
ABSTRACT
Malignant pleural effusion (MPE) is a common complication of lung cancer. Accumulating evidence has suggested that circular RNAs (circRNAs) play important roles in oncogenesis and progression of cancer. However, the expression pattern of circRNAs in MPE remains largely unknown and awaits investigation. The study was designed to elucidate the potential roles of differentially expressed circRNAs in MPE. Herein, we detected a total of 1350 differentially expressed circRNAs and 1727 differentially expressed mRNAs in lung adenocarcinoma-associated malignant pleural effusion (LA-MPE) compared with tuberculous pleural effusion (TPE) by Clariom D Human Microarray. Among the top 5 up-regulated circRNAs (hsa_circ_0067705, hsa_circ_0025542, hsa_circ_0072793, hsa_circ_0084927, and hsa_circ_0085386), four were verified significantly up-regulated in LA-MPE by qRT-PCR and hsa_circ_0085386 had an increasing trend. CircRNA-miRNA-mRNA network for the top 5 up-regulated circRNAs was constructed and pathway analysis indicated that the enriched mRNA targets involved in PI3K-Akt signaling pathway, Axon guidance, Regulation of actin cytoskeleton and Rap1 signaling pathway were potentially regulated by these aberrantly expressed circRNAs. We generated specific circRNA profiles in LA-MPE for the first time. And analysis of circRNA regulatory network could provide evidence that circRNAs are important in MPE development because they participate in cancer-related pathways by sequestering miRNAs. Our findings suggested that aberrantly expressed circRNAs may be involved in the development of LA-MPE.
Introduction
Malignant pleural effusion (MPE) is a common, debilitating complication of a number of cancers, and is defined as the accumulation of a great deal of exudates in the pleural cavity, accompanied by the presence of malignant cells. The majority of MPE is due to metastatic disease, most commonly lung cancer, especially lung adenocarcinoma [Citation1]. The presence of MPE implies an advanced-stage disease with reduced life expectancy and quality [Citation2,Citation3], and it is associated with high morbidity and mortality [Citation4]. Though the promising progress in cancer therapy, the treatment of MPE remains palliative, with median survival ranging from 3 to 12 months [Citation5]. The high mortality of MPE patients is largely ascribed to a lack of reliable biomarkers for disease detection and treatment, and most importantly the underlying biology is poorly understood [Citation6]. Pleural fluid cytologic examination is commonly used for the diagnosis of MPE. However, the mean sensitivity of this method remains low (only ~ 60% with a range of 40–87%), and its accuracy is affected by sample preparation, cytologist expertise, and tumor types [Citation7]. Moreover, with China being heavily burdened with tuberculosis [Citation8], it is sometimes difficult to differentiate MPE from tuberculous pleural effusion (TPE). The current methods for treating MPE, including needle aspiration, indwelling pleural catheters, and pleurodesis, are ineffective [Citation1]. Hence, it is imperative to deepen the understanding of the pathogenesis of MPE and then identify effective diagnostic and therapeutic markers for MPE.
It is currently accepted that a combination of increased fluid production and impaired lymphatic outflow underlie the development of MPE, and considerable efforts have been invested in exploring the mechanism of tumor-host interplay [Citation9] and T cell subsets [Citation10–Citation14] in MPE formation. The advent of next-generation sequencing technologies has fueled the discoveries of new classes of non-coding RNAs that may trigger the development of MPE research. For instance, microRNAs have been reported to be promising candidates for MPE biomarkers [Citation15–Citation18]. Recently, another class of non-coding RNA molecules, circRNAs, has drawn increasing interests for their regulatory roles in cancer and other diseases. CircRNAs are usually abundantly expressed, with tissue- and developmental stage-specific patterns [Citation19–Citation21] and the covalently closed continuous loop structure confers them the resistance to RNases [Citation19,Citation20,Citation22], endowing them clear advantages as novel diagnostic markers in comparison to linear RNA [Citation20,Citation21]. In addition, it has been proposed that circRNAs can act as microRNA sponges, form RNA-protein complexes, and regulate target gene transcription and splicing [Citation23,Citation24]. All that matters is that circRNAs may play important roles in the carcinogenesis and progression of cancer, exhibiting distinctive expression profiles [Citation25–Citation28]. However, to date, the expression pattern and roles of circRNAs in MPE remain elusive.
In this study, we evaluated the expression profiles of circRNAs in MPE and TPE by Clariom D human microarray. We identified a number of differentially expressed circRNAs in MPE compared with TPE and constructed a predictive model for the circRNA-miRNA-mRNA regulatory network. We propose that aberrantly expressed circRNAs may be involved in the development of MPE by regulating target genes.
Materials and methods
Study subjects
The study protocol was approved by the Institutional Review Board for Human Studies, Beijing Luhe Hospital, Capital Medical University, Beijing, China. Informed consent was obtained from all subjects. A total of six pleural effusion (PE) patients were recruited for microarray assay. Three patients had MPE associated with recently diagnosed lung adenocarcinomata. MPE was diagnosed by the presence of malignant cells in pleural fluid and/or on a pleural biopsy specimen. The other three patients were diagnosed with TPE, as evidenced by the growth of Mycobacterium tuberculosis from PE or by demonstration of granulomatous pleuritis on a pleural biopsy specimen without evidence of other granulomatous diseases. After antituberculosis chemotherapy, disappearance of PE and clinical symptoms was observed in all patients with tuberculous pleurisy. At the time of sample collection, none of the patients had received any anticancer treatment, antituberculosis chemotherapy, corticosteroids, or other nonsteroid anti-inflammatory drugs. Patients were excluded if they had received any invasive procedures directed into the pleural cavity or if they had suffered chest trauma within 3 months prior to hospitalization. The demographic characteristics of the 6 patients are summarized in .
Table 1. Demographics of the study participants
Sample collection and processing
The PE samples were collected from each subject in heparin-treated tubes, using a standard thoracocentesis technique within 24 hours after hospitalization. The obtained PE specimens were immediately immersed in ice and were then centrifuged at 1200 rpm for 5 mins. The cell-free supernatant of PE was immediately frozen at -80°C after centrifugation for subsequent determination of soluble mediators. The cell pellets were resuspended in TRIzol reagent (Invitrogen, NY, USA) to extract the total RNA according to the manufacturer’s instructions. RNA samples were stored at -80°C before use.
CircRNA, miRNA and mRNA microarray expression profiling
The purity and concentration of extracted RNA were determined by OD260/OD280 using a NanoDrop ND-1000 instrument (NanoDrop Thermo, Wilmington, DE, USA). The integrity of the RNA was assessed by 1% formaldehyde denaturing gel electrophoresis.
Microarray profiling was conducted by Premedical Technology Corporation, Beijing, China. Sample labeling, microarray hybridization and washing were performed according to the manufacturer’s standard protocols. Briefly, each purified RNA sample was transcribed to double-strand cDNA, followed by cRNA synthesis and biotin-labeling. The labeled cRNAs were hybridized onto the Clariom D Human Assay Microarrays, which included transcriptome-wide gene- and exon-level expression profiles. After washing, the arrays were scanned using the GeneChip Scanner 3000 7G (Affymetrix, Santa Clara, CA, USA). The Affymetrix GeneChip Operating Software was used to analyze the acquired array images. Quantile normalization and subsequent data processing were performed using the R software package (R version 3.4.4). Differentially expressed RNAs between MPE and TPE were estimated by fold-change filtering combined with Student’s t-test. False discovery rate (FDR, <0.05) was calculated in order to correct the P-value. Transcripts with fold change in expression ≥ 2.0 and a p-value < 0.05 were considered significantly differentially expressed. Hierarchical clustering was performed to show the distinguishable genes expression pattern among samples. Differentially expressed RNAs were filtered and illustrated as a volcano plot. Principal component analysis was performed for differentially expressed circRNAs and mRNAs obtained from the microarray assays.
Validation of candidate circRNAs using quantitative reverse transcription real-time polymerase chain reaction (qRT-PCR)
The validation of selected candidate circRNAs was performed using qRT-PCR. Total RNA was reversely transcribed to cDNA according to the manufacturer’s instructions. Quantitative RT-PCR analysis and data collection were performed on ABI 7900HT Real-time PCR System (Applied Biosystems). The primers were synthesized by Premedical Technology Corporation (Beijing, China) (). RPS18, GAPDH or 18SrRNA were used as the references for normalization. All PCRs were triplicated. The relative expressions of circRNAs were calculated using the formula 2−(ΔCt MPE – ΔCt TPE).
Table 2. Specific circRNA primers used for quantitative RT-PCR analysis
Prediction of miRNAs and mRNAs for candidate circRNAs and computational analysis
Potential miRNA response elements (MREs) were searched for candidate circRNAs as miRNAs interact with circRNAs through MREs. MREs were predicted according to the cutoff value determined by the miRNA support vector regression (mirSVR) using miRanda [Citation29]. The putative target miRNAs of the candidate circRNAs were identified by the aforementioned method. The target genes of miRNAs were further predicted based on TargetScan [Citation30] and miRDB [Citation31] to study the potential functions of the circRNAs. The predicted miRNAs and mRNAs were then compared with detected miRNAs and mRNAs as determined by the same microarray assay respectively. To study gene ontology (GO) enrichment, we used the Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/) to determine the top functional annotations and classifications of significantly differentially expressed circRNA-targeted gene pairs. In addition, Clusterprofiler package was used to annotate and categorize the functions of the target genes for the differentially expressed circRNAs in the pathways. The GO and pathway enrichment analyses were visualized by R software packages.
Construction of circRNA-miRNA-mRNA interaction networks
CircRNA was used as a bait to enrich the circRNA-miRNA-mRNA network according to the target prediction software miRanda [Citation29], TargetScan [Citation30] and miRDB [Citation31]. To visualize their interactions, Cytoscape software was used to build the circRNA-miRNA-mRNA networks.
Statistical analysis
Unpaired Student’s t-tests were evaluated for significantly differentially expressed transcripts between MPE and TPE. Evaluation of statistical significance for the qRT-PCR results was performed by Student’s t-test using the SPSS Statistics 19.0 software package (IBM, Chicago, IL, USA). p < 0.05 was considered statistically significant. Plotting of the data was performed with Cytoscape, GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA), or R software package (R version 3.4.4).
Results
Demographic characteristics of the study subjects
We recruited 6 pleural effusion patients to perform microarray assay, including 3 lung adenocarcinoma associated malignant pleural effusion (LA-MPE) and 3 tuberculous pleural effusion (TPE) at Beijing Luhe Hospital, Capital Medical University, Beijing, China. The demographic characteristics of the 6 patients are summarized in . All the patients were diagnosed based on histopathological examination. Most (5/6) of the patients had a history of accompanying diseases that had been stabilized. Both the LA-MPE and TPE are exudates on the basis of Light’s criteria [Citation32]. The concentration of adenosine deaminase (ADA) in LA-MPE is much lower than that in TPE (p < 0.05).
Profiles of differentially expressed circRNAs and mRNAs in MPE and coding genes-mediated GO and pathway analysis
The Clariom D Human Array was used to profile circRNA and mRNA expressions for LA-MPE and TPE samples. In total, we detected 12,960 circRNAs (8,587 up-regulated, 4,373 down-regulated) and 20,666 mRNAs (13,343 up-regulated, 7,323 down-regulated). Among these, 1,350 circRNAs (817 up-regulated, 533 down-regulated) and 1,727 mRNAs (1,104 up-regulated, 623 down-regulated) were significantly differentially expressed (fold change ≥ 2.0 and p value < 0.05). Hierarchical clustering analysis on these circRNAs and mRNAs showed distinctive expression patterns for LA-MPE and TPE samples ()), suggesting the regulation of LA-MPE pathogenesis is distinct from TPE. The differentially expressed circRNAs and mRNAs between the two groups were identified through using volcano plot filtering ()). In both differentially expressed circRNAs and mRNAs, a majority of the transcripts (> 60%) were up-regulated in LA-MPE ()). The differentially expressed circRNAs are widely distributed on chromosomes, including sex chromosome ()).
Figure 1. Differential expression of circRNAs and mRNAs in MPE and TPE. (a, b) Hierarchical clustering analysis of differentially expressed circRNAs (a) and mRNAs (b) between MPE group and TPE group; each group included three individuals (FC (absolute value) ≥ 2.0, P < 0.05). Each column represents one sample, and each row represents a transcript. “Red” indicates high relative expression, and “Green” indicates low relative expression. (c, d) Volcano plots showing differentially expressed circRNAs (c) and mRNAs (d) in MPE. Red and green points represent up-regulated and down-regulated transcripts in MPE, respectively (FC (absolute value) ≥ 2.0, P < 0.05). MPE, Malignant Pleural Effusion; TPE, Tuberculous Pleural Effusion.
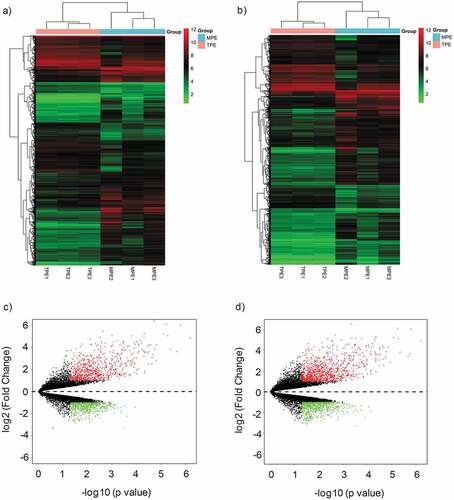
Figure 2. Characterizations of circRNAs and mRNAs expression in MPE compared with TPE. (a) Pie chart of aberrantly expressed circRNAs and mRNAs in MPE. (b) The distribution of differentially expressed circRNAs in human chromosomes. MPE, Malignant Pleural Effusion; TPE, Tuberculous Pleural Effusion.
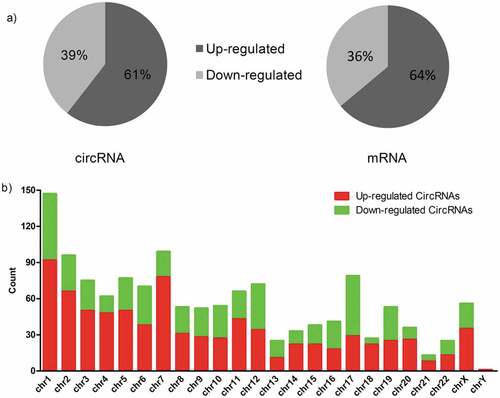
We performed gene ontology analysis for differentially expressed mRNAs. As for up-regulated mRNAs in MPE ()), the biological process analysis indicated that they were mainly involved in cell migration, extracellular matrix organization, cell-cell adhesion, and cell adhesion. The cellular component analysis showed that they were mainly involved in extracellular exosome, focal adhesion, cell surface, plasma membrane, and extracellular space. While the molecular function revealed that they mainly took part in cadherin binding involved in cell-cell adhesion, integrin binding, and protein binding. On the other hand, the GO enrichment analysis on the down-regulated mRNAs of MPE ()) showed that the biological process was primarily enriched in T cell activation and regulation of immune response. The major cellular components were plasma membrane, T cell receptor complex, immunological synapse, and cytoplasm. The molecular function enriched in SH3/SH2 adaptor activity.
Figure 3. GO and KEGG analysis of differentially expressed mRNAs in MPE. (a, b) GO annotation of up-regulated (a) and down-regulated (b) mRNAs. BP, biological process; CC, cellular component; MF, molecular function. (c, d) KEGG pathway enrichment analysis for up-regulated (c) and down-regulated (d) mRNAs. The related pathways were ranked according to GeneRatio (Selection Counts/Selection Size).
KEGG analysis showed that there were 26 pathways related to up-regulated mRNAs in MPE, including PI3K-Akt signaling pathway, proteoglycans in cancer, focal adhesion, and Rap1 signaling pathway. The related pathways were ranked according to GeneRatio. The top 3 related pathways were PI3K-Akt signaling pathway, proteoglycans in cancer and focal adhesion ()). Similarly, the down-regulated mRNAs were enriched in 37 pathways, such as Th17 cell differentiation, T cell receptor signaling pathway, Th1 and Th2 cell differentiation, Natural killer cell mediated cytotoxicity and so on. The top 3 related pathways were Cytokine-cytokine receptor interaction, Th17 cell differentiation and Natural killer cell mediated cytotoxicity according to GeneRatio ()).
Robustness of microarray assay and validation of the differentially expressed circRNA candidates
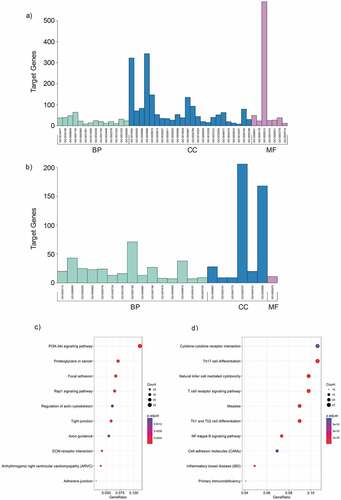
To confirm the validity of microarray assay, we evaluated whether the identified differentially expressed circRNAs and mRNAs can distinguish MPE from TPE. We performed principal component analysis (PCA) on the differential circRNAs obtained from microarray data and confirmed that MPE and TPE were divided into distinctive groups ()). Similarly, the PCA plot of the differentially expressed mRNAs showed that MPE were also separated from TPE ()). Hence, the differentially expressed circRNAs and mRNAs could distinguish MPE from TPE, firmly supporting the robustness of microarray assay for discovering specific circRNAs for MPE.
Figure 4. The principle component analysis (PCA) on the microarray data. (a) PCA was performed on the differentially expressed circRNAs obtained from microarray data. The MPE samples can be separated from the TPE samples. (b) PCA plot of the differentially expressed mRNAs.
To confirm the microarray results, we firstly randomly selected four circRNAs (hsa_circ_0003954, hsa_circ_0004086, hsa_circ_0050579, and hsa_circ_0001554, FC ≥ 2.0, P < 0.05) for qRT-PCR validation. The primers used for the qRT-PCR analysis are listed in . All samples were evaluated in triplicates with negative and positive controls. Consistent with the circRNA microarray result, two circRNAs (hsa-circ-0001554 and hsa-circ-0004086) were confirmed to be up-regulated in LA-MPE ()). Hsa-circ-0003954 and hsa-circ-0050579 failed qRT-PCR analysis with non-specific amplification. To further confirm the reliability of the microarray expression data and exclude technical reasons, we used an independent set of samples consisting of 9 LA-MPE and 9 TPE to verify the top 5 (relatively high fold change from the microarray assay) significantly up-regulated circRNAs (hsa_circ_0067705, hsa_circ_0025542, hsa_circ_0072793, hsa_circ_0084927 and hsa_circ_0085386). As shown in ) and (c), the relative expression of hsa_circ_0067705 and hsa_circ_0025542 was much higher in MPE than that in TPE, respectively. The relative expression of hsa_circ_0085386 in MPE had an increasing trend compared with that in TPE, though not statistically significant ()). Of note, hsa_circ_0072793 and hsa_circ_0084927 were detected in MPE group, but not in TPE group ()). Therefore, the top 5 significantly up-regulated circRNAs were selected for further analysis.
Figure 5. Validation of differentially expressed candidate circRNAs. The expression levels of circRNAs were detected using qRT-PCR. (a) Validation of the expression of has_circ_0001554 and has_circ_0004086. The heights of the columns represent the fold change (MPE/TPE) computed from the microarray data and the qPCR. (b, c, d, e, f) Validation of the expression of hsa_circ_0067705, hsa_circ_0025542, hsa_circ_0085386, hsa_circ_0072793, and hsa_circ_0084927. CircRNA expression was quantified using the 2−(ΔCt MPE – ΔCt TPE) method with normalization to 18SrRNA expression levels. Values are expressed as the mean ± SEM (n = 9). qRT-PCR, quantitative reverse transcription real-time polymerase chain reaction; SEM, standard error of the mean; MPE, Malignant Pleural Effusion; TPE, Tuberculous Pleural Effusion; ND, non-detected.
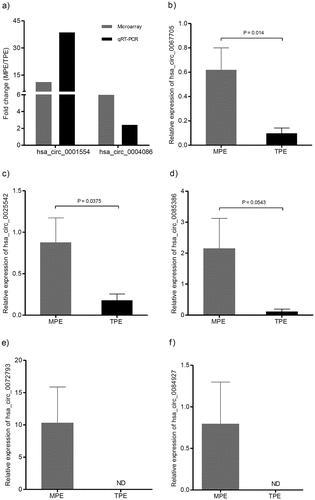
Prediction of the top 5 candidate circRNA targeted genes and bioinformatics analysis for the interaction network of circRNA-miRNA-mRNA
To predict the targets of the top 5 significantly up-regulated circRNAs (hsa_circ_0067705, hsa_circ_0025542, hsa_circ_0072793, hsa_circ_0084927 and hsa_circ_0085386), we firstly established the basic circRNA-miRNA connectivity with miRanda. As mentioned above, the majority of circRNAs and mRNAs were up-regulated, and pathway analysis showed up-regulated mRNAs were mainly enriched in cancer-related pathways. To construct competing endogenous RNA (ceRNA) network, the predicted miRNAs were subjected to the down-regulated miRNA expression filter using data from the same microarray. After being filtered, the intersection miRNAs were ranked according to the mirSVR scores, thereby identifying the 5 highest ranking miRNAs for further analysis. There were only three miRNA candidates for hsa_circ_0025542, namely, hsa-miR-298, hsa-miR-607, hsa-miR-4710. The 5 highest-ranking miRNA candidates binding targets of the other four circRNAs were listed below: 1) For hsa_circ_0067705: hsa-miR-298, hsa-miR-3185, hsa-miR-544a, hsa-miR-4675, and hsa-miR-6071; 2) For hsa_circ_0072793: hsa-miR-4432, hsa-miR-4522, hsa-miR-1261, hsa-miR-6126, and hsa-miR-3123; 3) For hsa_circ_0084927: hsa-miR-4319, hsa-miR-520e, hsa-miR-520b, hsa-miR-623, and hsa-miR-302e; 4) For hsa_circ_0085386: hsa-miR-4692, hsa-miR-3612, hsa-miR-4698, hsa-miR-4514, and hsa-miR-5695. Following miRNA target predictions, we proceeded to generate the miRNA-mRNA interaction network with TargetScan and miRDB. After cross validation with the up-regulated mRNA expression data from the same microarray, we finally obtained a total of 23 down-regulated miRNAs and 675 up-regulated mRNAs that were predicted to be regulated by the top 5 up-regulated candidate circRNAs. To gain insights into the functions of the top 5 up-regulated candidate circRNAs in LA-MPE, we conducted Gene Ontology and pathway enrichment analyses for the predicted target genes. The Gene Ontology analysis showed that the target genes were mainly enriched for the GO terms of protein binding, plasma membrane, and extracellular exosome ()). The pathway analysis revealed that the target genes were mainly enriched in PI3K-Akt signaling pathway, followed by Axon guidance, Regulation of actin cytoskeleton, and Rap1 signaling pathway ()). Finally, based on the predictions of the miRNA and mRNA targets for the top 5 up-regulated candidate circRNAs and microarray data, we constructed a ceRNA network of circRNA-miRNA-mRNA interactions with Cytoscape software delineating the putative regulations on these targets in the pathogenesis of LA-MPE ( and ).
Table 3. The top 5 up-regulated circRNAs targeted circRNA-miRNA-mRNA network
Figure 6. GO and KEGG analysis of the top 5 up-regulated circRNA targeted mRNAs. (a) GO enrichment for targeted mRNAs. All significant enriched GO terms were listed. (b) KEGG pathway enrichment for targeted mRNAs. Top 10 significant enriched pathway terms were listed.
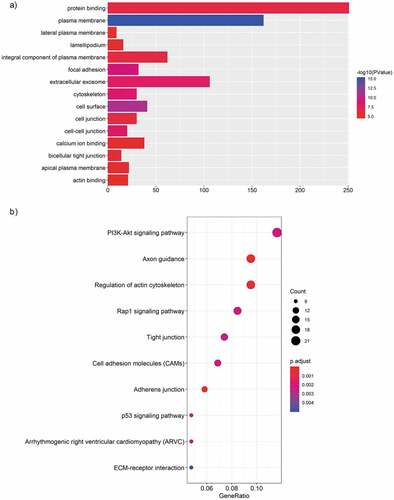
Figure 7. The circRNA-miRNA-mRNA network. 23 miRNA and 675 genes were targeted by the top 5 significantly up-regulated circRNAs. Diamond, circRNA; Rectangle, miRNA; Ellipse, mRNA; Red, up-regulated, Fold Change (FC) ≥ 2.0 and p-value < 0.05; Green, down-regulated, FC≤-2.0 and p-value < 0.05.
Discussion
Malignant pleural effusion is an advanced-stage manifestation of lung cancer, especially lung adenocarcinoma [Citation1,Citation2]. Despite its longstanding recognition, there is currently no effective treatment option beyond needle aspiration to relieve the symptom of shortness of breath. Displaying versatility in regulating many pathological processes, non-coding RNAs may serve as another layer of mechanisms for the pathogenesis of MPE and be promising targets for therapy. In the current study, we profiled differentially expressed circRNAs in LA-MPE compared with TPE and constructed a regulatory network for circRNA-miRNA-mRNA in LA-MPE. We have identified specific circRNAs that may regulate the LA-MPE pathogenesis.
Non-coding RNAs play multidimensional roles in regulating many physiopathological processes [Citation33]. Recently, many studies explored aberrantly expressed miRNAs in MPE and proposed their potentials as markers for diagnosis and/or prognostic predictions [Citation15–Citation18,Citation34]. CircRNAs possess covalently closed loop structures and lack free 3ʹ- and 5ʹ- ends, therefore are more stable than other RNA molecules. This makes them more attractive candidates for biomarkers than other types of RNA. To date, the expression pattern and role of circRNAs in the MPE pathogenesis remains largely unknown. Some studies investigated the expression profiles of circRNAs in peripheral blood to extrapolate their potential roles in certain diseases [Citation35–Citation38]. But the conclusions should be carefully extended to tissues where the diseases reside, because expression of circRNAs is usually tissue-specific. To explore the role of circRNAs in MPE, we profiled circRNAs expression in LA-MPE and TPE clinical samples with microarrays. We identified 1,350 significantly differentially expressed circRNAs in LA-MPE, suggesting circRNAs may be important in regulating the pathogenesis of LA-MPE. What was noteworthy was that a majority of differentially expressed circRNAs were up-regulated. This result was in accordance with the clustering results on the differentially expressed mRNAs, where most of the genes were up-regulated in LA-MPE ()). The fact that most genes were up-regulated in LA-MPE suggests that numerous pathways may promote the development and progression of MPE. Indeed, KEGG pathway analysis on differentially expressed up-regulated mRNAs confirmed that cancer-related pathways of PI3K-Akt signaling pathway, Proteoglycans in cancer, Focal adhesion, and Rap1 signaling pathway were enriched in LA-MPE.
The aberrantly expressed circRNAs may regulate the expression of parental genes and play important roles in the pathological processes of diseases. And a considerable number of studies have indicated that circRNAs can act as “miRNA sponges” to regulate gene expression [Citation39–Citation41]. As reported by the present study, most circRNAs and mRNAs were up-regulated, we speculated that circRNAs may regulate the expression of target genes by ceRNA mechanism. Based on the miRNA target predication software, we initially predicted miRNA targets of the top 5 up-regulated and validated circRNAs and then subjected them to down-regulated MPE-associated miRNAs,indicating up-regulated circRNAs might inhibit the expression of down-regulated miRNAs. Among the 23 potential miRNAs, some have been reported as important regulaters in many kinds of cancers [Citation42–Citation48]. For example, L Bao et al revealed that down-regulation of miR-298 increased P-Glycoprotein expression and induced doxorubicin resistance in sensitive breast cancer cells, suggesting miR-298 was associated with the chemoresistant mechanisms of metastatic human breast cancer by directly modulating P-Glycoprotein expression [Citation42]. Similarly, a study by S Wei et al showed that miR-623 was down-regulated in lung adenocarcinoma and suppressed the invasion and metastasis targeting Ku80 through ERK/JNK inactivation mediated downregulation of MMP-2/9 [Citation48]. No functional data is yet available for these miRNAs in MPE and may be considered promising candidates for future study. Subsequently, to further evaluate the biological roles of the top 5 up-regulated circRNAs, we predicted the target genes of these 23 down-regulated miRNAs. After subjecting predicted mRNAs to up-regulated LA-MPE associated mRNAs from the same microarray, we finally constructed a circRNA-miRNA-mRNA network consisting of 5 up-regulated circRNAs, 23 down-regulated miRNAs and 675 up-regulated mRNAs ().
Functional annotation of target genes can provide insights of the biological functions of the differentially expressed circRNAs in MPE. KEGG analysis showed that there were 14 pathways related to the top 5 up-regulated circRNAs, including PI3K-Akt signaling pathway, Regulation of actin cytoskeleton, and Rap1 signaling pathway. PI3K-Akt signaling pathway plays an important role in tumorigenesis and regulates critical cellular functions including survival, proliferation and metabolism [Citation49]. Regulation of actin cytoskeleton is an essential step in cell migration during activation of the epithelial-mesenchymal transition program, which is associated with metastatic properties of cancer cells [Citation50]. And Rap1 signaling pathway has also been reported to be involved in tumorigenesis and tumor progression [Citation51]. All these results strongly indicate that these top 5 up-regulated circRNAs may be of importance in the development and progression of MPE. At the same time, we also performed GO analysis, illustrating the biological process, cellular component, and molecular function of the target genes of these 5 up-regulated circRNAs. These GO results showed that target genes of candidate circRNAs were mainly enriched in GO terms of protein binding, plasma membrane, and extracellular exosome, further suggesting that circRNAs may be involved in MPE development by regulating miRNA and their target genes.
As a conclusion, we identified multiple circRNAs which are differentially expressed in LA-MPE for the first time, proposed that the circRNA-miRNA-mRNA axis may be the possible molecular regulatory mechanism of MPE, and hoped the study can provide a reference for the broad analysis of circRNAs in MPE. Nevertheless, some limitations must be considered in this study. First, despite the appreciable effort of extending samples, the sample size was still limited and future studies should endeavor to recruit larger number of pleural effusion samples to confirm our results. Second, though the predicted genes of circRNAs were all subjected to LA-MPE associated microarray data and intersection genes were selected for analysis, the exact molecular mechanisms of candidate circRNAs needed to be elaborated in follow-up studies. Third, though we detected the expression profile of circRNAs in cell pellets in pleural effusion, its expression in cell-free supernatant need to be explored. However, studies showed that exonic circRNAs is not stable in serum, with a half-life of less than 15s [Citation24].
Disclosure of Potential Conflicts of Interest
The authors have no conflicts of interest to declare.
Acknowledgments
We thank all investigators who were involved in this study. We express our thanks to our colleagues Dong Wei, Chunxue Ran, and Congfeng Li.
The Corresponding author Ziliang Hou thanks his wife Shengjiao Wang for her dedication and love. The acceptance of this paper will be a gift for their unborn child.
Additional information
Funding
References
- Penz E, Watt KN, Hergott CA, et al. Management of malignant pleural effusion: challenges and solutions. Cancer Manag Res. 2017;9:229–241.
- Sugiura S, Ando Y, Minami H, et al. Prognostic value of pleural effusion in patients with non-small cell lung cancer. Clin Cancer Res. 1997;3(1):47–50.
- Goldstraw P, Crowley J, Chansky K, et al. The IASLC lung cancer staging project: proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM classification of malignant tumours. J Thorac Oncol. 2007;2(8):706–714.
- Roberts ME, Neville E, Berrisford RG, et al. Management of a malignant pleural effusion: british thoracic society pleural disease guideline 2010. Thorax. 2010;65(Suppl 2):ii32–40.
- Antony VB, Loddenkemper R, Astoul P, et al. Management of malignant pleural effusions. Eur Respir J. 2001;18(2):402–419.
- Psallidas I, Kalomenidis I, Porcel JM, et al. Malignant pleural effusion: from bench to bedside. Eur Respir Rev. 2016;25(140):189–198.
- Hooper C, Lee YC, Maskell N. Investigation of a unilateral pleural effusion in adults: british thoracic society pleural disease guideline 2010. Thorax. 2010;65(Suppl 2):ii4–17.
- Zhu S, Xia L, Yu S, et al. The burden and challenges of tuberculosis in China: findings from the global burden of disease study 2015. Sci Rep. 2017;7(1):14601.
- Stathopoulos GT, Kalomenidis I. Malignant pleural effusion: tumor-host interactions unleashed. Am J Respir Crit Care Med. 2012;186(6):487–492.
- Chen YQ, Shi HZ, Qin XJ, et al. CD4+CD25+ regulatory T lymphocytes in malignant pleural effusion. Am J Respir Crit Care Med. 2005;172(11):1434–1439.
- Ye ZJ, Zhou Q, Gu YY, et al. Generation and differentiation of IL-17-producing CD4+ T cells in malignant pleural effusion. J Immunol. 2010;185(10):6348–6354.
- Ye ZJ, Zhou Q, Yin W, et al. Interleukin 22-producing CD4+ T cells in malignant pleural effusion. Cancer Lett. 2012;326(1):23–32.
- Ye ZJ, Zhou Q, Yin W, et al. Differentiation and immune regulation of IL-9- producing CD4+ T cells in malignant pleural effusion. Am J Respir Crit Care Med. 2012;186(11):1168–1179.
- Lin H, Tong ZH, Xu QQ, et al. Interplay of Th1 and Th17 cells in murine models of malignant pleural effusion. Am J Respir Crit Care Med. 2014;189(6):697–706.
- Han HS, Yun J, Lim SN, et al. Downregulation of cell-free miR-198 as a diagnostic biomarker for lung adenocarcinoma-associated malignant pleural effusion. Int J Cancer. 2013;133(3):645–652.
- Shin YM, Yun J, Lee OJ, et al. Diagnostic value of circulating extracellular miR-134, miR-185, and miR-22 levels in lung adenocarcinoma-associated malignant pleural effusion. Cancer Res Treat. 2014;46(2):178–185.
- Xie L, Wang T, Yu S, et al. Cell-free miR-24 and miR-30d, potential diagnostic biomarkers in malignant effusions. Clin Biochem. 2011;44(2–3):216–220.
- Xie L, Chen X, Wang L, et al. Cell-free miRNAs may indicate diagnosis and docetaxel sensitivity of tumor cells in malignant effusions. BMC Cancer. 2010;10:591.
- Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. Rna. 2013;19(2):141–157.
- Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–338.
- Salzman J, Chen RE, Olsen MN, et al. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9(9):e1003777.
- Suzuki H, Zuo Y, Wang J, et al. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006;34(8):e63.
- Hsiao KY, Sun HS, Tsai SJ, et al. New member of noncoding RNA with novel functions. Exp Biol Med (Maywood). 2017;242(11):1136–1141.
- Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32(5):453–461.
- Hsiao KY, Lin YC, Gupta SK, et al. Noncoding effects of circular RNA CCDC66 promote colon cancer growth and metastasis. Cancer Res. 2017;77(9):2339–2350.
- Chen J, Li Y, Zheng Q, et al. Circular RNA profile identifies circPVT1 as a proliferative factor and prognostic marker in gastric cancer. Cancer Lett. 2017;388:208–219.
- Li H, Hao X, Wang H, et al. Circular RNA expression profile of pancreatic ductal adenocarcinoma revealed by microarray. Cell Physiol Biochem. 2016;40(6):1334–1344.
- Xuan L, Qu L, Zhou H, et al. Circular RNA: a novel biomarker for progressive laryngeal cancer. Am J Transl Res. 2016;8(2):932–939.
- Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012;13(4):271–282.
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20.
- Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43(Databaseissue):D146–152.
- Light RW, Macgregor MI, Luchsinger PC, et al. Pleural effusions: the diagnostic separation of transudates and exudates. Ann Intern Med. 1972;77(4):507–513.
- Yang JH, Qu LH. DeepBase: annotation and discovery of microRNAs and other noncoding RNAs from deep-sequencing data. Methods Mol Biol. 2012;822:233–248.
- Wang T, Lv M, Shen S, et al. Cell-free microRNA expression profiles in malignant effusion associated with patient survival in non-small cell lung cancer. PloS one. 2012;7(8):e43268.
- Jiang G, Ma Y, An T, et al. Relationships of circular RNA with diabetes and depression. Sci Rep. 2017;7(1):7285.
- Ouyang Q, Wu J, Jiang Z, et al. Microarray expression profile of circular RNAs in peripheral blood mononuclear cells from rheumatoid arthritis patients. Cell Physiol Biochem. 2017;42(2):651–659.
- Cui X, Niu W, Kong L, et al. hsa_circRNA_103636: potential novel diagnostic and therapeutic biomarker in major depressive disorder. Biomark Med. 2016;10(9):943–952.
- Wu N, Jin L, Cai J. Profiling and bioinformatics analyses reveal differential circular RNA expression in hypertensive patients. Clin Exp Hypertens. 2017;39(5):454–459.
- Zhong L, Wang Y, Cheng Y, et al. Circular RNA circC3P1 suppresses hepatocellular carcinoma growth and metastasis through miR-4641/PCK1 pathway. Biochem Biophys Res Commun. 2018;499(4):1044–1049.
- Zeng K, Chen X, Xu M, et al. CircHIPK3 promotes colorectal cancer growth and metastasis by sponging miR-7. Cell Death Dis. 2018;9(4):417.
- Ma X, Yang X, Bao W, et al. Circular RNA circMAN2B2 facilitates lung cancer cell proliferation and invasion via miR-1275/FOXK1 axis. Biochem Biophys Res Commun. 2018;498(4):1009–1015.
- Bao L, Hazari S, Mehra S, et al. Increased expression of P-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am J Pathol. 2012;180(6):2490–2503.
- Zhu Z, Wang S, Zhu J, et al. MicroRNA-544 down-regulates both Bcl6 and Stat3 to inhibit tumor growth of human triple negative breast cancer. Biol Chem. 2016;397(10):1087–1095.
- Slattery ML, Mullany LE, Sakoda LC, et al. The PI3K/AKT signaling pathway: associations of miRNAs with dysregulated gene expression in colorectal cancer. Mol Carcinog. 2018;57(2):243–261.
- Kanlikilicer P, Rashed MH, Bayraktar R, et al. Ubiquitous release of exosomal tumor suppressor miR-6126 from ovarian cancer cells. Cancer Res. 2016;76(24):7194–7207.
- Chu J, Li Y, Fan X, et al. MiR-4319 suppress the malignancy of triple-negative breast cancer by regulating self-renewal and tumorigenesis of stem cells. Cell Physiol Biochem. 2018;48(2):593–604.
- Li S, Zhang H, Ning T, et al. MiR-520b/e regulates proliferation and migration by simultaneously targeting EGFR in gastric cancer. Cell Physiol Biochem. 2016;40(6):1303–1315.
- Wei S, Zhang ZY, Fu SL, et al. Hsa-miR-623 suppresses tumor progression in human lung adenocarcinoma. Cell Death Dis. 2016;7(9):e2388.
- Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9(6):667–676.
- Sousa-Squiavinato ACM, Rocha MR, Barcellos-de-Souza P, et al. Cofilin-1 signaling mediates epithelial-mesenchymal transition by promoting actin cytoskeleton reorganization and cell-cell adhesion regulation in colorectal cancer cells. Biochim Biophys Acta Mol Cell Res. 2018;pii: S0167–4889(18)30429–4.
- Okada T, Sinha S, Esposito I, et al. The Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT by restraining Ras-MAPK signalling. Nat Cell Biol. 2015;17(1):81–94.