
ABSTRACT
Background: Atherosclerosis (AS) is a major risk factor for cardiovascular disease. microRNAs play a key role in gene regulation in the formation and development of atherosclerotic plaques. Herein, the role and target gene of miR-185 in AS were explored.
Materials and methods: Cell viability, migration and invasion were examined by cell counting kit-8 (CCK-8) and transwell assay. The relative luciferase activity was measured by luciferase reporter assay. The levels of miR-185, STIM1, vascular endothelial growth factor (VEGF) and matrix metalloprotein-9 (MMP-9) were evaluated by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and Western blot.
Results: The results revealed that ox-LDL decreased miR-185 expression, and enhanced STIM1 expression in MOVAS cells, as well promoted cell viability, migration and invasion. 3ʹ-UTR of STIM1 contained miR-185 binding site according to the Targetscan. miR-185 silencing or STIM1 overexpression promoted the viability, migration and invasion of ox-LDL-induced MOVAS cells. miR-185 overexpression or STIM1 silencing had the opposite effect. Besides, miR-185 silencing up-regulated the levels of VEGF and MMP-9 in vitro, and increased the lesions of arterial wall tissues and STIM1 positive rate in vivo. However, STIM1 silencing reversed these effects.
Conclusions: Sum up, STIM1 was a potential target gene of miR-185 in AS. Knockdown of miR-185 facilitated the progression of AS through enhancing cell proliferation, migration and invasion via targeting STIM1. The research provides a novel view of miR-185/STIM1 axis function in AS development, and this targeting method may prevent and treat AS.
Introduction
The WHO study found that the incidence of coronary heart disease (CHD) is the highest among cardiovascular diseases [Citation1,Citation2]. The famous GRACE study shows that although the drug and interventional therapy of CHD have achieved great progress and achieved good results [Citation3]. However, there is no fundamental improvement in the final outcome of myocardial infarction, mainly due to the high incidence of myocardial infarction and high mortality. Myocardial infarction is mainly caused by the rupture of coronary atherosclerotic plaque [Citation4]. It is found that the prognosis of myocardial infarction is more determined by the composition and stability of atherosclerotic plaques, rather than the degree of stenosis [Citation5–Citation7]. Atherosclerosis (AS) is a major risk factor for cardiovascular disease. The main characteristics of AS are arterial intimal lipid deposition, smooth muscle cells and proliferation of fibrous matrix components [Citation8]. AS can gradually develop into atherosclerotic plaques, and the whole process involves a variety of cell changes, including macrophages, dendritic cells, vascular smooth muscle cells and endothelial cells, as well as various proinflammatory substances, including adhesion molecules, chemokines and cytokines [Citation9].
miRNAs are a newly discovered class of endogenous noncoded RNA, which is commonly found in multicellular eukaryotes. It can play an important role in the biological metabolism, tumorigenesis and immune inflammation by inhibiting the expression of its target molecules [Citation10–Citation14]. Recent studies have found that in the formation of AS, a variety of miRNAs regulate the biological processes of related cells mainly through the complex regulatory network between its target genes, such as cell proliferation, apoptosis, metastasis and the production of inflammatory cytokines, affecting the functional structure of the vascular wall and the stability of the plaque [Citation15,Citation16]. miR-185 is located at chromosome 22q11.21, and its precursor is composed of 82 bp nucleotides [Citation17]. miR-185 has been shown to play a role in tumor suppressor genes in many tumors, and can inhibit the proliferation of lung cancer, breast cancer, ovarian cancer, colorectal cancer and renal cell carcinoma [Citation18–Citation22]. Nevertheless, the influence of miR-185 in the pathological mechanism of AS is not clear.
Ca2+ release activation Ca2+ channel (CRAC) is the most important cell receptor-dependent Ca2+ channel, and it can feel Ca2+ release in cell calcium library and cause calcium influx. It exists in various cells and plays an important role in many physiological functions [Citation23]. In 2005, Roos and other scholars used RNAi scanning technology to screen 170 genes of S2 cells in Drosophila melanogaster, of which one gene encoded protein could significantly affect the changes in intracellular Ca2+, which is stromal interaction molecule 1 (STIM1) [Citation24]. STIM1 is located in the cell endoplasmic reticulum, and it senses calcium concentration in the endoplasmic reticulum [Citation25]. Meanwhile, STIM1 is the “key” to activate calcium channels on the cell membrane. STIM1 is widespread in nonexcitable cells, including tumor cells and vascular smooth muscle cells (VSMCs) [Citation26–Citation28].
In our work, we constructed the AS model in vivo and in vitro. The role and target of miR-185 in AS model in vivo and in vitro were explored. To be specific, the expression of miR-185 and STIM1, proliferation and metastasis, and the corresponding factors were investigated in vitro. Additionally, the lesion area and STIM1 levels were examined in vivo.
Materials and methods
Cell culture and transfection
Mouse aortic vascular smooth muscle cells (MOVAS) were obtained from ATCC. Cells were maintained in dulbecco’s modified eagle medium (DMEM, Solarbio, Beijing, China), containing 10% fetal bovine serum (FBS; MRC, Jiangsu, China), and 100 × penicillin-streptomycin mixed solution (Leagene, Beijing, China) in 37°C incubator with 95% humidified and 5% CO2 (MR80W, SheYanYiQi, Shanghai, China). After maintaining, cells were treated with 50 ng/mL ox-LDL (Yeasen, Shanghai, China) at 37°C for 48 h. miR-185 mimics, miR-185 inhibitor and miRNA negative control (200 nM) were purchased from GenePharma (Shanghai, China). Ox-LDL-pretreated cells were transfected with unspecific scrambled siRNA, STIM1 siRNA, pcDNA3.1(+) and pcDNA3.1(+)-STIM1 plasmids (GenePharma, Shanghai, China) by lipofectamine™ 2000 (Thermo, Beijing, China) at 37°C for 24 h (h).
Cell counting kit-8 assay
Cell counting kit-8 (CCK-8; Beyotime, Shanghai, China) assay was applied to detect cell viability. The 2 × 103cells/ml of cells in the logarithmic phase were inoculated in the 96-well plates and maintained in the incubator for 24 h. Then, cells were treated with ox-LDL for 24 and 48 h. Ox-LDL-pretreated cells were transfected with above plasmids for 24 and 48 h. CCK8 reagent was added into each well. Cells were cultured in an incubator for 2 h. Microplate reader (SuPerMax 3000AL; Flash, Shanghai, China) was carried out to assess the absorbance at 450 nm.
Transwell assay
The migration and invasion of MOVAS cells were assessed using transwell analysis. Cell invasion was tested by BD matrigel, but it was not used for cell migration. BD matrigel was poured into the upper chamber of Transwell for 20 min and then DMEM was poured into the BD matrigel. Transwell was transferred to the culture dish. DMEM with 15% FBS was poured into the lower chamber. The treated cells were digested by trypsin, and suspended, and cell suspension was cultured in the upper chamber at 37°C for 24 h. Cells were stained with crystal violet for 20 min at 37°C. Cells were observed by the inverted fluorescence microscope (MF31, Mshot, Guangzhou, Guangdong, China).
Luciferase reporter assay
Luciferase activity was detected by double luciferase report system detection kit (Promega). miR-185 mimics and miRNA negative control were co-transfected with STIM1-3ʹUTR or STIM1-3ʹUTR mutant plasmids (400 ng) into MOVAS cells by lipofectamine™ 2000 for 48 h. Cells were lysed using 1 × PLB at 37°C for 20 min (min). Suspension was transferred into the black enzyme plate. Cells were exposed to LARII and stop&Glo® reagent. Luciferase activity was detected by GloMax® Discover Multimode Microplate Reader (Promega).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNAs of cells were collected and lysed with TRIzol (Invitrogen). Reverse transcription was used to synthesize cDNA from 1 μg RNA by Hieff™ First Strand cDNA Synthesis Kit (Yeasen). The primers are listed in . cDNA was amplified by SYBR Green qPCR Master Mix Mix (MedChenExpress, New Jersey, USA). The condition of amplification was as follows: pre-denaturation: 92°C for 30 s (s); denaturation: (92°C for 5 s, 58°C for 30 s) for 30 cycles; elongation: 72°C for 30 min. The internal controls were U6 and β-actin. The gene expression was quantified by the formula 2−ΔΔCT.
Table 1. The sequences of primers.
Western blot assay
Total proteins were collected from the treated cells by RIPA buffer (Solarbio, Beijing, Chian) and quantified using BCA protein assay (Thermo). Each protein was loaded into sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and bound to the PVDF membrane (Reno, Hangzhou, China). The membrane was soaked in TBST-soluble 5% dried skimmed milk at 37°C for 1 h, and then the membrane was bound to the corresponding primary antibodies (anti-STIM1, ab62031, 1:800; anti-vascular endothelial growth factor (VEGF), ab32152, 1:700; anti-matrix metalloprotein 9 (MMP-9), ab194314, 1:700; anti-β-actin, ab8226, 1:600; Abcam) at 4°C overnight, accompanied by bound to the corresponding secondary antibodies (Goat anti-rabbit IgG, ab150077, 1:8000; Goat anti-mouse, IgG, ab6785, 1:7000; Rabbit anti-mouse IgG, ab6726, 1:7000; Abcam) at 37°C for 1 h. The blots were detected by ECL detection reagent (Yeasen).
Animal model and aortic tissue collection
Forty-two C57 mice (6–8 wk old, 20 ± 5 g) were provided from Rochen (Shanghai, China). The mice were kept in cages (5 mice per cage) with the free food and water under a comfortable environment (Illumination: 12 h light/dark cycle, temperature: 22 ± 5°C, humidity: 40–70%). The mice were divided into 2 groups: control group (6 mice were kept in normal rearing), model group (36 mice were fed with atherogenic diet (1.25% cholesterol and 15% fat) for 12 wk. After the establishment of the atherosclerosis model in mice, the model group was further divided into six groups: model group (six mice were intravenously injected with saline), NC (six mice were intravenously injected with unspecific scrambled siRNA treated cell suspension), mock group (six mice were intravenously injected with antagomir treated cell suspension), INMO group (six mice were intravenously injected with antagomir-185 and unspecific scrambled siRNA treated cell suspension), INsiSTIM1 group (six mice were intravenously injected with antagomir-185 and siRNA STIM1 treated cell suspension), siSTIM1 NC group (six mice were intravenously injected with antagomir and siRNA STIM1 treated cell suspension).
The mice were sacrificed using euthanasia. The aortic tissue was extracted and embedded in tissue OCT-freeze medium (Rebiosci, Shanghai, China). The embedded tissue was sliced by slicer (CM1520, Leica, Germany) for hematoxylin-eosin (HE) staining and immunohistochemistry (IHC) assays. Animal experiments had been approved by the Ethics Committee.
HE staining assay
The embedded tissue slices were dewaxed with xylene for 10 min, anhydrous ethanol for 2 min, 95% ethanol for 1 min, 80% ethanol for 1 min and 75% ethanol for 1 min. Then, the slices were washed using distilled water for 2 min. Hematoxylin (Solarbio, Beijing, China) was used to stain the slices. The slices were differentiated by hydrochloric acid ethanol for 30 s, washed by distilled water for 15 min and stained by eosin (Salarbio) for 2 min. The slices were dehydrated with 95% ethanol for 3 min, anhydrous ethanol for 2 min and xylene for 2 min. The slices were sealed using resinene. The slices were observed under the inverted microscope. The lesions of arterial wall tissues were defined as following: accumulation of visible foam cells and forming a local uplift, segmental lesions, formation of plaques under the intima, visible tissue necrosis in plaque, cholesterol crystal, tissue calcification.
IHC assay
The embedded tissue slices were washed by distilled water for 3 times. The slices were soaked in the citrate buffer (pH = 6.0) for 30 min. The hydrogen peroxide reagent was dripped to the slices for 10 min. The slices were sealed using goat serum for 20 min. The anti-STIM1 were added to the slices at 4°C for 24 h. Then, the slices were incubated with the secondary antibodies (Mouse anti-goat IgG-HRP, sc-2354, 1:6000, Santa Cruz Biotechnology) at room temperature for 20 min. The slices were stained by DAB staining solution (Leica) for 15 min. The slices were washed by distilled water for 3 times. The slices were dyed by hematoxylin. The slices were observed under the inverted fluorescence microscope.
Statistical analysis
IBM SPSS Statistics 20 was used for statistical analysis. The data were shown as mean ± standard deviation (SD). The differences among groups were assessed by one-way analysis of variance (ANOVA). P < 0.05 was presented as a statistical difference.
Results
miR-185 was low expressed and STIM1 was high expressed in ox-LDL treated MOVAS cells
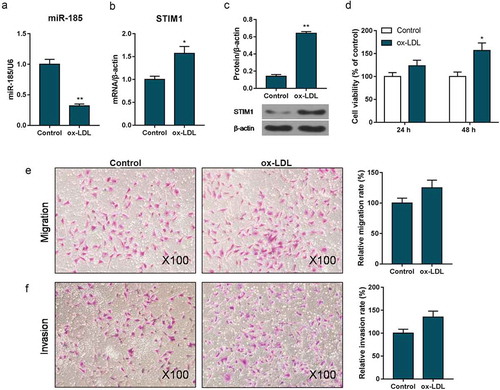
To explore the influence of ox-LDL in MOVAS cells, the expression of miR-185 and STIM1, the viability, migration and invasion were performed. The RT-qPCR and Western blot data showed that when MOVAS cells were treated with ox-LDL, miR-185 level was inhibited while STIM1 expression was enhanced, compared to control group (). The CCK-8 revealed that cell viability was heightened in ox-LDL exposed MOVAS cells at 48 h (). Besides, the transwell results observed that the abilities of migration and invasion were significantly enhanced, when cell was exposed to ox-LDL ().
Figure 1. miR-185 was low expressed and STIM1 was high expressed in ox-LDL treated MOVAS cells. MOVAS cells were subjected to ox-LDL. (a-c) The expression of miR-185 and STIM1were detected by RT-qPCR (a, b) and Western blot (c). CCK-8 was applied to measure cell viability. (e, f) Transwell assay was carried out to analyze cell migration (e) and invasion (f). *P< 0.05, **P< 0.01, versus control.
Inhibition of miR-185 enhanced the abilities of proliferation, migration and invasion in ox-LDL induced MOVAS cells
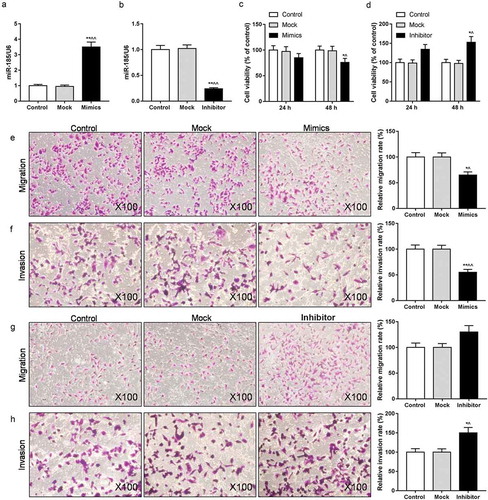
To study the impact of miR-185 on the viability and metastasis of ox-LDL induced MOVAS cells, CCK-8 and transwell assays were used. miR-185 expression was up-regulated in miR-185 mimics-transfected cells, but down-regulated in miR-185 inhibitor-transfected cells ((a,b)). As CCK-8 data shown, miR-185 overexpression suppressed cell viability, and inhibition of miR-185 increased cell viability (). The transwell assay result observed that the abilities of cell migration and invasion were depressed in miR-185 mimics, whereas enhanced in miR-185 inhibitor ().
Figure 2. Inhibition of miR-185 enhanced the abilities of proliferation, migration and invasion in ox-LDL-induced MOVAS cells. MOVAS cells were subjected to ox-LDL, and then transfected with miR-185 mimics and miR-185 inhibitor. (a, b) The miR-185 level was assessed by RT-qPCR. (c, d) Cell viability was analyzed by CCK-8. (e-h) Transwell was used to analyze cell migration (e, f) and invasion (g, h). *P< 0.05, **P< 0.01, versus control. ^P< 0.05, ^^P< 0.01, versus mock.
SITM1 was a target gene of miR-185
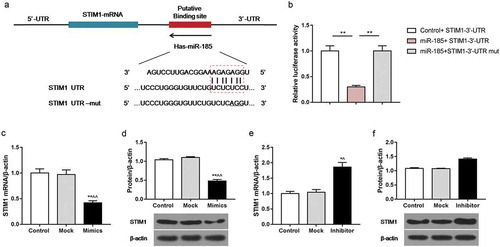
To identify whether the STIM1 was a target gene of miR-185, Targetscan website and luciferase activity assay were performed. The prediction results of Targetscan website revealed the binding site of STIM1 target gene to miR-185 (). The luciferase activity assay data showed that in comparison with control + STIM1-3ʹ-UTR group, the relative luciferase activity was repressed in miR-185 + STIM1-3ʹ-UTR group. However, when cells were treated with miR185 mimics and STIM1-3ʹ-UTR mut, there was no obvious change in relative luciferase activity (). In addition, the RT-qPCR and Western blot results observed that in comparison with ox-LDL-treated cells, the mRNA and protein levels of STIM1 were decreased in cells exposed to ox-LDL and miR-185 mimics, while were increased in cells subjected to ox-LDL and miR-185 inhibitor ().
Figure 3. SITM1 was a target gene of miR-185. MOVAS cells were subjected to ox-LDL, and then transfected with miR-185 mimics, miR-185 inhibitor, STIM1-3ʹ-UTR and STIM1-3ʹ-UTR mut. (a) Targetscan website predicted the binding site of STIM1 target gene to miR-185. (b) The luciferase reporter assay was performed to assess the relative luciferase activity. (c-f) The mRNA and protein expression of STIM1 were detected by RT-qPCR (c, e) and Western blot (d, e). *P< 0.05, **P< 0.01, versus control. ^P< 0.05, ^^P< 0.01, versus mock.
STIM1 silencing attenuated the abilities of proliferation, migration and invasion in ox-LDL induced MOVAS cells
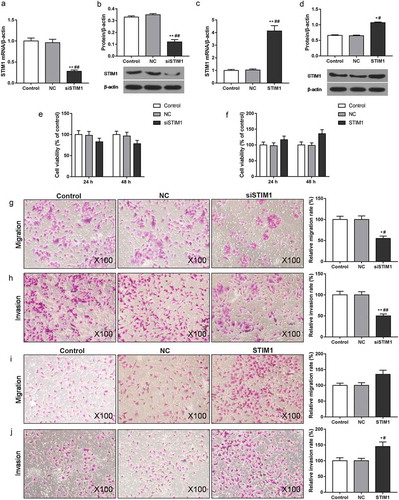
In order to investigate the effect of STIM1 on the viability and metastasis of ox-LDL induced MOVAS cells, CCK-8 and transwell assays were used. When cells were transfected with STIM1 siRNA plasmid, the STIM1 expression was down-regulated (). The mRNA and protein levels of STIM1 were up-regulated in cells exposed to pcDNA3.1(+)-STIM1 plasmid (). As CCK-8 data shown, silence of STIM1 inhibited cell viability while STIM1 overexpression promoted cell viability (). The transwell assay results found that siSTIM1 decreased the capacities of migration and invasion, but STIM1 increased cell migration and invasion ().
Figure 4. STIM1 silencing attenuated the abilities of proliferation, migration and invasion in ox-LDL induced MOVAS cells. MOVAS cells were subjected to ox-LDL, and then transfected with unspecific scrambled siRNA, STIM1 siRNA, pcDNA3.1(+) and pcDNA3.1(+)-STIM1 plasmids. (a-d) The levels of STIM1 were detected by RT-qPCR (a, c) and Western blot (b, d). (e, f) Cell viability was tested by CCK-8. (g-j) Cell migration (g, i) and invasion (h, j) were evaluated using transwell assay. *P< 0.05, **P< 0.01, versus control. #P< 0.05, ##P< 0.01, versus NC.
Inhibition of miR-185 enhanced the abilities of proliferation, migration and invasion in ox-LDL induced MOVAS cells by targeting STIM1
To test whether miR-185 silencing promotes cell proliferation and metastasis by targeting STIM1, the Rescue experiment was executed. The CCK-8 data showed that cell viability was increased in inhibitor+NC group, compared to NC group. In comparison with inhibitor+NC group, cell viability was decreased in inhibitor+siSTIM1 group (). Similarly, the relative migration and invasion rates of cells were enhanced in cells exposed to miR-185 inhibitor, in relative to NC group. However, in comparison with inhibitor+NC group, the rates of cell migration and invasion were depressed in inhibitor+siSTIM1 ().
Figure 5. Inhibition of miR-185 enhanced the abilities of proliferation, migration and invasion in ox-LDL induced MOVAS cells by targeting STIM1. (a) CCK-8 was used to test cell viability. (b-e) Transwell assay was carried out to analyze cell migration (b, d) and invasion (c, e). *P< 0.05, versus control. #P< 0.05, versus NC. ^P< 0.05, versus mock. &P< 0.05, &&P< 0.01, versus inhibitor+NC.
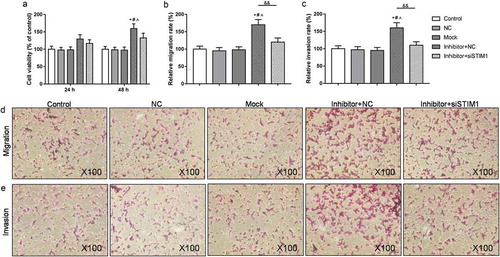
Inhibition of miR-185 up-regulated the expression of VEGF and MMP-9 by targeting STIM1 in ox-LDL induced MOVAS cells
To examine the molecular mechanism of miR-185 and STIM1 on ox-LDL induced MOVAS cells, the levels of STIM1, VEGF, MMP-9 and miR-185 were assessed by RT-qPCR and Western blot. As RT-qPCR revealed, the mRNA levels of STIM1, VEGF and MMP-9 were strengthened, and miR-185 expression was repressed in cells exposed miR-185 inhibitor, compared to NC group. Nevertheless, when cells were transfected with miR-185 inhibitor and STIM1 siRNA plasmid together, STIM1, VEGF, MMP-9 and miR-185 mRNA levels were inhibited, compared with inhibitor+NC (). At the same time, the protein expression of STIM1, VEGF and MMP-9 in each group were in line with the mRNA expression ().
Figure 6. Inhibition of miR-185 up-regulated the expression of VEGF and MMP-9 by targeting STIM1 in ox-LDL induced MOVAS cells. (a-d) The mRNA expression of STIM1, VEGF, MMP-9 and miR-185 were assessed by RT-qPCR. (e-g) The protein levels of STIM1, VEGF and MMP-9 were determined by Western blot. *P< 0.05, **P< 0.01, versus control. #P< 0.05, ##P< 0.01, versus NC. ^P< 0.05, ^^P< 0.01, versus mock. &P< 0.05, &&P< 0.01, versus inhibitor+NC.
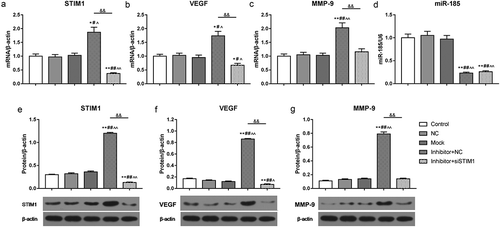
Inhibition of miR-185 augmented atherosclerosis by targeting STIM1 in vivo
To further analyze the effect of miR-185 and STIM1 on atherosclerosis in mice, the lesions of arterial wall tissues were evaluated by HE staining assay. The data revealed that in comparison with control group, the lesions of arterial wall tissues were increased in atherosclerosis model mice. The lesions of arterial wall tissues were further enhanced in INMO group. However, the lesions of arterial wall tissues were decreased in InsiSITM1 and siSTIM1 NC group, compared to INMO group (). Subsequently, we examined whether miR-185 silencing enhanced atherosclerosis in mice by targeting STIM1, the level of STIM1 was assessed by IHC assay. The results showed that STIM1 positive rate was increased in model group, in relative to control group. When model mice were injected with miR-185 inhibitor treated-cell suspension, STIM1 positive rate was further increased. Nevertheless, when model mice were injected with miR-185 inhibitor and STIM1 siRNA plasmid treated-cell suspension, STIM1 positive rate was decreased, compared to INMO group. Meanwhile, in comparison with INsiSTIM1 group, STIM1 positive rate further suppressed in siSTIM1 NC group ().
Figure 7. Inhibition of miR-185 augmented atherosclerosis by targeting STIM1 in vivo. (a) The lesions of arterial wall tissues were detected by HE staining assay. (b) STIM1 positive rate was measured by IHC assay. *P< 0.05, **P< 0.01, versus control. ^P< 0.05, ^^P< 0.01, versus mock. &P< 0.05, versus INMO. #P< 0.05, ##P< 0.01, versus INsiSTIM1.
Discussion
Oxidized low-density lipoprotein (ox-LDL) is the main risk factor inducing AS [Citation29,Citation30]. Ox-LDL can be able to enter cells through a receptor pathway. In this pathway, the cholesterol in ox-LDL can be absorbed by macrophages, making it a “foam” cell and adhering to the arterial wall, gradually forming atherosclerotic plaques and blocking the corresponding blood vessels [Citation31,Citation32]. It has been found that the stimulation of ox-LDL to endothelial cells, dendritic cells, macrophages and vascular smooth muscle cells can cause a series of changes in the cell, such as the apoptosis of endothelial cells, the migration of vascular smooth muscle cells and so on [Citation33–Citation36]. The proliferation and metastasis of VSMCs play a pivotal role in the formation of atherosclerosis and the stability of plaques [Citation37–Citation39]. Therefore, we chose the mouse aortic vascular smooth muscle cells (MOVAS) as the object of study and used ox-LDL to induce MOVAS cells for building AS model in vitro. We found that ox-LDL promoted the proliferation, migration and invasion of MOVAS cells.
More and more studies have proved that miRNAs have been involved in the development of AS, which control the proliferation, apoptosis, metastasis and inflammatory response of AS cells [Citation16,Citation40,Citation41]. miR-9 has been confirmed to repress the proliferation of VSMCs [Citation42]. Yang F et al. have proved that miR-22 suppressed proliferation and migration of VSMCs [Citation43]. However, the effect of miR-185 on AS is unknown. Jiang H et al. have confirmed that miR-185 can control cholesterol homeostasis via regulating low-density lipoprotein receptor expression [Citation44]. miR-185 has been shown to depress proliferation and metastasis of various cancer cells, including breast cancer, colon cancer, pancreatic cancer and so on [Citation45–Citation47]. Hence, we suspected that miR-185 might modulate the proliferation and metastasis of MOVAS cells. As we expected, we found that miR-185 was significantly down-regulated in ox-LDL-induced MOVAS cells. miR-185 overexpression decreased cell viability, migration and invasion, whereas miR-185 silencing had the opposite effect. The result proved that inhibition of miR-185 enhanced the abilities of proliferation and metastasis in AS cells.
It is well known that STIM1 is implicated in regulating the pathogenesis of AS by controlling the biological function of endothelial and smooth muscle cells [Citation48,Citation49]. Hou J et al. have illustrated that miR-185 targeted regulation of STIM1 gene to inhibit angiogenesis of human microvascular endothelial cells [Citation50]. Base on these evidences, we guessed that STIM1 might be a potential target gene of miR-185 in AS. In our study, we found that 3ʹ-UTR of STIM1 contained miR-185 binding site according to the Targetscan. Besides, the relative luciferase activity was significantly depressed in miR-185 + STIM1-3ʹ-UTR group, while not changed in miR-185 + STIM1-3ʹ-UTR mut group, compared to control+ STIM1-3ʹ-UTR group. The result suggested that STIM1 was a target gene for miR-185 in AS. Moreover, the data showed that STIM1 was high expressed in ox-LDL-induced MOVAS cells. miR-185 overexpression down-regulated STIM1 level, contrarily, miR-185 silencing had the opposite effect. The result proved that miR-185 and STIM1 had a negative correlation in AS. Furthermore, Yang D et al. have elucidated STIM1 silencing attenuates the proliferation, migration and invasion of colorectal cancer cells [Citation51]. Potier M et al. have validated that knockdown of STIM1 suppresses the viability and metastasis of [Citation52]. Similarly, our data substantiated that STIM1 silencing repressed the proliferation, migration and invasion of AS cells. On the contrary, STIM1 overexpression had the opposite effect. Subsequently, we used the Rescue experiment to further verify that miR-185 silencing promoted the proliferation and metastasis of AS cells through targeting STIM1 gene.
Vascular endothelial growth factor (VEGF) is a highly specific vascular endothelial growth factor, which promotes the increase of vascular permeability, extracellular matrix degeneration, vascular endothelial cell migration, proliferation and angiogenesis [Citation53]. A study found that 1,25-Dihydroxyvitamin D3 increased the viability of VSMCs via increasing VEGF levels [Citation54]. Schlich reported that VEGF might link adipose inflammation to increased VSMC proliferation [Citation55]. Dorafshar suggested that VEGF appeared to have a vascular protective function by directly inhibiting VSMC proliferation [Citation56]. Zhang found that nesfatin-1 promoted VSMC proliferation, migration and neointimal hyperplasia via elevating MMP2/MMP-9 levels and inhibiting PPARγ gene expression [Citation57]. MMP-9 has been proved to be involved in angiogenesis by releasing VEGF [Citation58]. Kim J et al. have found that Human sLZIP accelerates AS by inducing MMP-9 expression and VSMCs migration [Citation59]. Hence, we suspected that the molecular mechanism of miR-185 and STIM1 on AS cells might regulate VEGF and MMP-9. As we expected, the data revealed that miR-185 silencing enhanced the levels of VEGF and MMP-9 in ox-LDL-treated MOVAS cells. Nevertheless, when ox-LDL-treated cells were exposed to miR-185 inhibitor and STIM1 siRNA, the levels of VEGF and MMP-9 were inhibited, compared to inhibitor + NC group. The phenomenon testified that miR-185 silencing promoted AS by targeting STIM1 and up-regulating VEGF and MMP-9 expression.
To further investigate the role of miR-185 and STIM1 in AS cells in vivo, the lesions of arterial wall tissues and STIM1 level were tested by HE staining and IHC assays. The result found that miR-185 silencing markedly increased the lesions of arterial wall tissues, while knockdown of STIM1 obviously decreased the relative area. In addition, consistent with in vitro results, miR-185 silencing significantly up-regulated the expression of STIM1, and STIM1 silencing down-regulated STIM1 level in vivo. These results further indicated that the expression of miR-185 and STIM1 had a negative correlation in AS, and miR-185 silencing augmented the progression of AS by targeting STIM1. Strictly speaking, there are still shortcomings in this study. As we all know, atherosclerosis goes through the following processes, including lipid deposition in the intima; proliferation of VSMCs and fibrous components; and breakdown of the internal tissue of the plaque and its binding to the deposited lipid [Citation60,Citation61]. In vivo experiment, we could find the effects of miR-185 and STIM1 on the whole aortic wall, while, we only investigated the VSMCs in vitro. Though the proliferation and migration of VSMCs are key steps in the progression of AS, it was still incomplete and circumscribed [Citation62]. The next step of our study is going to further explore the molecular mechanisms of AS in vivo.
All in all, above results proved that STIM1 was a potential target gene of miR-185 in AS. The relationship between miR-185 and STIM1 was negatively correlated in AS. Knockdown of miR-185 facilitated the progression of AS through enhancing cell proliferation, migration and invasion via targeting STIM1. The research provides a novel view of miR-185/STIM1 axis function in AS development, and this targeting method may prevent and treat AS.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Hung SH, Su C-H, Lin H-C, et al. A sialoadenectomy is associated with an increased risk of coronary heart disease: a three-year follow-up study. PLoS One. 2018;13(6):e0199135.
- Whalley B, Thompson DR, Taylor RS. Psychological interventions for coronary heart disease: cochrane systematic review and meta-analysis. Int J Behav Med. 2014;21(1):109–121.
- Pieper KS, Gore JM, FitzGerald G, et al. Validity of a risk-prediction tool for hospital mortality: the global registry of acute coronary events. Am Heart J. 2009;157(6):1097–1105.
- Abbate R, Cioni G, Ricci I, et al. Thrombosis and acute coronary syndrome. Thromb Res. 2012;129(3):235–240.
- Fernandez-Ortiz A, Badimon JJ, Falk E, et al. Characterization of the relative thrombogenicity of atherosclerotic plaque components: implications for consequences of plaque rupture. J Am Coll Cardiol. 1994;23(7):1562–1569.
- Roth L, Rombouts M, Schrijvers DM, et al. Chronic intermittent mental stress promotes atherosclerotic plaque vulnerability, myocardial infarction and sudden death in mice. Atherosclerosis. 2015;242(1):288–294.
- Shah PK, Falk E, Badimon JJ, et al. Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques. Potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation. 1995;92(6):1565–1569.
- Gao M, Xin G, Qiu X, et al. Establishment of a rat model with diet-induced coronary atherosclerosis. J Biomed Res. 2016;31(1):47–55.
- Badimon L, Vilahur G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J Intern Med. 2014;276(6):618–632.
- Bernardo BC, Ooi JYY, Lin RCY, et al. miRNA therapeutics: a new class of drugs with potential therapeutic applications in the heart. Future Med Chem. 2015;7(13):1771–1792.
- Karnati HK, Panigrahi MK, Gutti RK, et al. miRNAs: key players in neurodegenerative disorders and epilepsy. J Alzheimers Dis. 2015;48(3):563–580.
- Mehrgou A, Akouchekian M. Therapeutic impacts of microRNAs in breast cancer by their roles in regulating processes involved in this disease. J Res Med Sci. 2017;22:130.
- Rocci A, Hofmeister CC, Pichiorri F. The potential of miRNAs as biomarkers for multiple myeloma. Expert Rev Mol Diagn. 2014;14(8):947–959.
- Shaffi, SK, Galas, D, Etheridge, A, et al. Role of microRNAs in renal parenchymal diseases – a new dimension. Int J Mol Sci. 2018;19(6):1797
- Kumar S, Kim CW, Simmons RD, et al. Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs. Arterioscler Thromb Vasc Biol. 2014;34(10):2206–2216.
- Yilmaz SG, Isbir S, Kunt AT, et al. Circulating microRNAs as novel biomarkers for atherosclerosis. In Vivo. 2018;32(3):561–565.
- Kim JO, Grady B, Page K, et al. miR-185 plays an anti-hypertrophic role in the heart via multiple targets in the calcium-signaling pathways. PLoS One. 2015;10(3):e0122509.
- Dong-Xu W, Jia L, Su-Juan Z. MicroRNA-185 is a novel tumor suppressor by negatively modulating the Wnt/beta-catenin pathway in human colorectal cancer. Indian J Cancer. 2015;52(Suppl 3):E182–5.
- Huang, Q, Sun Y, Ma X, et al. Androgen receptor increases hematogenous metastasis yet decreases lymphatic metastasis of renal cell carcinoma. Nat Commun. 2017;8(1):918.
- Li S, Ma Y, Hou X, et al. MiR-185 acts as a tumor suppressor by targeting AKT1 in non-small cell lung cancer cells. Int J Clin Exp Pathol. 2015;8(9):11854–11862.
- Xiang Y, Ma N, Wang D, et al. MiR-152 and miR-185 co-contribute to ovarian cancer cells cisplatin sensitivity by targeting DNMT1 directly: a novel epigenetic therapy independent of decitabine. Oncogene. 2014;33(3):378–386.
- Zou Q, Wu H, Fu F, et al. RKIP suppresses the proliferation and metastasis of breast cancer cell lines through up-regulation of miR-185 targeting HMGA2. Arch Biochem Biophys. 2016;610:25–32.
- Kimura M, Nishi K, Higashikawa A, et al. High pH-sensitive store-operated Ca(2+) entry mediated by Ca(2+) release-activated Ca(2+) channels in rat odontoblasts. Front Physiol. 2018;9:443.
- Roos J, DiGregorio PJ, Yeromin AV, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169(3):435–445.
- Perni S, Dynes JL, Yeromin AV, et al. Nanoscale patterning of STIM1 and Orai1 during store-operated Ca2+ entry. Proc Natl Acad Sci U S A. 2015;112(40):E5533–42.
- Shi J, Miralles F, Kinet J-P, et al. Evidence that Orai1 does not contribute to store-operated TRPC1 channels in vascular smooth muscle cells. Channels (Austin). 2017;11(4):329–339.
- Xia J, Wang H, Huang H, et al. Elevated Orai1 and STIM1 expressions upregulate MACC1 expression to promote tumor cell proliferation, metabolism, migration, and invasion in human gastric cancer. Cancer Lett. 2016;381(1):31–40.
- Zhou JB, Sun -Y-Y, Zheng Y-L, et al. A study on blocking store-operated Ca2+ entry in pulmonary arterial smooth muscle cells with xyloketals from marine fungi. Acta Pharm. 2017;67(4):557–567.
- Gao LN, Zhou X, Lu Y-R, et al. Dan-Lou prescription inhibits foam cell formation induced by ox-LDL via the TLR4/NF-kappaB and PPARgamma signaling pathways. Front Physiol. 2018;9:590.
- Tang, F, Yang TL, Zhang Z, et al. Retraction notice to “MicroRNA-21 suppresses ox-LDL-induced human aortic endothelial cells injuries in atherosclerosis through enhancement of autophagic flux: involvement in promotion of lysosomal function” [Experimental Cell Research 359 Issue 2 (2017)374-383]. Exp Cell Res. 2018;369(1):177.
- Chaabane C, Coen M, Bochaton-Piallat ML. Smooth muscle cell phenotypic switch: implications for foam cell formation. Curr Opin Lipidol. 2014;25(5):374–379.
- Zhang K, Meng X, Kong J, et al. Simvastatin increases Prolyl-4-Hydroxylase alpha1 expression in atherosclerotic plaque and ox-LDL-stimulated human aortic smooth muscle cells via p38 MAPK and ERK1/2 signaling. J Mol Cell Cardiol. 2013;65:43–50.
- Ganesan R, Henkels KM, Wrenshall LE, et al. Oxidized LDL phagocytosis during foam cell formation in atherosclerotic plaques relies on a PLD2-CD36 functional interdependence. J Leukoc Biol. 2018;103(5):867–883.
- Hofmann A, Brunssen C, Poitz DM, et al. Lectin-like oxidized low-density lipoprotein receptor-1 promotes endothelial dysfunction in LDL receptor knockout background. Atheroscler Suppl. 2017;30:294–302.
- Obermayer G, Afonyushkin T, Binder CJ. Oxidized low-density lipoprotein in inflammation-driven thrombosis. J Thromb Haemost. 2018;16(3):418–428.
- Pan J, Lu L, Wang X, et al. AIM2 regulates vascular smooth muscle cell migration in atherosclerosis. Biochem Biophys Res Commun. 2018;497(1):401–409.
- Li M, Liu Q, Lei J, et al. MiR-362-3p inhibits the proliferation and migration of vascular smooth muscle cells in atherosclerosis by targeting ADAMTS1. Biochem Biophys Res Commun. 2017;493(1):270–276.
- Wang B, Ge Z, Cheng Z, et al. Tanshinone IIA suppresses the progression of atherosclerosis by inhibiting the apoptosis of vascular smooth muscle cells and the proliferation and migration of macrophages induced by ox-LDL. Biol Open. 2017;6(4):489–495.
- Zhang X, Shi H, Wang Y, et al. Down-regulation of hsa-miR-148b inhibits vascular smooth muscle cells proliferation and migration by directly targeting HSP90 in atherosclerosis. Am J Transl Res. 2017;9(2):629–637.
- Li S, Sun W, Zheng H, et al. Microrna-145 accelerates the inflammatory reaction through activation of NF-kappaB signaling in atherosclerosis cells and mice. Biomed Pharmacother. 2018;103:851–857.
- Yokoyama, Y, Mise, N, Suzuki Y, et al. MicroRNAs as potential mediators for cigarette smoking induced atherosclerosis. Int J Mol Sci. 2018;19(4): 1097.
- Ham O, Lee S-Y, Song B-W, et al. Small molecule-mediated induction of miR-9 suppressed vascular smooth muscle cell proliferation and neointima formation after balloon injury. Oncotarget. 2017;8(55):93360–93372.
- Yang F, Chen Q, He S, et al. miR-22 is a novel mediator of vascular smooth muscle cell phenotypic modulation and neointima formation. Circulation. 2018;137(17):1824–1841.
- Jiang H, Zhang J, Du Y, et al. microRNA-185 modulates low density lipoprotein receptor expression as a key posttranscriptional regulator. Atherosclerosis. 2015;243(2):523–532.
- Wang R, Tian S, Wang H-B, et al. MiR-185 is involved in human breast carcinogenesis by targeting Vegfa. FEBS Lett. 2014;588(23):4438–4447.
- Xia D, Li X, Niu Q, et al. MicroRNA-185 suppresses pancreatic cell proliferation by targeting transcriptional coactivator with PDZ-binding motif in pancreatic cancer. Exp Ther Med. 2018;15(1):657–666.
- Zhang W, Sun Z, Su L, et al. miRNA-185 serves as a prognostic factor and suppresses migration and invasion through Wnt1 in colon cancer. Eur J Pharmacol. 2018;825:75–84.
- Rodriguez-Moyano, M, Diaz, I, Dionisio, N, et al. Urotensin-II promotes vascular smooth muscle cell proliferation through store-operated calcium entry and EGFR transactivation. Cardiovasc Res. 2013;100(2):297–306.
- Wang LY, Zhang J-H, Yu J, et al. Reduction of store-operated Ca(2+) entry correlates with endothelial progenitor cell dysfunction in atherosclerotic mice. Stem Cells Dev. 2015;24(13):1582–1590.
- Hou J, Liu L, Zhu Q, et al. MicroRNA-185 inhibits angiogenesis in human microvascular endothelial cells through targeting stromal interaction molecule 1. Cell Biol Int. 2016;40(3):318–328.
- Yang D, Dai X, Li K, et al. Knockdown of stromal interaction molecule 1 inhibits proliferation of colorectal cancer cells by inducing apoptosis. Oncol Lett. 2018;15(6):8231–8236.
- Potier M, Gonzalez JC, Motiani RK, et al. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. Faseb J. 2009;23(8):2425–2437.
- Gillies MC, Daien V, Nguyen V, et al. Re: comparison of age-related macular degeneration treatments trials (CATT) Research Group, et al.: five-year outcomes with anti-vascular endothelial growth factor treatment of neovascular age-related macular degeneration: the comparison of age-related macular degeneration treatments trials (Ophthalmology 2016; 123:1751-1761). Ophthalmology. 2017;124(3):e31–e32.
- Cardus A, Parisi E, Gallego C, et al. 1,25-Dihydroxyvitamin D3 stimulates vascular smooth muscle cell proliferation through a VEGF-mediated pathway. Kidney Int. 2006;69(8):1377–1384.
- Schlich R, Willems M, Greulich S, et al. VEGF in the crosstalk between human adipocytes and smooth muscle cells: depot-specific release from visceral and perivascular adipose tissue. Mediators Inflamm. 2013;2013:982458.
- Dorafshar AH, Angle N, Bryer-Ash M, et al. Vascular endothelial growth factor inhibits mitogen-induced vascular smooth muscle cell proliferation. J Surg Res. 2003;114(2):179–186.
- Zhang JR, Lu Q-B, Feng W-B, et al. Nesfatin-1 promotes VSMC migration and neointimal hyperplasia by upregulating matrix metalloproteinases and downregulating PPARgamma. Biomed Pharmacother. 2018;102:711–717.
- Butoi E, Gan AM, Tucureanu MM, et al. Cross-talk between macrophages and smooth muscle cells impairs collagen and metalloprotease synthesis and promotes angiogenesis. Biochim Biophys Acta. 2016;1863(7 Pt A):1568–1578.
- Kim J, Ko J. Human sLZIP promotes atherosclerosis via MMP-9 transcription and vascular smooth muscle cell migration. Faseb J. 2014;28(11):5010–5021.
- Borow KM, Nelson JR, Mason RP. Biologic plausibility, cellular effects, and molecular mechanisms of eicosapentaenoic acid (EPA) in atherosclerosis. Atherosclerosis. 2015;242(1):357–366.
- McMahon M, Skaggs B. Pathogenesis and treatment of atherosclerosis in lupus. Rheum Dis Clin North Am. 2014;40(3):475–95, viii.
- Gao S, Xu L, Zhang Y, et al. Salusin-alpha inhibits proliferation and migration of vascular smooth muscle cell via Akt/mTOR signaling. Cell Physiol Biochem. 2018;50(5):1740–1753.