
ABSTRACT
In our previous work, gene PPP1R11 (protein phosphatase 1 regulatory subunit 11) was significantly expressed in pigs after Streptococcus suis 2 (SS2) challenged. This study firstly confirmed that SS2 induced significant expression of PPP1R11 gene in porcine alveolar macrophage (PAM) cells, and apoptosis of PAM cells were observed. After that, the core promoter of porcine PPP1R11 was identified and its transcription factor AREB6 which significantly regulated PPP1R11. We also characterized that the PPP1R11 gene is a target of miR-34a. Further, we found that PPP1R11 helped to inhibit apoptosis of PAM cells under SS2 infecting, through transcription factor AREB6 was negatively correlated with apoptosis whereas miR-34a was positively correlated. Those findings provide a functional connection among the transcription factor AREB6, miR-34a, PPP1R11 gene and apoptosis of PAM cells in the pathogenesis of the SS2 infection.
Introduction
Streptococcus suis 2 (S. suis 2 or SS2) is an important swine pathogen and emerging zoonotic agent, causing arthritis, pneumonia, septicemia, meningitis and even acute death, and has broken out several times worldwide, and been isolated from a wide range of mammalian species [Citation1,Citation2]. In our previous work, PPP1R11 (protein phosphatase 1 regulatory subunit 11) gene was significantly expressed in pigs after SS2 challenged[Citation3]. The PPP1R11 gene is a member of the PPP (protein phosphatases) superfamily, which encodes protein inhibitor 3 (Inh3) to inhibit PP1, which regulates diverse cellular processes, such as cell cycle, transcription, protein synthesis and metabolism [Citation4–Citation7]. The role of PPP1R11 in immune response has been paid more attention than in other functions [Citation8,Citation9]. Recently, PPP1R11 was found as an uncharacterized RING finger E3 ligase, which attenuates TLR (Toll-like receptor) 2 signaling in response to Staphylococcus aureus infection by targeting TLR2 for proteasomal degradation[Citation9].
AREB6 [Atp1a1 (Na, K-ATPase α1 subunit) regulatory element binding factor 6] is a zinc finger-homeodomain transcription factor (is also known as Zinc finger E-box binding homeobox 1, ZEB1) that induce epithelial–mesenchymal transition (EMT), which plays important roles in both normal physiological and pathological processes [Citation10–Citation12]. For example, AREB6 was identified as a repressor on the immunoglobulin heavy-chain enhancer[Citation13]; and as a negative transcription factor for gene interleukin 2 (IL-2) to turn off the IL-2 gene transcription just after T-cell activation [Citation14,Citation15].
MiRNAs (microRNAs) are noncoding small RNA molecules capable of regulating gene expression at the post-transcriptional level[Citation16]. Some host miRNAs have been reported to involve in the host-bacteria crosstalk [Citation17,Citation18], and miRNAs play important roles in modulating macrophages response toward pathogens, e.g. dynamic regulators of macrophage polarization and plasticity [Citation19,Citation20]. Macrophages are the first defense cells in the body which are exposed to pathogenic microorganisms[Citation21], like SS2[Citation22]. In literatures, alveolar macrophage (AM) cells were employed to study the function of genes, especially in pathogen infection [Citation22–Citation27].
Apoptosis is considered as a major defense mechanism of the body. Multiple pathogens induce macrophage apoptosis as a mode of immune evasion[Citation28]. The resistance or susceptibility to viruses or bacteria was strongly linked to apoptosis [Citation29,Citation30]. In the case of S. suis, reports about apoptotic effects are limited [Citation31–Citation33]. Zeng & Lu (2003) demonstrated the muramidase released protein (MRP) which is a virulence factor of SS2 induced HEp-2 cell apoptosis. [Citation31] Tenenbaum, et al. (2006) indicated that S. suis causes cell death in PCPEC (porcine choroid plexus epithelial cell) predominant by necrosis, although apoptosis may be involved in the process of PCPEC cell death[Citation33]. By comparing S. suis wild strain with suilysin-deficient mutant in porcine respiratory epithelial cells under ALI (air-liquid interface cultures) conditions, Meng et al. (2016) demonstrated that suilysin contributes to apoptosis, and the cytolytic activity of suilysin is crucial for this effect [Citation34].
The aim of this study focused on the PPP1R11 gene, try to explain the mechanism underlying its differential expressions in pigs after SS2 infection. The results demonstrated that transcription factor AREB6 binds to and activates PPP1R11 promoter, and the PPP1R11 is a target of miR-34a. AREB6 and miR-34a both supported to regulate porcine AM (PAM) cell apoptosis through PPP1R11.
Materials and methods
Animal samples and bacteria SS2 and cells
The pig samples and bacteria SS2 were harvested according to previous study, [Citation3] which experimental protocols were approved by the Laboratory Animal Monitoring Committee of Hubei Province in China (no. Y2017-0612) and was guided to perform. PAM (porcine alveolar macrophage) cells were isolated from pigs aged 30 ~ 40 days which were tested for being pathogen-free: i.e., free from porcine respiratory and reproductive syndrome virus, porcine circovirus type 2 (PCV2), PCV3, Pseudorabies virus and SS2. The isolation processing of PAMs were: the lung was washed 3 ~ 4 times with fetal bovine serum (PBS, pH 7.2) supplemented with 2% fetal bovine serum (Fisher Scientific, Waltham, MA, USA). The cell pellets were resuspended and mixed with prechilled GIBCO RPMI-1640 medium (Fisher Scientific) containing 40% FBS (Hyclone Laboratories Inc., USA). The number of the prepared PAMs reached 10 [Citation8],10 [Citation9]/ml with >95% viability. Aliquots of PAMs were frozen and stored in liquid nitrogen before use. Porcine kidney 15 cells (PK cells -) were obtained from the cell database of Wuhan University. Cells were cultured in F12 (Gibco, Waltham, USA) or DMEM (Gibco) supplied with 10% fetus bovine serum (Gibco) at 37°C in a humidified atmosphere of 5% CO2. The cells were plated and grown until they were 70–80% confluent at the time of use.
Plasmid construction and dual-luciferase reporter assays
Deletion fragments from the potential promoter region of the porcine PPP1R11 gene were amplified and double-digested with KpnI and HindIII, then cloned into the pGL3-basic vector (Promega). In addition, the 3′-UTR of PPP1R11 was amplified, double-digested with PmeI and XhoI, and then cloned into the pmirGLO vector (Promega, USA). Mutated binding site was generated using a Fast Site-Directed Mutagenesis Kit (TIANGEN, China) and mutagenic primers (PPP1R11-mut-F1: CCTTTGTTCTCACTCGGTAACTGCCTGTCCTGGGATCCAGTTATCCTGG; PPP1R11-mut-R1: GACAGGCAGTTACCGAGTGAGAACAAAGGGAAGAGATTGGGACCAAGATGC; PPP1R11-mut-F2: CCAGCTTTCACTCGGTGGGTCCTAGTCAGATTCCAGGCAACCTCG; PPP1R11-mut-R2: GACTAGGACCCACCGAGTGAAAGCTGGGGCTGTTGGAAACCCAGTTTAAC).
The miRNA or siRNA was transfected into PAM cells and PK cells using Lipofectamine RNAi MAX (Invitrogen, USA). Plasmids were transfected into the PAM cells using Lipofectamine 3000 (Invitrogen), while Lipofectamine 2000 (Invitrogen) was used when transfecting plasmids into the PK cells. The plasmid transfection amount was 2.5 μg, and the luciferase activities were measured with a PerkinElmer 2030 Multilabel Reader (PerkinElmer, USA).
qPCR analysis
Reverse transcription was performed using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, USA). Random hexamers, oligo(dT) or miRNA-specific stem-loop primers were added to initiate cDNA synthesis. qPCR (quantitative PCR) was performed on a BioRad CFX384 system (Bio-Rad, USA) using the iTaq Universal SYBRGreen Supermix (Bio-Rad). The qPCR primers were as follows: PPP1R11-F, CGGAGCCTAACCATCAAAC; PPP1R11-R, CCCTCCTCTTCCTCCTCAT. All PCR reactions were performed in triplicate. By employing the 2−ΔΔCt method, gene expression levels were normalized to the expression of β-actin and, mRNA expression levels were normalized to the expression of U6 using Gene Expression Macro software (Bio-Rad). Quantitative PCR results were used by the t-test two-tailed analysis.
Western blot analysis
Cell protein lysates were generated using RIPA Lysis Buffer (Beyotime, China) and were added to PMSF (Beyotime) at a ratio of 100:1. Cellular proteins were extracted 48 h posttransfection, the number of cells was 10 [Citation6]. Proteins were separated by SDS-PAGE and were transferred into polyvinylidene fluoride membranes (Millipore, USA). Primary antibodies specific for PPP1R11 (1:500, 20,263–1-AP, Proteintech, China), AREB6 (1:1000; A5600, ABclonal, USA), and β-actin (1:2000; AC006, ABclonal, USA) were used for immunoblotting. An Image Quant LAS4000 mini (GE Healthcare Life Sciences, USA) was used to detect protein expression.
Core promoter analysis
To determine the core promoter, multiple fragments were systematically deleted. The 2000 bp upstream from the first start codon of the PPP1R11 gene was set as a target region for the core promoter of interest. The targeted deletion was located upstream of the promoter at −2000 bp, −1500 bp, −1000 bp, −700 bp, −400 bp, −225 bp and −200 bp, respectively. Briefly, the work process as, the deleted core promoters were amplified and cloned into a vector. Subsequently, the successful plasmid bearing the deleted core promoter was transfected into PK cell. The luciferase activity was then determined upon transfection. Seven forward primers and one common reverse primer are shown in for generating deletion fragments, and the restriction enzyme sites (KpnI and HindIII) and protective bases were added into primers.
Table 1. PCR primers for the deletion fragment of the porcine PPP1R11 promoter
Transcription factor analysis
Based on the core promoter result, the BIOBASEsoftware was used to predict the transcription factor and its binding sites, and then EMSA (electrophoretic mobility shift assay) and ChIP (chromatin immunoprecipitation) were used to confirm the binding of the transcription factor within the core promoter region.
Electrophoretic mobility shift assays (EMSA)
The nuclear proteins of PK cells were extracted with the Nucleoprotein Extraction Kit (Beyotime). Oligos corresponding to the AREB6 binding sites of the PPP1R11 core promoter were synthesized and annealed into double strands. The DNA binding activity of the candidate transcription factor protein was detected by the LightShift® Chemiluminescent EMSA Kit (Pierce, USA). The 10 μg ovarian follicle nuclear extract was added to 20 fmol Biotin-labeled double-stranded oligonucleotides, 1× binding buffer, 2.5% Glycerol, 5 mM MgCl2, 50 ng Poly (dI•dC), 0.1 mM EDTA and 0.05% NP-40. The control group was added 2 pmol of unlabeled competitor oligonucleotides. The mixtures were then incubated at 24°C for 20 min. The reactions were analyzed by electrophoresis in 5.5% polyacrylamide gels in 0.5 × TBE buffer at 180 V for 35 min, and then transferred to a nylon membrane. The dried nylon was scanned with a GE ImageQuant LAS4000 mini (GE-Healthcare, USA).
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using the EZ-ChIP™ Kit (Millipore, USA). Briefly, after crosslinking the chromatin with 1% formaldehyde at 37°C for 10 min and neutralizing it with glycine for 5 min at room temperature, PK cells were washed with cold PBS, scraped and collected on ice. Then, cells were harvested, lysed and sonicated. Nuclear lysates were sonicated 20 times for 10 s with 20 min intervals in ice water using Scientz-IID (Scientz, China). An equal amount of chromatin was immuno-precipitated at 4°C overnight with at least 1.5 μg of AREB6 (GTX105278, GeneTex, USA) and normal mouse IgG (Millipore) antibodies. Immuno-precipitated products were collected after incubation with Protein A + G coated magnetic beads. The beads were washed, and the bound chromatin was eluted in ChIP elution buffer. Then, the proteins were digested with Proteinase K for 4 h at 45°C. DNA from the immune-precipitated complex was amplified through PCR. The primer sequences were as follows: AREG6-ChIP-F, 5′-GGACGCCTCTTCCGCCAAAT-3′; AREG6-ChIP-R, 5′-AGCCAGGTTCCTCCACCTTCTTT-3′.
MiRNA analysis
The software TargetScan (http://www.targetscan.org/) was used to predict the miRNAs targeting PPP1R11, obtained two potential candidates: miR-181 and miR-34a. As the miR-181 has no effect on the activity of the fluorescent carrier after the pmiRGLO vector is inserted into the PPP1R11 3′-UTR, it is not a potential miRNA to target the gene PPP1R11, whereas results showed that miRNA-34a has a targeted relationship with the PPP1R11-3′-UTR region.
The pig PPP1R11 3ˈ-UTRs were amplified, double-digested with PmeI and XhoI, and then cloned into the pmirGLO vector (Promega, USA). Binding site mutants were generated using a Fast Site-Directed Mutagenesis Kit (TIANGEN, China) and mutagenic primers. The primer sequences were as follows: PPP1R11-3′UTR-F, GGGTTTAAACTTGATCCCTTCTCCAGC; PPP1R11-3′-UTR-R, CCGCTCGAGAGACCTCACAATCCCACA; PPP1R11-3′-UTR-mut-F, CCAGCTTTCACTCGGTGGGTCCTAGTCAGATTCCAGGCAACCTCG; PPP1R11-3′-UTR-mut-R, GACTAGGACCCACCGAGTGAAAGCTGGGGCTGTTGGAAACCCAGTTTAAC. The synthesized siRNA-PPP1R11 (50nM) and miRNA-34a mimics (50nM) (RiboBio, Guangzhou, Guangdong, China) were transfected into cells with Lipofectamine RNAiMAX (Invitrogen). After 24 h and 48 h, cells were harvested for qPCR assays and western blot assays, respectively.
Apoptosis analysis
PAMs were transfected with miR-34a mimics, NC (negative control) mimics, miR-34a inhibitor, NC inhibitor, pCDNA3.1-PPP1R11, pCDNA3.1-AREB6, pCDNA3.1, siRNA-PPPR11, siRNA-AREB6 or siRNA NC and harvested 48 h after transfection. Fluorescence-activated cell sorting (FACS) was used to measure apoptosis. The experiments were performed according to the manufacturer’s protocol of the Annexin V-FITC Apoptosis Detection Kit (Invitrogen, USA). Apoptosis analysis was analyzed by software FlowJo v10.0.7.
Statistical analysis
All results are presented as the mean ± SD. Each experiment had three biological replicates, in each of which contains three technical repeats. Two-tailed t-test was used when two groups were compared. Significant differences were evaluated using an independent-samples t-test. P< 0.05 was considered to be statistically significant.
Results
PPP1R11 regulated the apoptosis of PAM cells challenged SS2
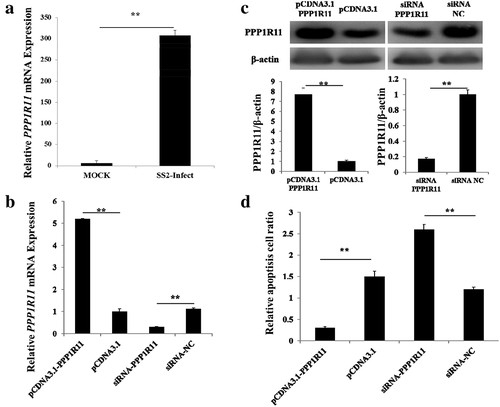
We firstly tested the PPP1R11 gene expression in PAM cells after SS2 challenged. The qPCR analysis showed that PPP1R11 in PAM cells was significantly up-regulated when compared the SS2 challenged samples to SS2 unchallenged samples ()). Then, we tested whether PPP1R11 can affect the apoptosis of PAM cells. pCDNA3.1-PPP1R11, pCDNA3.1, siRNA-PPP1R11 or siRNA-NC was transfected into PAMs. PPP1R11 mRNA and protein expression levels were significantly increased when PAM cells were transfected with pCDNA3.1-PPP1R11 or suppressed when transfected with pCDNA3.1, siRNA-PPP1R11 or siRNA NC (, ). As shown in ), PPP1R11 overexpression suppressed apoptosis of PAM cells and PPP1R11 inhibition promoted PAM cell apoptosis.
Figure 1. PPP1R11 regulated the apoptosis of porcine alveolar macrophage (PAM) cells. (a) PPP1R11 mRNA levels were detected at 24 h after being challenged with SS2 (0.5 moi). (b) qPCR was used to detect PPP1R11 in PAM cells at 24 h after transfection with pcDNA3.1-PPP1R11, and pcDNA3.1 as control; with siRNA-PPP1R11, and siRNA NC, respectively. (c) Western blot and a densitometric analysis using software imageJ to detect PPP1R11 protein levels at 48 h after PAM cells transfection. (d) PAMs were transfected with pcDNA3.1-PPP1R11, pcDNA3.1, siRNA-PPP1R11 or siRNA negative control (NC), harvested and stained with anti-annexin V-propidium iodide, and analyzed by FACS at 48 h post transfection. The results were expressed as the mean ± S.E.M. (three independent replicates per group). ** p < 0.01.
Transcription factor AREB6 regulates expression of PPP1R11
To date, there have been no reports about porcine PPP1R11; thus, we identified its core promoter and regulated elements. A 2000 bp contig in 5ʹ-flanking region of the porcine PPP1R11 gene was amplified by PCR. To determine the promoter region, seven promoter deletions were introduced upstream of the luciferase gene. Luciferase activity analysis in PK cells revealed that the 5ʹ-flanking sequence from −225 to −200 bp was important for its transcriptional activity ()).
Figure 2. Determination of the core promoter of PPP1R11 gene. (a) This shows the luciferase activity changes with the seven deletion fragments in PK cells. The left panel shows the name of pGL3-based vector for each deletion mutant. The right panel indicates the relative activities of these deletion constructs. Luciferase activity was analyzed at 24 h after transfection. (b) Prediction results of the transcription factor for the PPP1R11 core promoter by software BIOBASE. (c) Luciferase activities of the four candidate transcription factors-USP, p54, C/EBP and PAX4-through point mutation of their binding site of the PPP1R11 core promoter.
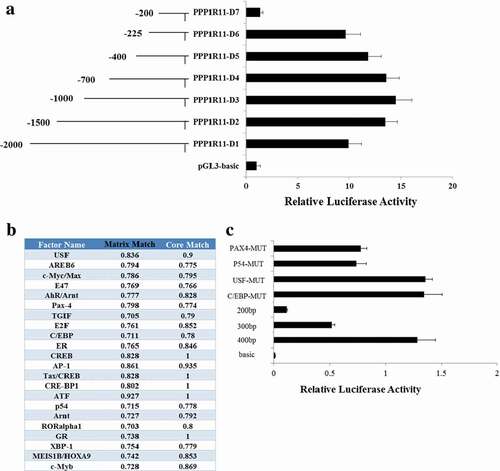
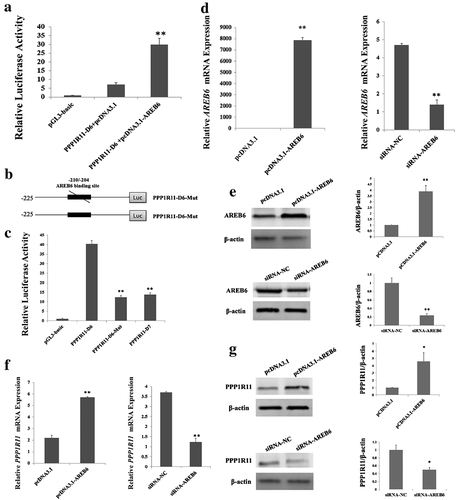
To further identify the transcription factor which binds to the core promoter of PPP1R11, the software BIOBASE was used to predict the transcription factor and its binding sites within the region −225 to −200 bp; the predicted results are shown in ). From those results, we have selected five candidates: Pax4 (paired box 4), p54, USF (upstream stimulating factor), C/EBP (CCAAT-enhancer-binding protein) and AREB6. We mutated the binding sites of those five candidates and transfected them into PK cells to detect fluorescence, and found there was no significant difference from their corresponding wild-type in four candidates: i.e., Pax4, p54, USF and C/EBP ()). Those indicated that the four transcription factors are not key transcription factors regulating the PPP1R11 gene. We found that the fluorescence activity increased significantly after the transfection of the AREB6 overexpression vector into PK cells compared to the PGL3-basic and pCDNA3.1 no-load vectors ()), then identified point mutations in the AREB6-binding sites of the PPP1R11 promoter ()) and observed that its fluorescence activity changed significantly ()). Therefore, we concluded that AREB6 may be a key transcription factor regulating the PPP1R11 gene. Further, we overexpressed and knocked-down expression of AREB6 by the transfection with pCDNA3.1-AREB6 and siRNA-AREB6 (, ), then checked the mRNA and protein expression levels of the target PPP1R11, and found that the expression of PPP1R11 was increased when AREB6 was overexpressed and was decreased by knockdown of AREB6 (, )). Those results further demonstrated that AREB6 is a key transcription factor for PPP1R11.
Figure 3. Verification of the binding effect of AREB6 on the PPP1R11 core promoter region. (a) Luciferase activity was analyzed at 24 h after PK cells were transfected with pGL3-basic; PPP1R11-D6 + pcDNA3.1; and PPP1R11-D6 + pcDNA3.1-AREB6 constructs. (b, c) Point mutation in the AREB6-binding sites of the PPP1R11 promoter; luciferase activity was analyzed at 24 h after transfection. (d) qPCR was used to detect AREB6 at 24 h after PK cells transfected with pcDNA3.1-AREB6, pcDNA3.1 as control; siRNA-AREB6, and siRNA NC, respectively. (e) Western blot and a densitometric analysis by using software imageJ to detect AREB6 protein levels in PK cells at 48 h after transfection. (f) qPCR was used to detect PPP1R11 mRNA levels at 24 h posttransfection in PK cells. (g) Western blot and densitometric analysis using software imageJ showed PPP1R11 protein levels at 48 h after PK cells transfection. The results are expressed as the mean ± S.E.M. (three independent replicates per group). ** p < 0.01.
Transcription factor AREB6 binds to PPP1R11 promoter
ChIP analysis was performed to investigate whether AREB6 binds to the pig PPP1R11 gene promoter in vitro. A 244 bp DNA region from samples immunoprecipitated with an anti-AREB6 antibody was amplified, and no band from the anti-IgG or anti-OCT1 antibody precipitates was observed ()). The results indicated that AREB6 binds to the pig PPP1R11 gene promoter region in vitro.
Figure 4. ChIP and EMSA showed that AREB6 could bind to the PPP1R11 promoter. (a) ChIP assay. The DNA fragments interacting with AREB6 protein were pulled down by anti-HA antibodies. DNA isolated from immunoprecipitated material was amplified by PCR. The “Input” was the positive control, “IgG” and “OCT1” were the negative control group, and “AREB6” was the experimental group. Agarose gel electrophoresis showed that the DNA fragments were distributed in a range of 200–1000 bp and reached the ChIP test requirement. Total chromatin was used as the input. (b) EMSAs. Lane 1 is a negative control which only added the 5′ biotin-labeled probe containing the AREB6 binding site. Therefore, only the band of the free probe was shown. In contrast, lane 2 is the experiment group, which added the 5′ biotin-labeled probe containing the AREB6 binding site, and simultaneously added the nuclear extraction matters, where the moving band of the DNA- and nucleoprotein-complex could not be seen; lane 3 and lane 4 were both experimental controls. Lane 3 added the 50 × 5′ unlabeled probe containing the AREB6 binding site to competitively bind the AREB6 protein, so there was no band of the DNA- and protein-complex, whereas lane 4 added the 5′ unlabeled probe containing the AREB6 binding site that had been mutated, and therefore it still was able to observe the migration bands of the DNA- and protein-complex. The specific DNA-protein complex and DNA-protein-antibody complex bands are indicated by arrows.
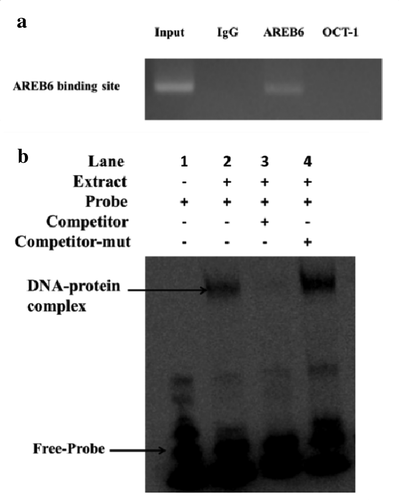
EMSA was used to further check the binding of AREB6 to PPP1R11 promoter in vitro, a strong gel-shift signal was detected when the AREB6-binding site probe was added in the PK cells nuclear extracts in lane 2. However, there was almost no gel-shift signal for the probes of the second or third AREB6-binding sites. The shift band signal generated using the probe for the AREB6-binding site was decreased when excess unlabeled competitor probe was added in lane 3, but was not affected by the same amount of mutant competitor probe in lane 4 ()).
MiR-34a directly targets PPP1R11
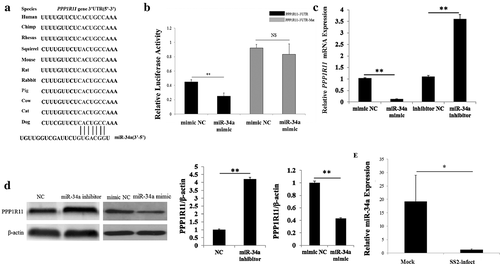
Our prediction results showed that miR-34a may be a candidate miRNA to regulate PPP1R11. The miR-34a-binding seed sequences in PPP1R11 3′-UTR were also highly conserved in mammals ()). A luciferase reporter system was used to determine the miR-34a binding site of PPP1R11 3′-UTR ()). After transfection of the miRNA-34a mimic and pmirGLO-PPP1R11-3′-UTR to PK cells, the fluorescence activities were significantly decreased, indicating that there is a target relationship between them. However, luciferase activity was unchanged when we co-transfected miR-34a mimics and pmirGLO-PPP1R11-3ˈUTR-Mut into PK cells ()). The qPCR and western blot results showed that miR-34a significantly inhibited the expression of PPP1R11 at the mRNA level and the protein level (, ). We further investigated whether the expression of miR-34a changed when SS2 infected PAM cells, the results showed that miR-34a was significantly downregulated in PAM cells infected with SS2 ()); meanwhile, the expression of PPP1R11 was upregulated ().
Figure 5. Verification of miR-34a targeting PPP1R11. (a) miR-34a binding site sequences in the PPP1R11 3′-UTR in 11 different species. (b) Luciferase activity was analyzed in PK cells at 24 h after co-transfected with pmiRGLO-PPP1R11-3′-UTR or pmiRGLO-PPP1R11-3′-UTR-Mut and miR-34a mimics or NC mimics. pmirGLO-PPP1R11-3′ UTR luciferase reporter gene with the miR-34a mimic or NC were co-transfected into PK cells; the results showed the miR-34a significantly inhibited luciferase activity. On the contrary, miR-34a had no significant effect on the dual luciferase activity when PPP1R11 3′-UTR was mutated. (c, d): Detection effect of miR-34a on PPP1R11. qPCR and Western Blot analysis were used to detect PPP1R11 mRNA levels and protein levels after PK cells were transfected with miR-34a mimics, NC mimics, miR-34a inhibitor, and inhibitor NC, respectively. Then, a densitometric analysis of by software imageJ. (e) qPCR was used to detect miR-34a levels at 24 h after PAM cells were infected with SS2 (0.5 moi). The results are expressed as the mean ± S.E.M. (three independent replicates per group). * p < 0.05; ** p < 0.01.
miR-34a and AREB6 regulate the apoptosis of PAM cells
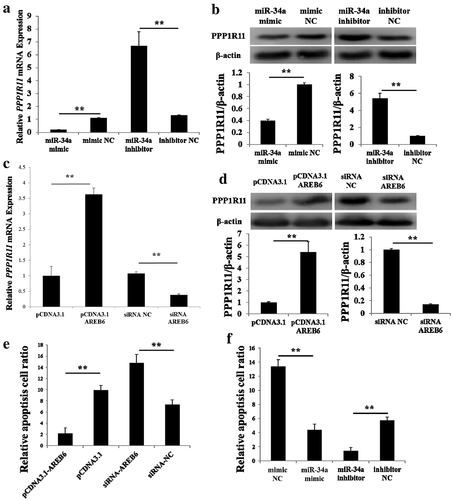
To further confirm their regulatory relationships above, we performed similar experiments in primary PAM cells. qPCR analyses revealed that PPP1R11 mRNA expression level was significantly downregulated after miR-34a mimic was transfected into PAM cells, whereas inhibition of miR-34a significantly increased PPP1R11 mRNA expression level in PAM cells ()). Western blot analyses showed that miR-34a mimic significantly inhibited PPP1R11 protein expression level, whereas inhibition of miR-34a significantly increased PPP1R11 protein expression level in PAM cells ()). Further, qPCR and Western blot data from PAM cells with overexpression and knockdown of AREB6 revealed that transcription factor AREB6 positively regulated the expression of PPP1R11 (, )). Our results demonstrated that PPP1R11 expression was significantly regulated by miR-34a and the transcription factor AREB6. Thus, miR-34a and AREB6 may affect PAM apoptosis through regulate PPP1R11. Subsequently, we investigated the effect of miR-34a and AREB6 on the apoptosis of PAM cells. As expected, miR-34a is positively correlated with apoptosis whereas AREB6 is negatively correlated (, ). These results indicated that miR-34a and AREB6 regulate PAM cells apoptosis through PPP1R11.
Figure 6. miR-34a and AREB6 regulate the apoptosis of PAM cells by PPP1R11. (a) qPCR was used to detect PPP1R11 in PAM cells at 24 h after transfected with miR-34a mimics, NC mimics, miR-34a inhibitor or NC inhibitor. (b) Western blot and a densitometric analysis by using software imageJ to detect PPP1R11 protein levels in PAM cells at 48 h after transfected with miR-34a mimics, NC mimics, miR-34a inhibitor or NC inhibitor. (c) qPCR was used to detect PPP1R11 in PAM cells at 24 h after transfected with pcDNA3.1-AREB6, pcDNA3.1, siRNA-AREB6 or siRNA NC. (d) Western blot and a densitometric analysis by using software imageJ to detect PPP1R11 protein levels in PAM cells at 48 h after transfected with pcDNA3.1-AREB6, pcDNA3.1, siRNA-AREB6 or siRNA NC. (e) The PAMs were transfected with pcDNA3.1-AREB6, pcDNA3.1, siRNA-AREB6 or siRNA NC, were harvested at 48 h and stained with anti-annexin V-propidium iodide, and then analyzed by FACS. (f) The PAMs were transfected with miR-34a mimics, NC mimics, miR-34a inhibitor or NC inhibitor, and harvested at 48 h and stained with anti-annexin V-propidium iodide, and then analyzed by FACS. The results are expressed as the mean ± S.E.M. (three independent replicates per group). * p < 0.05; ** p < 0.01.
Discussion
This study confirmed that SS2 significantly induced PPP1R11 gene expression in PAM cells, as our previous work showed its significant expressions in pigs after SS2 infection. [Citation3] Then we identified the core promoter and transcription factor of porcine gene PPP1R11 in PK cells. PK cells are one of the cell lines, little used to explore the Streptococcus-host cell interactions[Citation35]. In contrast, PAM cells are often used to study the Streptococcus-host cell interactions [Citation22,Citation24–Citation27,Citation36]. As PAM cells are isolated from the lung of sacrificed piglets, therefore we used PK cells to study the normal regulation of PPP1R11 gene, then to further study the function performances in PAM cells challenged with SS2.
Because there has no report about transcription factor of porcine PPP1R11, and here from the predicted candidates of transcription factors, we selected five candidates which have relatively higher fractional number ()) and have been commonly reported upon in the literature. Transcription factor USF regulates many different genes and numerous biological processes directly[Citation37], the C/EBP has been found to regulate IL-6 and IL-1β response through an NF-κB response element, which is a marker pathway that regulates immune responses[Citation38], PAX4 is a member of the PAX family of transcription factors, play important roles in development, pathology, and cancer[Citation39], and p54 has been shown to promote castration-resistant prostate cancer growth[Citation40]. From the above evidence, the PPP1R11expression was regulated by transcription factor AREB6 in both PK and PAM cells. Several papers have reported that transcription factors are involved in the pathological process [Citation10,Citation40,Citation41]. AREB6 was also confirmed to physically bind to the PPP3CC promoter. The elucidation of the AREB6-PPP3CC axis reveals novel insights into the molecular mechanisms underlying the progression of glioma and provides a rationale for the development of clinical anticancer intervention strategies based on targeting the AREB6-PPP3CC axis[Citation42]. Those are supported our results that AREB6 is the core promoter of PPP1R11 expression and involved in SS2 pathogenesis.
Previous papers indicated that miRNAs play a vital role in the process of bacterial infection[Citation43]. Up to date, few miRNAs for PPP1R11 and for SS2 pathogen, as well as interactions between bacterial small RNAs (sRNAs) and host miRNAs have been reported [Citation44–Citation47]. We identified and confirmed that PPP1R11 is a target gene of miR-34a. miR-34a may promote apoptosis and may play a function in aging[Citation48]. Here we found that miR-34a increases apoptosis of PAM cells and PPP1R11 suppresses apoptosis of PAM cells. Thus, miR-34a might involve regulating PAM cell apoptosis through targeting PPP1R11.
In conclusion, this study identified the core promoter of porcine gene PPP1R11 and its regulation elements, including transcription factors AREB6 and miR-34a, which significantly regulated PPP1R11. And that PPP1R11 could inhibit PAM cell apoptosis, further found that miR-34a is positively correlated with apoptosis whereas AREB6 is negatively correlated with apoptosis. Our research indicates that PPP1R11 is involved in the pathogenesis of the SS2 infection by suppressing PAM cell apoptosis and this process is regulated by AREB6 and miR-34a. In future, promising work would be interaction PPP1R11 with PP1, inflammation analysis and related aspects, such as IRF-7 or IFN-alpha.
Author Contributions
G.L., S.M. and QS.M. designed the study; J.W., M.Q. and H.W. performed lab work and statistical analysis; X.P. and G.L. drafted the manuscript; J.W., S.M. and QS.M. Reviewed the manuscript; all authors approved the paper.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Goyette-Desjardins G, Auger JP, Xu J, et al. Streptococcus suis, an important pig pathogen and emerging zoonotic agent-an update on the worldwide distribution based on serotyping and sequence typing. Emerg Microbes Infect. 2014;3:e45.
- Khan US, Atanasova KR, Krueger WS, et al. Epidemiology, geographical distribution, and economic consequences of swine zoonoses: a narrative review. Emerg Microbes Infect. 2013;2:e92.
- Gaur U, Xiong YY, Luo QP, et al. Breed-specific transcriptome response of spleen from six to eight week old piglet after infection with Streptococcus suis type 2. Mol Biol Rep. 2014;41:7865–7873.
- Moorhead GB, Trinkle-Mulcahy L, Ulke-Lemee A. Emerging roles of nuclear protein phosphatases. Nat Rev Mol Cell Biol. 2007;8:234–244.
- Ceulemans H, Bollen M. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol Rev. 2004;84:1–39.
- Han Y, Song XX, Feng HL, et al. Mutations of t-complex testis expressed gene 5 transcripts in the testis of sterile t-haplotype mutant mouse. Asian J Androl. 2008;10:219–226.
- Cheng L, Pilder S, Nairn AC, et al. PP1gamma2 and PPP1R11 are parts of a multimeric complex in developing testicular germ cells in which their steady state levels are reciprocally related. PLoS One. 2009;4:e4861.
- Wang L, Zhao J, Ren J, et al. Protein phosphatase 1 abrogates IRF7-mediated type I IFN response in antiviral immunity. Eur J Immunol. 2016;46:2409–2419.
- McKelvey AC, Lear TB, Dunn SR, et al. RING finger E3 ligase PPP1R11 regulates TLR2 signaling and innate immunity. Elife. 2016;5:e18496.
- Ikeda K, Halle JP, Stelzer G, et al. Involvement of negative cofactor NC2 in active repression by zinc finger-homeodomain transcription factor AREB6. Mol Cell Biol. 1998;18:10–18.
- Cassandri M, Smirnov A, Novelli F, et al. Zinc-finger proteins in health and disease. Cell Death Discov. 2017;3:17071.
- Zhang P. Sun Y and Ma L. ZEB1: at the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle. 2015;14:481–487.
- Genetta T, Ruezinsky D, Kadesch T. Displacement of an E-box-binding repressor by basic helix-loop-helix proteins: implications for B-cell specificity of the immunoglobulin heavy-chain enhancer. Mol Cell Biol. 1994;14:6153–6163.
- Williams TM, Moolten D, Burlein J, et al. Identification of a zinc finger protein that inhibits IL-2 gene expression. Science. 1991;254:791–1794.
- Becker JC, Brabletz T, Kirchner T, et al. Negative transcriptional regulation in anergic T cells. Proc Natl Acad Sci USA. 1995;92:375–2378.
- Lee RC, Feinbaum RL, Ambros V, et al. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854.
- Duval M, Cossart P, Lebreton A. Mammalian microRNAs and long noncoding RNAs in the host-bacterial pathogen crosstalk. Semin Cell Dev Biol. 2017;65:11–19.
- Keck J, Gupta R, Christenson LK, et al. MicroRNA mediated regulation of immunity against gram-negative bacteria. Int Rev Immunol. 2017;36(5):287–299.
- Abdalla AE, Duan X, Deng W, et al. MicroRNAs play big roles in modulating macrophages response toward mycobacteria infection. Infect Genet Evol. 2016;45:378–382.
- Self-Fordham JB, Naqvi AR, Uttamani JR, et al. MicroRNA: dynamic regulators of macrophage polarization and plasticity. Front Immunol. 2017;8:1062.
- Bonnardel J, Guilliams M. Developmental control of macrophage function. Curr Opin Immunol. 2018;50:64–74.
- Pan YY, Shi YF, Ding LQ, et al. The role played by alveolar macrophages in development of pneumonia caused by Streptococcus suis 2. Chin Vet Med. 2012;42(08):791–795. in Chinese.
- Jobin MC, Gottschalk M, Grenier D. Upregulation of prostaglandin E2 and matrix metalloproteinase 9 production by human macrophage-like cells: synergistic effect of capsular material and cell wall from Streptococcus suis. Microbiol Pathogen. 2006;40:29–34.
- Graveline R, Segura M, Radzioch D. TLR2-dependent recognition of Streptococcus suis is modulated by the presence of capsular polysaccharide which modifies macrophage responsiveness. Int Immunol. 2007;19:375–389.
- de Greeff A, Benga L, Wichgers Schreur PJ, et al. involvement of NF-kB and MAP-kinases in the transcriptional response of alveolar macrophages to Streptococcus suis. Vet Microbiol. 2010;41(1–2):59–67.
- Bin L, Luping D, Bing S, et al. Transcription analysis of the porcine alveolar macrophage response to Mycoplasma hyopneumoniae. PLoS One. 2014;9(8):e101968.
- Zhen Y, Wang F, Liang W, et al. Identification of differentially expressed non-coding rna in porcine alveolar macrophages from tongcheng and large white pigs responded to PRRSV. Sci Rep. 2018;8(1):15621.
- Ucker DS. Exploiting death: apoptotic immunity in microbial pathogenesis. Cell Death Differ. 2016;23(6):990–996.
- Lee S, Hirohama M, Noguchi M, et al. Influenza A virus infection triggers pyroptosis and apoptosis of respiratory epithelial cells through type I IFN signaling pathway in a mutually exclusive manner. J Virol. 2018;27:1885–1889.
- Rai P, Parrish M, Tay IJ, et al. Streptococcus pneumoniae secretes hydrogen peroxide leading to DNA damage and apoptosis in lung cells. Proc Natl Acad Sci USA. 2015;112(26):E3421–30.
- Zeng Q, Lu C. Fusion and apoptosis of epithelial cells induced by muramidase released protein of Streptococcus suis type 2. Wei Sheng Wu Xue Bao. 2003;43: 407–412. in Chinese.
- Lin X, Huang C, Shi J, et al. Investigation of pathogenesis of H1N1 influenza virus and swine Streptococcus suis serotype 2 co-infection in pigs by microarray analysis. PLoS ONE. 2015;10:e0124086.
- Tenenbaum T, Essmann F, Adam R, et al. Cell death, caspase activation, and HMGB1 release of porcine choroid plexus epithelial cells during Streptococcus suis infection in vitro. Brain Res. 2006;1100:1–12.
- Meng F, Wu NH, Seitz M, et al. Efficient suilysin-mediated invasion and apoptosis in porcine respiratory epithelial cells after streptococcal infection under air-liquid interface conditions. Sci Rep. 2016;6:26748.
- Jiang CG, Liu DQ, Cai XH. Adhesion kinesics of streptococcus suis type 2 to PK-15 Cells. Chin Anim Husbandry Vet Med. 2008;35(09):27~29.
- Li B, Du L, Xu X, et al. Transcription analysis on response of porcine alveolar macrophages to co-infection of the highly pathogenic porcine reproductive and respiratory syndrome virus and Mycoplasma hyopneumoniae. Virus Res. 2015;196:60–69.
- Balcerzyk A, Niemiec P, Iwanicki T, et al. Upstream stimulating factor 1 (USF-1) gene polymorphisms and the risk, symptoms, and outcome of pediatric ischemic stroke. J Stroke Cerebrovasc Dis. 2018;27(7):1885–1889.
- Srivastava RAK, Cornicelli JA, Markham B, et al. Gemcabene, a first-in-class lipid-lowering agent in late-stage development, down-regulates acute-phase C-reactive protein via C/EBP-delta-mediated transcriptional mechanism. Mol Cell Biochem. 2018;449(1–2):167–183.
- Hayashi S, Rocancourt D, Buckingham M, et al. Lack of in vivo functional compensation between Pax family groups II and III in rodents. Mol Biol Evol. 2011;28(10):2787–2798.
- Yamamoto R, Osawa T, Sasaki Y, et al. Overexpression of p54(nrb)/NONO induces differential EPHA6 splicing and contributes to castration-resistant prostate cancer growth. Oncotarget. 2018;9:10510–10524.
- Sánchez-Tilló E, Siles L, de Barrios O, et al. Expanding roles of ZEB factors in tumorigenesis and tumor progression. Am J Cancer Res. 2011;1(7):897–912.
- Wang H, Zhao S, Chen B, et al. Repression of the expression of PPP3CC by ZEB1 confers activation of NF-κB and contributes to invasion and growth in glioma cells. Jpn J Clin Oncol. 2018;48(2):175–183.
- Kim JK, Lee HM, Park KS, et al. MIR144* inhibits antimicrobial responses against Mycobacterium tuberculosis in human monocytes and macrophages by targeting the autophagy protein DRAM2. Autophagy. 2017;13:423–441.
- Dutta T, Srivastava S. Small RNA-mediated regulation in bacteria: A growing palette of diverse mechanisms. Gene. 2018;656:60–72.
- Dutcher HA, Raghavan R. Origin, evolution, and loss of bacterial small RNAs. Microbiol Spectr. 2018;6(2):RWR-0004–2017.
- Nitzan M, Rehani R, Margalit H. Integration of bacterial small RNAs in regulatory networks. Annu Rev Biophys. 2017;46:131–148.
- Zorgani MA, Quentin R, Lartigue MF. Regulatory RNAs in the less studied streptococcal species: from nomenclature to identification. Front Microbiol. 2016;7:1161.
- Deng X, Zheng H, Li D, et al. MicroRNA-34a regulates proliferation and apoptosis of gastric cancer cells by targeting silent information regulator 1. Exp Ther Med. 2018;15:3705–3714.