014;6(268):268ra179.
- Singh M, Jensen MD, Lerman A, et al. Effect of low-dose rapamycin on senescence markers and physical functioning in older adults with coronary artery disease: results of a pilot study. J Frailty Aging. 2016;5(4):204–207.
- Blagosklonny MV. Does rapamycin slow down time? Oncotarget. 2018;9(54):30210–30212.
- Pospelova TV, Bykova TV, Zubova SG, et al. Rapamycin induces pluripotent genes associated with avoidance of replicative senescence. Cell Cycle. 2013;12(24):3841–3851.
- Chen T, Shen L, Yu J, et al. Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell. 2011;10(5):908–911.
- Wu Y, Li Y, Zhang H, et al. Autophagy and mTORC1 regulate the stochastic phase of somatic cell reprogramming. Nat Cell Biol. 2015;17(6):715–725.
- Aarts M, Georgilis A, Beniazza M, et al. Coupling shRNA screens with single-cell RNA-seq identifies a dual role for mTOR in reprogramming-induced senescence. Genes Dev. 2017;31(20):2085–2098.
- Albers MW, Williams RT, Brown EJ, et al. FKBP-rapamycin inhibits a cyclin-dependent kinase activity and a cyclin D1-Cdk association in early G1 of an osteosarcoma cell line. J Biol Chem. 1993;268(30):22825–22829.
- Gaben AM, Saucier C, Bedin M, et al. Rapamycin inhibits cdk4 activation, p 21(WAF1/CIP1) expression and G1-phase progression in transformed mouse fibroblasts. Int J Cancer. 2004;108(2):200–206.
- Chatterjee A, Mukhopadhyay S, Tung K, et al. Rapamycin-induced G1 cell cycle arrest employs both TGF-_ and Rb pathways. Cancer Lett. 2015;360:134–140.
- Mukhopadhyay S, Frias MA, Chatterjee A, et al. The enigma of rapamycin dosage. Mol Cancer Ther. 2016;15(3):347–353.
- Efeyan A, Ortega-Molina A, Velasco-Miguel S, et al. Induction of p53-dependent senescence by the MDM2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer Res. 2007;67(15):7350–7357.
- Korotchkina LG, Leontieva OV, Bukreeva EI, et al. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY). 2010;2(6):344–352.
- Leontieva O, Gudkov A, Blagosklonny M. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010;9:4323–4327.
- Kim YY, Jee HJ, Um JH, et al. Cooperation between p21 and Akt is required for p53-dependent cellular senescence. Aging Cell. 2017;16(5):1094–1103.
- Huang B, Deo D, Xia M, et al. Pharmacologic p53 activation blocks cell cycle progression but fails to induce senescence in epithelial cancer cells. Mol Cancer Res. 2009;7(9):1497–1509.
- Korotchkina LG, Demidenko ZN, Gudkov AV, et al. Cellular quiescence caused by the Mdm2 inhibitor nutlin-3a. Cell Cycle. 2009;8(23):3777–3781.
- Lane DP, Verma C, Fang CC. The p53 inducing drug dosage may determine quiescence or senescence. Aging (Albany NY). 2010;2(11):748.
- Sui X, Fang Y, Lou H, et al. p53 suppresses stress-induced cellular senescence via regulation of autophagy under the deprivation of serum. Mol Med Rep. 2014;11(2):1214–1220.
- Hirota Y, Daikoku T, Tranguch S, et al. Uterine-specific p53 deficiency confers premature uterine senescence and promotes preterm birth in mice. J Clin Invest. 2010;120(3):803–815.