
ABSTRACT
Caveolin-1 (Cav-1) is an integral membrane protein that plays an important role in proliferative and terminally differentiated cells. As a structural component of Caveolae, Cav-1 interacts with signaling molecules via a caveolin scaffolding domain (CSD) regulating cell signaling. Recent reports have shown that Cav-1 is a negative regulator in tumor metastasis. Therefore, we hypothesize that Cav-1 inhibits cell migration through its CSD. HeLa cells were engineered to overexpress Cav-1 (Cav-1 OE), Cav-1 without a functional CSD (∆CSD), or enhanced green fluorescent protein (EGFP) as a control. HeLa cell migration was suppressed in Cav-1 OE cells while ∆CSD showed increased migration, which corresponded to a decrease in the tight junction protein, zonula occludens (ZO-1). The migration phenotype was confirmed in multiple cancer cell lines. Phosphorylated STAT-3 was decreased in Cav-1 OE cells compared to control and ∆CSD cells; reducing STAT-3 expression alone decreased cell migration. ∆CSD blunted HeLa proliferation by increasing the number of cells in the G2/M phase of the cell cycle. Overexpressing the CSD peptide alone suppressed HeLa cell migration and inhibited pSTAT3. These findings suggest that Cav-1 CSD may be critical in controlling the dynamic phenotype of cancer cells by facilitating the interaction of specific signal transduction pathways, regulating STAT3 and participating in a G2/M checkpoint. Modulating the CSD and targeting specific proteins may offer potential new therapies in the treatment of cancer metastasis.
KEYWORDS:
Introduction
Caveolins are structural components of Caveolae, which are small membrane invaginations enriched in glycosphingolipids and cholesterol [Citation1,Citation2]. Caveolins interact with lipids and are involved in cholesterol transport [Citation3,Citation4]. Of the three isoforms of caveolin (Cav-1, −2, and −3), Cav-1 is the dominant isoform in non-muscular tissues [Citation5]. Cav-1 is an integral membrane protein that is central to scaffolding and signal transduction in Caveolae [Citation6–Citation9]. Mutant and truncated caveolin proteins have been shown to dramatically decrease cholesterol content and dysregulate cellular growth patterns [Citation10–Citation13]. Cav-1 expression is downregulated during detachment and migration in normal cells but causes hyper-proliferation when overexpressed in tumor cells [Citation14]. Such data suggest that Cav-1 may act differently depending upon the expression of the signaling proteins in the cells and the signal cascade being triggered.
Cav-1 has differential roles in cancer biology and is suggested to act both as a tumor promotor and suppressor, a concept that is controversial [Citation15–Citation17]. Divergent effects are likely cell type-specific and depend on downstream interactions with signaling networks [Citation15,Citation16]. There is evidence to suggest that Cav-1 expression may correlate to tumor grade and stage and may undergo a switch in expression with more severe tumor pathology [Citation17]. Cav-1 has the potential to regulate diverse cancer-associated processes such as cell transformation, migration, survival, and death [Citation18]. Cav-1 has also been shown to have a critical role in tumor progression and metastasis [Citation14,Citation19,Citation20].
Caveolins have two domains, namely a membrane binding domain containing 3 C-terminal cysteines that can be palmitoylated for membrane anchoring and a caveolin scaffolding domain (CSD) which is an aromatic residue-rich motif [Citation21,Citation22]. The CSD binds directly with cholesterol and is involved in cholesterol transport and also provides a platform for the Caveolar organization of functional proteins to impact signaling [Citation9,Citation23]. The CSD is a key domain for Cav-1 interactions with other signaling proteins such as adenylyl cyclase (AC), heterotrimeric Gα and Gβγ, Src, PI3 kinase (PI3K), endothelial nitric oxide synthase (eNOS, NOS 3), protein kinase A (PKA), protein kinase C (PKC), and mitogen-activated protein kinase (MAPK, ERK) [Citation8,Citation24]. CSD provides an inhibitory binding site for protein phosphatases 1 and 2A and maintains the Akt activity for potential cell survival in pancreatic ductal adenocarcinoma (PDAC) [Citation25] and prostate cancer [Citation26]. On the other hand, the CSD’s inhibitory modulation of cell signaling proteins (i.e., Gi2α, eNOS, Src family kinases, EGF-R and PKCα), that are involved in cell proliferation suggests its behavior as a negative regulator of cell proliferation and survival [Citation27]. However, Cav-1 level of expression is critical for its dual behavior together with CSD playing an important role in regulating activation of tyrosine kinases as well as serine and threonine kinases that are involved in various signaling pathways leading to cell growth and proliferation [Citation28–Citation30]. More importantly, CSD modulates the activities of different ion channels. It interacts with calcium channel TRPC1 for regulating Ca+2 influx through store-operated cation channels affecting cellular processes such as cell proliferation and tumor invasion. These cellular mechanisms are critically regulated by extracellular Ca+2 concentrations and Ca+2 influx [Citation31–Citation34]. Cav-1 scaffolding domain also interacts with voltage-gated sodium channel type X and potassium channel (Kv1.3) regulating their surface localization for various cellular mechanisms [Citation35,Citation36]. Though many studies have focused on the importance of the CSD in cell biology, specifically with the interaction of specific binding partners, the impact of this domain on cell physiology is less well understood.
Based on these previous findings of Cav-1 importance in cell and cancer biology, we tested the hypothesis that the scaffolding domain of Cav-1 is essential in regulating the migration and proliferation of tumor cells.
Results
Stable overexpression of Cav-1 and Cav-1 lacking the scaffolding domain (Cav-1δCSD) in HeLa cells
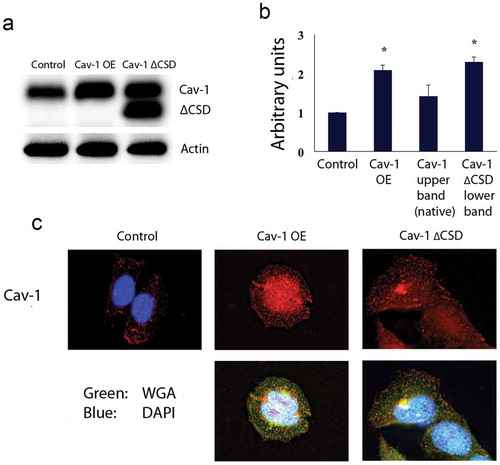
Hela cells were genetically engineered to stably overexpress Cav-1 or Cav-1 with CSD deletion (Cav-1 OE, Cav-1ΔCSD, respectively). Stable expression of enhanced green fluorescent protein (EGFP) was used as a comparative control. Cav-1 protein expression increased two-fold in Cav-1 OE and Cav-1ΔCSD (lower band detected by the antibody-upper band is native Cav-1) cells compared to the control cells (). Cav-1 localization (, upper panels) (WGA, wheat germ agglutinin, , lower panels) were confirmed by immunohistochemistry and found not to be different between the various stable cell lines.
Figure 1. Hela cells stably expressing variants of Cav-1. (A) Western blot detecting Control, Cav-1 OE and Cav-1 ∆CSD indicates increased expression of Cav-1 and Cav-1 ∆CSD. The Cav-1 antibody results in a native Cav-1 band (upper) and a Cav-1 ∆CSD band (lower) for cells expressing Cav-1 ∆CSD. Actin was used as a loading control. (B) Densitometric quantification of Cav-1 expression in stable cell lines. Cav-1 expression increased twofold in Cav-1 OE or Cav-1 ∆CSD expressing cells compared with control cells. *P< 0.05 vs. Control cells by one-way ANOVA. (C) Immunochemistry of expression of Cav-1 OE and Cav-1 ∆CSD shows normal localization. Wheat germ agglutinin, a cell membrane stain, was used to confirm normal cell morphology.
The scaffolding domain of Cav-1 plays a critical role in reducing cell migration
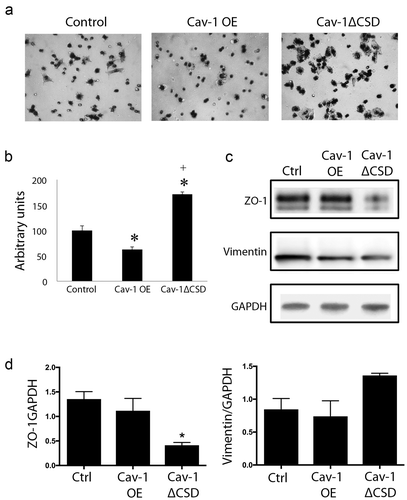
The migration assay revealed that overexpression of Cav-1 with an in intact CSD led to a suppression of cellular migration (), whereas Cav-1∆CSD significantly promoted cell migration compared to both Cav-1 OE and control cells (). Thus, the CSD of caveolin may play a critical role in preventing cell migration. Epithelial–mesenchymal transition markers were further analyzed to determine if the CSD utilizes such transition as a key feature of developing a migratory phenotype. Zonula occludens (ZO-1) is a tight junction protein critical to cell-cell communication and adhesion, whereas, vimentin is an intermediate filament that serves as a marker of mesenchymal cells associated with a migratory phenotype [Citation37]. We observed a significant decrease in ZO-1 and a trend towards an increase in vimentin in Cav-1∆CSD cells compared to control and Cav-1 OE cell ().
Figure 2. Cav-1 overexpression decreased cell migration via CSD. (A) Representative images of hematoxylin stained migrated cells. (B) Quantification of migrated cells in control, Cav-1 OE and Cav-1 ∆CSD cells. The migration was increased in Cav-1 ∆CSD cells while Cav-1 overexpression inhibited cell migration compared with control cells. *, + P< 0.05 vs. Control, Cav-1 OE cells, respectively (one-way ANOVA). (C and D) Epithelial–mesenchymal transition markers were analyzed to determine if the CSD alters this transition to induce migratory phenotype. Zonula occludens (ZO-1) and vimentin expression were assessed. Cav-1∆CSD significantly decreased ZO-1 and showed a trend towards an increase in vimentin expression in cells compared to control and Cav-1 OE. *, + P< 0.05 vs. Control (one-way ANOVA).
Cav- 1 regulates molecular signaling linked to migration via its scaffolding domain
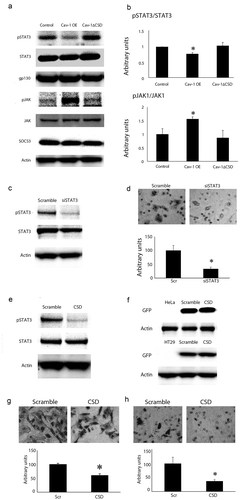
We next explored biochemical changes that may account for this hypermigratory phenotype of Cav-1∆CSD. Phosphorylated STAT-3 was reduced in Cav-1 OE compared to the control and Cav-1∆CSD cells; however, activated JAK was higher in Cav-1 overexpressing cells (). These significant changes in Cav-1 OE cells were absent in Cav-1∆CSD cells suggesting that an intact CSD is critical to expression or activation of specific molecular signaling partners. No significant changes were observed in gp130 or SOCS3 expression among the three cell types. To specifically probe the role of STAT3 in HeLa cell migration, we utilized STAT3 targeted siRNA in control cells. STAT3 specific siRNA treatment reduced the amount of activated (phospho) STAT3 compared to scrambled control (). Migration assays showed that downregulation of pSTAT3 caused a two-fold decrease in the migration of the HeLa cells (), an effect similar to Cav-1 OE cells that show decreased migration and phospho STAT3 expression. To further confirm the role of the CSD in regulating cancer cell migration, we overexpressed only the CSD (scrambled as a control) with a GFP tag. Overexpression of CSD peptide alone dramatically suppressed of p STAT3 expression in HeLa cells (). Expression of CSD or scrambled peptide was confirmed in HeLa and HT29 colon cancer cells (which normally do not express any caveolin) using GFP specific antibody (). In both of these cells, overexpression of CSD alone resulted in a significant suppression of cell migration (, HeLa and 3H, HT29). These data show the importance of CSD regulated STAT3 phosphorylation in the migration of multiple cancer cell types.
Figure 3. STAT3 phosphorylation was decreased in Cav-1 overexpression cells and overexpression of CSD alone decreases STAT3 phosphorylation and cell migration. (A) Representative western blot analyses of STAT-3, phospho-STAT3, gp130, JAK and phosphor-JAK, and SOCS3 in control, Cav-1 OE and Cav-1 ∆CSD cells. Actin was used as a loading control. (B) Densitometric quantification of phosphor/total STAT-3 and JAK (other proteins were unchanged) in control, Cav-1 OE and Cav-1 ∆CSD cells. Phospho-STAT3 was decreased whereas phosphor-JAK was increased in Cav-1 OE cells compared with control and Cav-1 ∆CSD cells. *P< 0.05 vs. LacZ. (one-way ANOVA). (C) Representative western blot analyses of phosphor/total STAT3 following STAT3 siRNA treatment (scrambled vector was used as control). (D) Representative images of hematoxylin stained migrated cells (top) and quantification of migrated cells (bottom) in scrambled control vs STAT3 siRNA treated cells. Loss of STAT3 decreased migration. *, + P< 0.05 vs. Scr (one-way ANOVA). (E) pEGFP tagged SCD or scrambled vector were transiently expressed in HeLa or HT-29 colorectal cancer cell line. GFP was observed in scrambled and CSD vector-treated cells. (F) Representative western blot analyses of CSD and scrambled vector with EGFP in HeLa cells for phospho/total STAT3. CSD treatment decreased pSTAT3. CSD expression led to decreased migration in HeLa (G) and HT29 (H) cells.
Cav-1 regulates cell proliferation and cycle via the scaffolding domain
To investigate if the change in migration patterns of the HeLa cells were not due to the effects of proliferation, we measured thymidine [3H] incorporation. Remarkably, we saw that there was no significant difference in the Cav-1 OE cells; however, serum-induced proliferation was blunted in Cav-1∆CSD cells (). Since Cav-1 can regulate the cell cycle through its scaffolding domain, we next investigated the phase of the cell cycle that is regulated by Cav-1. Cells were synchronized by serum starvation, treated with propidium iodide (PI), and subsequently sorted according to their DNA content. Our results showed that Cav-1∆CSD resulted in a higher number of cells in G2 phase compared to both control and Cav-1 OE cells (). To further understand the G2/M arrest, we also analyzed the active state of Cdc2, which is involved in the G2/M transition. Cdc2 is a universally conserved protein that mediates the transition from G2 to M phase. Cav-1 ∆CSD cells had high Cdc2 inhibitory phosphorylation in cells compared to Cav-1 OE or control cells (). No significant changes were observed in cyclin B1.
Figure 4. CSD contribute to cell proliferation and cell cycle arrest. (A) Proliferation assessed by [3H]-thymidine incorporation, was reduced in Cav1 ∆CSD cells compared with control and Cav-1 OE cells. *P< 0.05 vs. LacZ. (one-way ANOVA). (B) Representative image of FACS. The DNA content of propidium iodide-stained nuclei were analyzed by FACSCalibur flow cytometry, as described in Materials and Methods. (C) The percentage of cells in phase G1, S and G2/M. *P< 0.05 vs. Control cells. (one-way ANOVA). (D) Representative western blot analyses of Cdc2 and phospho-Cdc2 in Control, Cav-1 OE and Cav-1 ∆CSD cells.
![Figure 4. CSD contribute to cell proliferation and cell cycle arrest. (A) Proliferation assessed by [3H]-thymidine incorporation, was reduced in Cav1 ∆CSD cells compared with control and Cav-1 OE cells. *P< 0.05 vs. LacZ. (one-way ANOVA). (B) Representative image of FACS. The DNA content of propidium iodide-stained nuclei were analyzed by FACSCalibur flow cytometry, as described in Materials and Methods. (C) The percentage of cells in phase G1, S and G2/M. *P< 0.05 vs. Control cells. (one-way ANOVA). (D) Representative western blot analyses of Cdc2 and phospho-Cdc2 in Control, Cav-1 OE and Cav-1 ∆CSD cells.](/cms/asset/c827e505-d44f-43b2-ae63-1a27ee640e16/kccy_a_1618118_f0004_b.gif)
Transient overexpression of Cav-1 and Cav-1δCSD in multiple cancer cell lines regulates cell migration
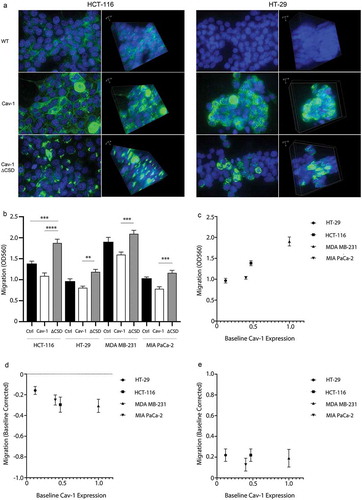
To confirm the observed role of caveolin on cell migration, we created lentiviral vectors of the various constructs to study transient expression in several cancer cell lines that have variable expression of Cav-1, such as MDA-MB-231 breast adenocarcinoma cells, MIA PaCa-2 pancreatic adenocarcinoma cells, HCT-116 colorectal carcinoma cells, and HT-29 colorectal adenocarcinoma cells. Fluorescence microscopy of Cav-1 in HCT-116 and HT-29 colon cancer cells revealed punctate expression of Cav-1 in these both cell types expressing the full-length form whereas the expression of Cav-1∆CSD was more focal (). The migration assays revealed that transient overexpression of Cav-1 consistently led to a significant decrease in cell migration whereas overexpression of Cav-1∆CSD increased migration (). When we correlated migration as a function of basal expression of Cav-1 in these various cells we observed a linear relationship of increased basal migration with increased endogenous Cav-1 expression in the various cell types (). However, when exogenous Cav-1 or Cav-1∆CSD added the correlation to baseline Cav-1 proteins expression was no longer observed but rather dependent on the exogenous expression ofcaveolin where wild-type Cav-1 () suppressed and Cav-1∆CSD () increased migration when normalized to the baseline migration response in the respective cell line. Such data confirm the importance of the CSD in the migration phonotype across various cell types irrespective of endogenous Cav-1 expression.
Figure 5. Cell migration in multiple cancer cells is impacted by exogenous Cav-1 and Cav-1∆CSD. (A) HCT-116 and HT-29 cells were grown on laminin-coated 35 mm glass bottom dishes, transduced with either Cav1 or Cav1∆CSD, and stained with both Cav1 (green) and DAPI (blue). Left panels at 2-D images whereas the right panels represent 3-D Z-stacked images of multiple sections. (B) HCT-116, HT-29 cells, MDA-MB-231 cells, and MIA PaCa-2 cells (~150,000/well, n = 5) were transduced with either Cav1 or Cav1∆CSD and migration was assessed using an 8uM Boyden chamber insert, followed by cell staining and calorimetric quantification (OD 560 nm). Scatter plots of cell migration (y-axis) as a function of baseline caveolin-1 protein expression (x-axis) under baseline conditions (C) as well as baseline-normalized conditions following exogenous Cav-1 (D) and Cav-1∆CSD (E) expression.
Discussion
Caveolins are dynamic proteins in many cells critical for stress adaptation and cell survival [Citation38]. Since caveolins are crucial for membrane structure and function, these proteins are involved in various pathologies [Citation39]. The role of Cav-1 in cancer is multifaceted and likely has dual functionality, as Cav-1 has been proposed to be a tumor promotor or suppressor depending upon the cancer cell type [Citation40]. Our results show the role of Cav-1 in cell migration and proliferation is dependent on the CSD and its role in the activation and expression of signaling proteins, such as JAK/STAT3: the CSD is key in determining the proliferative vs migratory phenotype of a variety of cancer cells and this can be enhanced in cancer cells that do not normally expression caveolin.
The caveolin scaffolding domain regulates an array of signaling pathways by providing a platform for localizing various receptors and signaling mediators and effectors important to cell signaling [Citation16]. Our studies show that loss of Cav-1 CSD enhances cell migration possibly through the regulation of STAT-3. Recent studies have identified that Cav-1 regulates and promotes cell growth by indirectly affecting STAT3 expression, where STAT3 acts as a positive regulator of cell progression and survival [Citation41]. STAT3, a proto-oncogene, is activated by the JAK/STAT pathway and has been shown to be constitutively activated in many cancer cell types including endometrial and cervical cancers [Citation42]. In in vitro studies, Cav-1 knockdown in MIA-PaCa-2 cell lines inhibited tumor progression by negatively regulating the JAK/STAT-3 pathway [Citation43]. In breast cancers, Cav-1 overexpression led to tumor invasion and metastasis by inhibiting STAT3 signaling [Citation44]. However, STAT3 also has the potential to be a tumor suppressor [Citation45] suggesting that its interaction with signaling proteins may impact its overall function. Suppression of cytokine activity by inhibiting JAK/STAT signaling through CSD was also determined in previous studies [Citation46]. It has been speculated that the CSD of Cav-1 can act as a pseudosubstrate for STAT3 and has the potential to negatively regulate the activation of STAT3 [Citation47].
In our studies, up-regulation of the CSD region alone in cells led to decreased STAT3 phosphorylation suggesting a direct regulation of STAT3 by caveolin dependent on the CSD. However, activated/upregulated STAT3 is of major concern as a heterogenic modulator of cell migration and invasion in various cancers [Citation48]. Activated focal adhesion kinase (FAK) mediated activation of STAT3 has been shown to derive anchorage-independent growth and invasion in ovarian carcinoma cells [Citation49]. Transient expression of Cav-1∆CSD in HCT116 and HT29 cells led to focal localization of the modified Cav-1 possibly suggesting an interaction of this CSD loss with the leading edge of cells contributing to the migratory phenotype. Previous experiments have shown that activated STAT3 bound directly to the Cav-1 promoter can inhibit its transcription [Citation44]. Conversely, Cav-1 was seen to negatively regulate the activation of STAT3 and invasion of brain-metastatic cancer cells [Citation50].
Caveolin-1 with is dual effects in cancer has a critical role in cell migration, metastasis, and invasion [Citation51]. Cav-1 was previously reported to induce high motility rates in metastatic cells [Citation52]. On the other hand, Cav-1 gene disruption can also induce metastasis and invasiveness. Induction of CSD into highly metastatic mammary carcinoma cell lines inhibits invasion via reduced secretion of MMP-2 and MMP-9 [Citation5]. In recent experiments, the introduction of Cav-1 ∆CSD in normal cells, showed retarded Ca+2 signaling pathways resulting in a number of pathologies [Citation34]. Deletion mutants and point mutations in CSD resulted in muscular dystrophies, tissue remodeling abnormalities, cancer progression including invasiveness and cancer cell migration whereas increased expression of full-length caveolin and expression of CSD in different cancer cell lines led to inhibition of cell migration [Citation53–Citation55]. In addition, it was observed in previous studies that different reactive oxygen species act as positive tumor regulators and have different effects on Cav-1 mediated cell migration and invasion suggesting another aspect of the dual role of caveolin [Citation56].
FAK is also necessary for cell invasion and migration as it plays an important role in cell surface signaling interactions via multiple signaling pathways [Citation57]. FAK might be activated by cellular Src forming dual kinase complex. The FAK/Src complex is associated with tumorigenesis, epithelial to mesenchymal transition and in orchestrating anchorage-independent growth, cell migration and invasion [Citation58]. However, although we saw a decrease in Z0-1 with deletion of the CSD, we did not observe any significant changes in the activation of Src or FAK (data not shown). Such signaling aspects need further exploration and elucidation.
Along with cell migration, another unique characteristic of cancer cells is their proliferative phenotype. Cav-1 has an important role in interacting with the cell cycle signaling pathways leading to mitotic cell division [Citation59,Citation60]. Overexpression of Cav-1 has a negative regulatory effect on cell cycle at G0/G1 phase [Citation61–Citation63]. We observed that HeLa cells expressing the CSD deletion had a larger number of cells in the G2/M phase compared to the WT and Cav-1 OE. We speculate that Cav-1 scaffolding domain may play a role in arresting the cells at the G2/M checkpoint and does not allow them to progress to mitosis. Nuclear translocation of Cav-1 during cell proliferation via VEGF suggests a role for Cav-1 in regulating transcription of cell cycle genes [Citation64]. Cav-1 was determined experimentally to be localized in the nucleus in ovarian carcinoma and was involved in transcription regulation of cyclin D1 by binding to its promoter site, hence affecting cell proliferation [Citation65]. It is possible that the Cav-1 scaffolding domain might be involved in activation/phosphorylation of JNK. Cav-1 is known to regulate JNK mediated apoptosis in lung adenocarcinoma [Citation66], and hence inhibit G2/M transition and induce apoptosis and autophagy [Citation67]. G2/M transition is modulated by Cdc2 [Citation68,Citation69] and cyclin B1 [Citation70–Citation72]. We found that the cells expressing Cav-1∆CSD had elevated phospho-Cdc2, supporting a role for the Cav-1 scaffolding domain regulation of cell cycle kinetics through Cdc2.
In summary, caveolin is a dynamic signaling protein and emerging data suggests that specific domains of caveolin may contribute to many of these dynamic features involved in regulating cell physiology and morphology. The Cav-1 scaffolding domain may be one such domain that plays a critical role in the cancer cell cycle, migration, and proliferation; modulating the interaction of the CSD with specific proteins may offer potential new therapies in the treatment of cancer cell phenotypes and a means to target cancer biology as a therapeutic.
Methods
Antibodies, cell lines, chemicals/medias
Polyclonal antibody to Cav-1 (sc-894), polyclonal antibody to actin (sc-1616) and gp130 (sc-656) were purchased from Santa Cruz Biotechnology; additional antibodies were purchased from Cell Signaling Technology (GAPDH, 2118; pSTAT3, 9131; STAT3, 9132; pJAK, 3331; JAK, 3332; SOCS3,2923; pCdc2, 9111; Cdc2, 9116; cyclinB1, 4138) or AbCam (GFP, ab290; ZO-1, ab96587; vimentin, ab45939). Lipofectamine 2000 (11,668,027), Opti-MEM Reduced Serum Medium (31,985,062), and 1% penicillin/streptomycin (15,240,096) were from ThermoFisher Scientific. G-418 solution was purchased from Roche Diagnosis (4,727,878,001). Human cervical epithelial cancer cell line (HeLa) were purchased from ATCC (CCL-2).
Plasmid and vector constructs, generation of stable cell lines, siRNA, and cell culture
Mouse Cav-1 (456bp) sequence was cloned into an expression vector pEGFP-N1 (632,469) from Clontech Laboratories with EGFP deleted. The original vector containing EGFP was used as a control. Lentivirus expressing these constructs were also generated. For the CSD only and scrambled constructs, human CSD was cloned into the pEGFP-N1 vector (scrambled sequence for control), these vectors were used for transient transfections. For the generation of stable cells, HeLa cells were seeded (5 × 105/well) on 24-well plates without antibiotics and incubated at 37°C overnight. After 24 h of incubation 0.8 μg of pEGFP-N1, pEGFP-N1-Cav-1 (EGFP deleted) or pEGFP-N1-Cav-1∆CSD (EGFP deleted) plasmid was transfected into Hela cells using 2 μl of Lipofectamine 2000 in Opti-MEM Reduced Serum Medium following the manufacturer’s instructions. Transfected cells were selected with G418 at 800 μg/ml for 4 weeks, and the expression of Cav-1 protein in selected cell clones was determined by Western blotting. HeLa cell were cultured in Dulbecco’s Modified Eagle Medium (DMEM, 10–013-CV) containing 10% fetal bovine serum (35–010-CV) from Corning in a 95% air, 5% CO2 humidified atmosphere at 37°C. Stably transfected HeLa cells were supplemented with 800μg/ml G418. siRNA for STAT3 (sc-29,493) and control (sc-44,230) was purchased from Santa Cruz Biotechnology.
Immunoblot analysis
Immunoblotting was performed on transfected multiple cancer cell lysates following the protocol described [Citation73]. Cell extracts were obtained by lysing cells in lysis buffer (50 mM Tris·HCl, 150 mM NaCl; pH 7.4) supplemented with protease and phosphatase inhibitor cocktail (5872, Cell Signaling). After 30 min incubation on ice, the cells were homogenized by a 23-gauge needle, and the lysates were cleared of debris and unbroken cells by centrifugation (800 g, 5 min at 4°C). Protein concentrations were determined by the Bio-Rad protein assay (5,000,001, Bio-Rad Laboratories). Equal amounts of cell lysates (20 ug) were loaded. The proteins were separated by 10% SDS-PAGE (NW00102BOX, ThermoFisher Scientific) and transferred to a polyvinylidene difluoride membrane by electro-elution. Membranes were blocked in tris-buffered saline and 1% Tween containing 3% bovine serum albumin solution and incubated with primary antibody (1:2000) overnight at 4°C. Bound primary antibodies were visualized using secondary antibodies (1:5000) conjugated with horseradish peroxidase (rabbit, sc-2030; mouse, sc-2031 from Santa Cruz Biotechnology) and ECL reagent (RPN2236, Amersham Biosciences).
Immunocytochemistry and microscopy
Cells were fixed with 4% paraformaldehyde for 15 min at room temperature (RT) and washed three times with PBS. Cells were permeabilized with 0.1% Triton X-100 for 10 min at RT followed by washing three times with PBS. Cells were then blocked with 3% bovine serum albumin in PBS for 30 min at RT. The cells were incubated with respective primary antibodies in 3% bovine serum albumin in PBS overnight at 4°C. The following day, cells were washed three times in PBS and incubated with respective secondary antibodies in 3% bovine serum albumin in PBS for 1 hr at RT. After incubation, cells were washed three times and mounted on slides using VECTASHIELD HardSet Antifade Mounting Medium (H-1200, Vector Laboratories). For detection, secondary antibodies Alexa Fluor 594 (R37119) or wheat germ agglutinin 488 conjugate (W11261) from ThermoFisher Scientific were used. Cells were imaged using the KEYENCE BZ-X710 fluorescent microscope equipped with a 40x lens (CFI Plan Apo; Nikon).
For HCT-116 and HT-29 studies, cells were grown on MatTek 35 mm dish with 14 mm glass bottom microwell coated with laminin at a concentration of 100 ug/mL. Cells were transduced with Lenti-Cav1 or Lenti-Cav1∆CSD for 24 hr. Virus was then removed and cells were incubated in serum-free McCoy’s 5A medium for 18 h at 37°C in a CO2 incubator (5% CO2). Cell culture media was then removed and replaced with 1 mL of 5% (w/v) formaldehyde solution in PBS and incubated for 15 min at room temperature. Fixative was then removed and washed 3X with PBS. Cells were then permeabilized with 1 mL of 0.5% TX-100 for 15 min at room temperature, followed by washing 3X with PBS. Cells were then blocked with 2 mL of 3% BSA for 1 h at room temperature and subsequently incubated in 1:500 antibody (Cav1 – Cell Signaling – Catalog #3267S) in 1% BSA in PBS in a humidified chamber overnight at 4°C. The solution was then decanted and the cells were washed 3X in PBS, 10 min each wash. Cells were then incubated with the secondary antibody (Goat anti Rabbit – Alexa Fluor 488) 1:1500 in 1% BSA for 1 h at room temperature in the dark. Secondary antibody solution was then decanted and washed 3X with PBS for 10 min each in the dark. Cells were then incubated with DAPI (1 ug/mL in PBS) for 5 min at room temperature in the dark and washed 3X with PBS for 10 min in the dark. Cells were then imaged using a 100X lens with oil on a Keyence BZ-X700.
Cell migration assay
The migration chambers were put in 24-well cell culture dish. In the upper chamber, 300 µl DMEM including cells (1.0x105/well) and 0.2% FBS was added. In the lower chamber, 750 µl DMEM with 10% FBS was added. Cells were then incubated at 37°C for 24 h. After 24-h HeLa cells were fixed by 2.5% glutaraldehyde at RT for 15 min. The cells were permeabilized by 0.5% triton X-100 at RT for 20 min. For staining, hematoxylin was added at RT for 15 min. After staining, non-migrated cells were scraped off from the migrated cells with cotton swabs. Then migrated cells were manually counted under a light microscope.
For studies on involving HCT-116, HT-29, MDA-MB-231, and MIA PaCa-2 cells, cells were transduced with lenti-Cav1 or lenti-Cav1∆CSD for 24 hr. Virus was then removed and cells were incubated in serum-free medium for 18 h at 37°C in a CO2 incubator (5% CO2). To assess migration in HCT-116, HT-29, MDA-MB-231, and MIA PaCa-2 cells, a QCM Chemotaxis Cell Migration Assay, 24-well (8 µm), colorimetric (Millipore Sigma), was used. Plates and reagents were brought to room temperature (23–25°C) prior to initiating assay. Cells were lifted with a 0.02% EDTA solution prepared in DPBS (w/o calcium and magnesium). A cell suspension was then prepared containing 0.5 × 106 cells/mL in cell culture medium + 0.2% FBS (chemoattractant-free media). Three hundred microliters of the prepared cell suspension was then placed inside each insert. Five hundred-microliter cell culture medium + 10% FBS was also placed into the lower chamber. The plate was then covered and incubated for 24 h at 37°C in an incubator (5% CO2). The cells and media from were then removed by pipetting out the remaining cell suspension and placing the migration insert into a clean well containing 400 µL of room temperature Cell Stain for 20 min. The insert was then rinsed by dipping into a beaker of water several times and then non-migratory cells layer from the interior of the insert were gently removed using two cotton-tipped swabs per insert. Insert was then air dried and transferred to a clean well containing 200 µL of extraction buffer for 15 min at room temperature, with gentle tilting of the insert back and forth. One hundred microliters of the dye mixture was then transferred to a 96-well microtiter and the optical density was measured at 560 nm.
Analysis of cellular DNA content by flow cytometry
Cells were plated in 6-well plates. After 48 h, cells were harvested by trypsinization and washed twice with ice-cold PBS, and then fixed with ice-cold 70% ethanol in DPBS (Dulbecco’s phosphate-buffered saline) at 4°C. Cells were then centrifuged and washed with staining buffer. After washing, the pellets were treated with 100 uL RNase A (1 mg/mL) for 30 min at 37°C. After incubation, 900 μL of staining buffer and 20 μL of propidium iodide (1 mg/mL) were added to each sample and incubated in the dark for 30 min. The samples were then analyzed with BD FACScan™ flow cytometry (BD Biosciences) using CellQuest Software (BD Biosciences).
[3h]thymidine incorporation
[3H]Thymidine incorporation was used to assess DNA synthesis as a measure of proliferation. Cells (3 x 105 per well) were seeded into a 6-well culture plate and serum-starved overnight. [3H]thymidine 1μCi (1 Ci = 37 GBq)/ml was added with or without 10% FBS and the cells were incubated for 24 h at 37°C. The cells were washed with cold PBS and 7.5% TCA and then dissolved in 0.5 M NaOH before liquid scintillation counting.
Statistical analysis
Statistical analyses were performed by one-way ANOVA followed by a Bonferroni’s post hoc test using GraphPad Prism 4 software (GraphPad Software, Inc.). All data are expressed as mean ± SE. statistical significance was defined as P < 0.05.
Acknowledgments
Funding for this project was provided by the National Institutes of Health (HL091071, AG052722) and the Veterans Administration (BX001963) to HHP.
Disclosure statement
HHP has equity as a founder in CavoGene LifeSciences Holdings, LLC.
Additional information
Funding
References
- Kathuria H, Cao YX, Ramirez MI, et al. Transcription of the caveolin-1 gene is differentially regulated in lung type I epithelial and endothelial cell lines. A role for ETS proteins in epithelial cell expression. J Biol Chem. 2004;279:30028–30036.
- Eberspacher E, Werner C, Engelhard K, et al. Long-term effects of hypothermia on neuronal cell death and the concentration of apoptotic proteins after incomplete cerebral ischemia and reperfusion in rats. Acta Anaesthesiol Scand. 2005;49:477–487.
- Smart EJ, Ying Y, Donzell WC, et al. A role for caveolin in transport of cholesterol from endoplasmic reticulum to plasma membrane. J Biol Chem. 1996;271:29427–29435.
- Busija AR, Patel HH, Insel PA. Caveolins and Cavins in the trafficking, maturation, and degradation of Caveolae: implications for cell physiology. Am J Physiol Cell Physiol. 2017;312:C459–C477.
- Williams TM, Lisanti MP. The caveolin genes: from cell biology to medicine. Ann Med. 2004;36:584–595.
- Patel HH, Insel PA. Lipid rafts and Caveolae and their role in compartmentation of redox signaling. Antioxid Redox Signal. 2009;11:1357–1372.
- Patel HH, Murray F, Insel PA. G-protein-coupled receptor-signaling components in membrane raft and Caveolae microdomains. Handb Exp Pharmacol. 2008;186:167–184.
- Patel HH, Murray F, Insel PA. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu Rev Pharmacol Toxicol. 2008;48:359–391.
- Bosch M, Marí M, Herms A, et al. Caveolin-1 deficiency causes cholesterol-dependent mitochondrial dysfunction and apoptotic susceptibility. Curr Biol. 2011;21:681-686.
- Pol A, Martin S, Fernandez MA, et al. Dynamic and regulated association of caveolin with lipid bodies: modulation of lipid body motility and function by a dominant negative mutant. Mol Biology Cell. 2004;15:99–110.
- Kuo CY, Lin YC, Yang JJ, et al. Interaction abolishment between mutant caveolin-1(Delta62-100) and ABCA1 reduces HDL-mediated cellular cholesterol efflux. Biochem Biophys Res Comm. 2011;414:337–343.
- Lacroix-Triki M, Geyer FC, Reis-Filho JS. Caveolin-1 P132L mutation in human cancers: 1 Caveat to be voiced. J Mol Diag: JMD. 2010;12:562–565.
- Schrauwen I, Szelinger S, Siniard AL, et al. A Frame-shift mutation in CAV1 is associated with a severe neonatal progeroid and lipodystrophy syndrome. PLoS One. 2015;10:e0131797.
- Chanvorachote P, Chunhacha P. Caveolin-1 regulates endothelial adhesion of lung cancer cells via reactive oxygen species-dependent mechanism. PLoS One. 2013;8:e57466.
- Bender F, Montoya M, Monardes V, et al. Caveolae and Caveolae-like membrane domains in cellular signaling and disease: identification of downstream targets for the tumor suppressor protein caveolin-1. Biol Res. 2002;35:151–167.
- Burgermeister E, Liscovitch M, Röcken C, et al. Caveats of caveolin-1 in cancer progression. Cancer Lett. 2008;268:187–201.
- Shatz M, Liscovitch M. Caveolin-1: a tumor-promoting role in human cancer. Int J Radiat Biol. 2008;84:177–189.
- Senetta R, Stella G, Pozzi E, et al. Caveolin-1 as a promoter of tumour spreading: when, how, where and why. J Cell Mol Med. 2013;17:325–336.
- Tse EY, Ko FC, Tung EK, et al. Caveolin-1 overexpression is associated with hepatocellular carcinoma tumourigenesis and metastasis. J Pathol. 2012;226:645–653.
- Patani N, Martin LA, Reis-Filho JS, et al. The role of caveolin-1 in human breast cancer. Breast Cancer Res Treat. 2012;131:1–15.
- Byrne DP, Dart C, Rigden DJ. Evaluating caveolin interactions: do proteins interact with the caveolin scaffolding domain through a widespread aromatic residue-rich motif? PLoS One. 2012;7:e44879.
- Hoop CL, Sivanandam VN, Kodali R, et al. Structural characterization of the caveolin scaffolding domain in association with cholesterol-rich membranes. Biochemistry. 2012;51:90–99.
- Tagawa A, Mezzacasa A, Hayer A, et al. Assembly and trafficking of Caveolar domains in the cell: Caveolae as stable, cargo-triggered, vesicular transporters. J Cell Biol. 2005;170:769–779.
- Bucci M, Gratton JP, Rudic RD, et al. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med. 2000;6:1362–1367.
- Hehlgans S, Eke I, Storch K, et al. Caveolin-1 mediated radioresistance of 3D grown pancreatic cancer cells. Radiother Oncol. 2009;92:362–370.
- Li L, Ren CH, Tahir SA, et al. Caveolin-1 maintains activated Akt in prostate cancer cells through scaffolding domain binding site interactions with and inhibition of serine/threonine protein phosphatases PP1 and PP2A. Mol Cell Biol. 2003;23:9389–9404.
- Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431–467.
- Razani B, Lisanti MP. Two distinct caveolin-1 domains mediate the functional interaction of caveolin-1 with protein kinase A. Am J Physiol Cell Physiol. 2001;281:C1241–50.
- Schmitz M, Kloppner S, Klopfleisch S, et al. Mutual effects of caveolin and nerve growth factor signaling in pig oligodendrocytes. J Neurosci Res. 2010;88:572–588.
- Schmitz M, Zerr I, Althaus HH. Effect of Cavtratin, a caveolin-1 scaffolding domain peptide, on oligodendroglial signaling cascades. Cell Molecular Neurobiol. 2011;31:991–997.
- Capiod T. Extracellular calcium has multiple targets to control cell proliferation. Adv Exp Med Biol. 2016;898:133–156.
- Kwiatek AM, Minshall RD, Cool DR, et al. Caveolin-1 regulates store-operated Ca2+ influx by binding of its scaffolding domain to transient receptor potential channel-1 in endothelial cells. Mol Pharmacol. 2006;70:1174–1183.
- Pani B, Ong HL, Brazer SC, et al. Activation of TRPC1 by STIM1 in ER-PM microdomains involves release of the channel from its scaffold caveolin-1. Proc Nat Acad Sci USA. 2009;106:20087–20092.
- Sundivakkam PC, Kwiatek AM, Sharma TT, et al. Caveolin-1 scaffold domain interacts with TRPC1 and IP3R3 to regulate Ca2+ store release-induced Ca2+ entry in endothelial cells. Am J Physiol Cell Physiol. 2009;296:C403–13.
- Perez-Verdaguer M, Capera J, Martinez-Marmol R, et al. Caveolin interaction governs Kv1.3 lipid raft targeting. Sci Rep. 2016;6:22453.
- Ohman E, Nilsson A, Madeira A, et al. Use of surface plasmon resonance coupled with mass spectrometry reveals an interaction between the voltage-gated sodium channel type X alpha-subunit and caveolin-1. J Prot Res. 2008;7:5333–5338.
- Scanlon CS, Van Tubergen EA, Inglehart RC, et al. Biomarkers of epithelial-mesenchymal transition in squamous cell carcinoma. J Dent Res. 2013;92:114–121.
- Schilling JM, Head BP, Patel HH. Caveolins as Regulators of Stress Adaptation. Mol Pharmacol. 2018;93:277–285.
- Lamaze C, Tardif N, Dewulf M, et al. The Caveolae dress code: structure and signaling. Curr Opin Cell Biol. 2017;47:117–125.
- Chen D, Che G. Value of caveolin-1 in cancer progression and prognosis: emphasis on cancer-associated fibroblasts, human cancer cells and mechanism of caveolin-1 expression (Review). Oncol Lett. 2014;8:1409–1421.
- Han F, Zhu HG. Caveolin-1 regulating the invasion and expression of matrix metalloproteinase (MMPs) in pancreatic carcinoma cells. J Surg Res. 2010;159:443–450.
- Chen CL, Hsieh FC, Lieblein JC, et al. Stat3 activation in human endometrial and cervical cancers. Br J Cancer. 2007;96:591–599.
- Chatterjee M, Ben-Josef E, Thomas DG, et al. Caveolin-1 is associated with tumor progression and confers a multi-modality resistance phenotype in pancreatic cancer. Sci Rep. 2015;5:10867.
- Chiu WT, Lee HT, Huang FJ, et al. Caveolin-1 upregulation mediates suppression of primary breast tumor growth and brain metastases by stat3 inhibition. Cancer Res. 2011;71:4932–4943.
- Zhang HF, Lai R. STAT3 in Cancer-Friend or Foe? Cancers (Basel). 2014;6:1408–1440.
- Jasmin JF, Mercier I, Sotgia F, et al. SOCS proteins and caveolin-1 as negative regulators of endocrine signaling. Trends Endocrinol Metabol. 2006;17:150–158.
- Geletu M, Mohan R, Arulanandam R, et al. Reciprocal regulation of the Cadherin-11/Stat3 axis by caveolin-1 in mouse fibroblasts and lung carcinoma cells. Biochim Biophys Acta. 2018;1865:794–802.
- Avalle L, Camporeale A, Camperi A, et al. STAT3 in cancer: A double edged sword. Cytokine. 2017;98:42–50.
- Xiao F, Dc C. FAK mediates STAT3 activation, migration and invasion in ovarian carcinoma cells. Cancer Res. 2014;74(19 Suppl):Abstractnr 3095.
- Parat MO, Riggins GJ. Caveolin-1, Caveolae, and glioblastoma. Neuro Oncol. 2012;14:679–688.
- Nunez-Wehinger S, Ortiz RJ, Diaz N, et al. Caveolin-1 in cell migration and metastasis. Curr Mol Med. 2014;14:255–274.
- Urra H, Torres VA, Ortiz RJ, et al. Caveolin-1-enhanced motility and focal adhesion turnover require tyrosine-14 but not accumulation to the rear in metastatic cancer cells. PLoS One. 2012;7:e33085.
- Hernandez-Deviez DJ, Howes MT, Laval SH, et al. Caveolin regulates endocytosis of the muscle repair protein, dysferlin. J Biol Chem. 2008;283:6476–6488.
- Shivshankar P, Halade GV, Calhoun C, et al. Caveolin-1 deletion exacerbates cardiac interstitial fibrosis by promoting M2 macrophage activation in mice after myocardial infarction. J Mol Cell Cardiol. 2014;76:84–93.
- Yang J, Zhu T, Zhao R, et al. Caveolin-1 inhibits proliferation, migration, and invasion of human colorectal cancer cells by suppressing phosphorylation of epidermal growth factor receptor. Med Sci Monit. 2018;24:332–341.
- Luanpitpong S, Talbott SJ, Rojanasakul Y, et al. Regulation of lung cancer cell migration and invasion by reactive oxygen species and caveolin-1. J Biol Chem. 2010;285:38832–38840.
- Zhao X, Guan JL. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011;63:610–615.
- Bolos V, Gasent JM, Lopez-Tarruella S, et al. The dual kinase complex FAK-Src as a promising therapeutic target in cancer. Onco Targets Ther. 2010;3:83–97.
- Scheel J, Srinivasan J, Honnert U, et al. Involvement of caveolin-1 in meiotic cell-cycle progression in Caenorhabditis elegans. Nat Cell Biol. 1999;1:127–129.
- Liu P, Rudick M, Anderson RG. Multiple functions of caveolin-1. J Biol Chem. 2002;277:41295–41298.
- Galbiati F, Volonte D, Liu J, et al. Caveolin-1 expression negatively regulates cell cycle progression by inducing G(0)/G(1) arrest via a p53/p21(WAF1/Cip1)-dependent mechanism. Mol Biol Cell. 2001;12:2229–2244.
- Fang K, Fu W, Beardsley AR, et al. Overexpression of caveolin-1 inhibits endothelial cell proliferation by arresting the cell cycle at G0/G1 phase. Cell Cycle (Georgetown, Tex). 2007;6:199–204.
- Torres VA, Tapia JC, Rodriguez DA, et al. caveolin-1 controls cell proliferation and cell death by suppressing expression of the inhibitor of apoptosis protein survivin. J Cell Sci. 2006;119:1812–1823.
- Feng Y, Venema VJ, Venema RC, et al. VEGF induces nuclear translocation of Flk-1/KDR, endothelial nitric oxide synthase, and caveolin-1 in vascular endothelial cells. Biochem Biophys Res Comm. 1999;256:192–197.
- Sanna E, Miotti S, Mazzi M, et al. Binding of nuclear caveolin-1 to promoter elements of growth-associated genes in ovarian carcinoma cells. Exp Cell Res. 2007;313:1307–1317.
- Joo JC, Hwang JH, Jo E, et al. Cordycepin induces apoptosis by caveolin-1-mediated JNK regulation of Foxo3a in human lung adenocarcinoma. Oncotarget. 2017;8:12211–12224.
- Wang R, Zhang Q, Peng X, et al. Stellettin B induces G1 arrest, apoptosis and autophagy in human non-small cell lung cancer A549 cells via blocking PI3K/Akt/mTOR pathway. Sci Rep. 2016;6:27071.
- Smits VA, Medema RH. Checking out the G(2)/M transition. Biochim Biophys Acta. 2001;1519:1–12.
- Kang D, Chen J, Wong J, et al. The checkpoint protein Chfr is a ligase that ubiquitinates Plk1 and inhibits Cdc2 at the G2 to M transition. J Cell Biol. 2002;156:249–260.
- Kawamoto H, Koizumi H, Uchikoshi T. Expression of the G2-M checkpoint regulators cyclin B1 and cdc2 in nonmalignant and malignant human breast lesions: immunocytochemical and quantitative image analyses. Am J Pathol. 1997;150:15–23.
- Canela N, Rodriguez-Vilarrupla A, Estanyol JM, et al. The SET protein regulates G2/M transition by modulating cyclin B-cyclin-dependent kinase 1 activity. J Biol Chem. 2003;278:1158–1164.
- Huang Y, Sramkoski RM, Jacobberger JW. The kinetics of G2 and M transitions regulated by B cyclins. PLoS One. 2013;8:e80861.
- Head BP, Patel HH, Roth DM, et al. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J Biol Chem. 2005;280:31036–31044.