
ABSTRACT
The deregulation of hematopoietic stem cell (HSC) transcriptional networks is a common theme in acute myelogenous leukemia (AML). Chromosomal translocations that alter the Mixed Lineage Leukemia 1 gene (MLL1, MLL, KMT2A) occur in infant, childhood and adult leukemia and at the same time, wild-type MLL1 is a critical regulator of HSC homeostasis. Typically, the endogenous, wild-type (WT) MLL1 and MLL fusion oncoproteins (MLL-FPs) remain both expressed in leukemia. WT and MLL-FPs activate overlapping sets of target genes, presenting a challenge for the selective therapeutic targeting of leukemic cells. We previously demonstrated that endogenous MLL1 is not required for the maintenance of MLL-FP-driven AML but is required for normal HSC homeostasis. Here we address the role of MLL-FPs in the initiation of leukemia in the absence of endogenous MLL1. We show that loss of endogenous Mll1 results in a rapid decrease in expression of shared HSC/leukemia target genes, yet MLL-AF9 restores the expression of most of these target genes in the absence of WT MLL1, with the critical exception of Mecom/Evi1. These observations underscore the sufficiency of MLL-fusion oncoproteins for initiating leukemia, but also illustrate that WT MLL1 target genes differ in their ability to be re-activated by MLL-FPs.
Introduction
The Mixed Lineage Leukemia 1 (MLL1, MLL, KMT2A) gene was first identified due to its involvement in chromosomal translocations in childhood acute leukemia and therapy-related secondary leukemia [Citation1–Citation3]. MLL1 translocations fuse the N-terminal portion of MLL1 in-frame with one of many known partners, resulting in oncogenic MLL-fusion proteins (MLL-FPs). Most MLL-FPs harbor the majority of the MLL1 N-terminal domains required for chromatin targeting, thus co-localization with WT MLL1 is expected and observed [Citation4,Citation5]. WT MLL1 is a histone H3 lysine 4 (H3K4) methyltransferase and performs an essential role in the development and maintenance of HSCs [Citation6–Citation8]. This is achieved in part by regulating a network of transcription factors, including Hoxa9, Eya1, Prdm16, and Mecom (encoding both MDS-Evi1 and Evi1) that each likely contributes to HSC maintenance [Citation8–Citation11]. Several of these HSCs-enriched targets, particularly Hoxa9, Eya1, and Evi1, are also upregulated by MLL-FPs and play an important role in AML pathogenesis [Citation12–Citation15]. In addition to target genes that are shared between WT MLL1 and MLL-FPs, there are also likely target genes unique to either protein [Citation16]. MLL-FPs do not retain the H3K4 methyltransferase activity and histone acetyltransferase interaction domains of full-length WT MLL1, but instead, most common fusions regulate target gene expression by ectopic recruitment of components of the super elongation complex (SEC) and the H3K79 methyltransferase Dot1L [Citation17,Citation18]. Collectively, these observations suggest a model in which MLL-FPs can hyper-activate a subset of an HSC program previously regulated by WT MLL1 [Citation14,Citation16].
Our recent studies have shown that loss of endogenous MLL1 in an established MLL-FP-driven leukemia does not affect the leukemia-initiating cell (LIC) frequency or leukemia progression kinetics [Citation19]. This was surprising, given the common view that WT MLL1 plays a critical role as a histone methyltransferase in regulating the expression of MLL-FP target genes [Citation20,Citation21]. Furthermore, we showed that the histone methyltransferase domain of endogenous MLL1 is not required for MLL-AF9 initiation and propagation of leukemia [Citation22]. However, we did not address whether MLL-FPs can initiate leukemic transformation in the absence of Mll1 after shared target genes/HSC regulators have been downregulated. Here we put our prior findings in context and add additional detail to the interrelationships between MLL1 and MLL-FPs by initiating transformation using Mll1-deficient hematopoietic stem/progenitor cells (HSPCs).
Results and discussion
We recently showed that loss of Mll1 in an established MLL-rearranged AML does not affect its clonogenic potential in vitro, potency in producing a fatal leukemia in a mouse model, or maintenance of an MLL-FP-dependent transcriptional program [Citation19]. These data relied on genetic deletion of the endogenous MLL1/Mll1 genes in a fully established leukemia (human cell line transformed mouse progenitors) [Citation19,Citation23]. Therefore, whether MLL-FPs were sufficient to re-activate an HSC-specific transcriptional program was not tested in this study. Loss of Mll1 in murine lineage-negative/Sca1+/c-Kit+ (LSK) cells in vivo or in vitro results in the rapid downregulation of Hoxa9, Meis1, Eya1, Mecom, and Prdm16, among other direct target genes ([Citation8] and see ). Most of these genes (except Prdm16) are also upregulated by MLL-FPs, thus we hypothesized that MLL-FPs introduced after downregulation of this transcriptional program may not be able to re-activate these genes without the presence of the wild-type endogenous MLL1, and would thus result in delayed leukemia onset in recipients.
To test this hypothesis, we induced deletion of Mll1 in sorted LSK cells in vitro using 4- hydroxytamoxifen (4-OHT), and target gene reduction was confirmed () and data not shown). Cells were then transduced with MLL-AF9 retrovirus for two additional days. Equal numbers of transduced control or Mll1Δ/Δ cells were injected into lethally irradiated recipients ()) or serially replated ()). As shown in ), Mll1Δ/Δ cells were transformed by MLL-AF9 and resulted in leukemia in all recipients with a similar latency as compared to control Mll1Δ/+ HSPCs. The leukemia percentage in the peripheral blood at 2 and 4 wk post-engraftment was not significantly different between Mll1Δ/+ and Mll1Δ/Δ groups ()). Selection pressure to retain non-deleted Mll1 alleles was not apparent since genomic PCR using leukemia cells from moribund recipients showed complete deletion of the floxed allele ()). Consistent with the in vivo studies, Mll1Δ/+ and Mll1Δ/Δ cells yielded comparable colony formation unit (CFU) frequency and total cell accumulation in vitro ()). Genomic PCR using cells prepared from the fourth plating also remained Mll1 deficient ()). Collectively, these data demonstrate that MLL-AF9 can transform Mll1-deficient HSPCs and result in a fully penetrant AML.
Figure 1. MLL1 is dispensable for leukemia initiation.
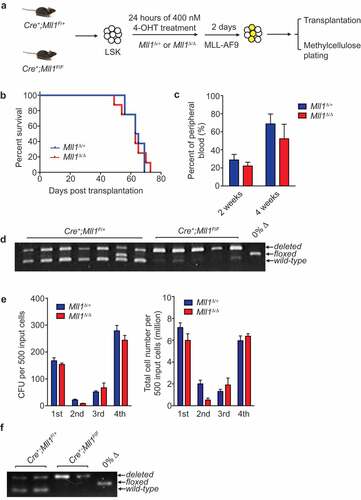
Previous studies have shown that Mll1 deletion results in rapid gene deregulation in normal HSPCs [Citation7,Citation8,Citation22,Citation24]. Furthermore, we showed that loss of Mll1 does not result in overall differentiation of purified HSCs, rather, the loss of a specific transcriptional program [Citation7,Citation8,Citation25] Using the same in vitro system, we tested whether MLL-FPs are capable of re-establishing expression of shared pro-leukemia target genes. First, we confirmed the reduced expression of Mll1-dependent genes in Mll1-deleted LSK cells 48 h after initiating deletion ()). In addition to the Mll1 transcript itself, direct target genes exhibited significantly reduced expression ()). These Mll1-deficient HSPCs were then transduced with MLL-AF9, transplanted, and leukemia was allowed to progress for 31 d in vivo ()). After expansion in recipients, leukemia cells remained Mll1 transcript-negative, whereas most target gene expression was restored to the level observed in controls ()). These data suggest that the fusion oncoprotein was capable of re-activating the majority of the shared HSC/leukemogenic program. However, Evi1 was a notable exception, which was not re-activated but was further reduced specifically in the Mll1Δ/Δ leukemia (), last graph). To confirm this finding at the clonal level, we performed qRT-PCR from individual clones selected from colonies shown in ). Each clone exhibited complete loss of Mll1 transcript, however the Hoxa9 expression level was typically as high as control clones, if not slightly higher. In wild-type cells, the level of Evi1 was variable depending on the clone picked, but Evi1high clones could no longer be observed in the Mll1-deficient cells, where the transcript was consistently not detectable (), Mll1Δ/Δ). Importantly, Evi1 expression was not reduced upon deletion of Mll1 (and Mll2) in established MLL-AF9-driven leukemia [Citation19]. The expression level of Evi1 in leukemia cells is thought to reflect the cell of origin and expression level at the time of transformation [Citation15,Citation26,Citation27]. Since the loss of Mll1 in this system does not result in differentiation generally [Citation8], these data suggest that wild-type MLL1 performs a function that directly and specifically maintains the Evi1/Mecom locus in a state responsive to super-activation by MLL-AF9. Therefore, although transformation of Mll1Δ/Δ HSPCs results in a fully penetrant leukemia that can progress with the same kinetics as wild-type-transformed cells (), it is likely that the cells adjust the leukemogenic program to compensate for the inability of MLL-FPs to upregulate some genes (in this example Evi1) in the absence of endogenous MLL1.
Figure 2. Mll1 loss affects target gene expression before the initiation of MLL-AF9 leukemia.
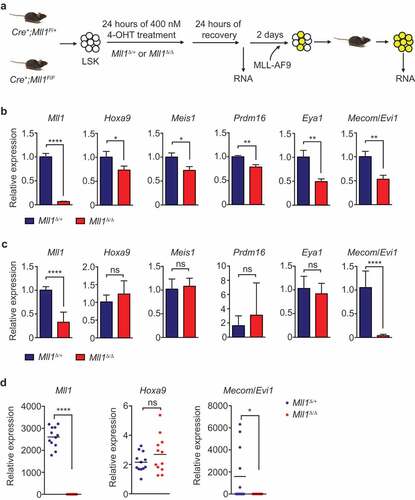
Given the strong dependency on genes such as Hoxa9, Meis1, and Eya1 on endogenous Mll1 in HSCs/HSPCs, it is surprising that MLL-AF9 can uniformly restore/super-activate expression of such target genes, given the absence of C-terminal domains. This observation suggests that expression of these genes never depended on functions of the MLL1 C-terminus in the first place. This is consistent with our observations that HSPCs lacking the SET domain of MLL1 (and therefore the histone methyltransferase activity) express MLL1 target genes at normal levels and can be transformed by MLL-FPs [Citation22]. Why the Mecom locus is an exception in that MLL-AF9 is incapable of re-activating its expression is unclear; it will be of interest to determine whether Mecom regulatory elements confer a requirement for biochemical activities that MLL-FPs cannot provide and whether Mecom represents an entire class of target genes that cannot be re-activated.
Overall, the data presented here and in Chen et al. [Citation19] demonstrate that inhibiting MLL1 activity alone is not likely to be beneficial as a therapeutic strategy in MLL-fusion driven AML. This may be due to the strong ability of MLL-FPs to over-ride the function of endogenous MLL1 through the ectopic recruitment of multiple transcriptional effector complexes [Citation28]. These data also show that MLL fusions do not act homogeneously on all target genes, a concept previously discussed by Garcia-Cuellar and colleagues [Citation29]. These observations illustrate the challenges in targeting particular effector domains of MLL-FPs, or particular downstream networks, as leukemia cells may be selected to adapt using alternative transcriptional effector interactions. Alternatively, these observations may also present combinatorial therapeutic opportunities, since enhanced reliance on particular genetic networks could result in new, predictable vulnerabilities.
Materials and methods
Animals
Animal studies were conducted in accordance with IACUC-approved animal protocols at Dartmouth College and the University of Colorado, Denver. The Mll1F/F mice [Citation7] were intercrossed with R26CreER (Cre+) mice from The Jackson Laboratories (#004847). C57BL/6 female mice between 6 and 12 wk from Jackson Laboratory were used as transplantation recipients. Deletion efficiency for Mll1 alleles was determined by semi-quantitative genomic PCR using the primers listed here: C9F4-TCTCTGAAGTAAGCCTTTCTTAG; C9R-CAGTGGACATTCCAACTCTTCAA; C9delR2-CACCCAGCATTGCAGAGTCAG.
Retroviral procedures, transformation, and engraftment of MLL-FP cells
The MSCV-MLL-AF9-YFP vector is a gift from Dr. Scott Armstrong. LSK cells were sorted and transduced as described [Citation8,Citation19] with retroviral supernatant produced by co-transfection in 293T cells. Transduced cells were either replated in M3434 medium (StemCell Technologies) or transplanted by retro-orbital injection together with 0.4 million unfractionated female C57Bl/6 bone marrow cells into lethally irradiated (950 rads) female C57Bl/6 recipients (The Jackson Laboratories). Animals were given Baytril-containing sterile water (0.1 mg/mL, Bayer) for 3 wk post-transplantation. The emergence of AML was confirmed by the presence of YFP+ blasts in the peripheral blood of recipients.
Quantitative real-time PCR (qRT-PCR)
Cells were lysed in TRIzol (Invitrogen) and RNA was purified with RNeasy Mini Kits (QIAGEN). cDNA was then produced using SuperScript III (Invitrogen) and qRT-PCR assays were performed using TaqMan Gene Expression Master mix (Applied Biosystems) using TaqMan assays listed here: Mll1_ex3 Forward: TTCTCGTCAAATAGCCCTGC; Mll1_ex3 Reverse: CTACTCTTGTCCTTCTCCACG; Mll1_ex3 Probe: TCTCTTCCCATGGTTCACCCCA.
G; Hoxa9 Forward: CCGAAAACAATGCCGAGAAT; Hoxa9 Reverse: CCGGGTTATTGGGATC
GAT; Hoxa9 Probe: AGAGCGGCGGAGACAAGCCCC; Meis1 Forward: GAGCAAGGTGATGG
CTTGGA; Meis1 Reverse: TGTCCTTATCAGGGTCATCATCG; Meis1 Probe: AACAGTGTAGC
TTCCCCCAGCACAGGT; Prdm16: Applied Biosystems Mm00712556_m1; Eya1: Applied Biosystems Mm00438796_m1; Mecom/Evi1: Applied Biosystems Mm00514814_m1; Gapdh: Applied Biosystems 4308313.
Drug treatment
4-OHT (Sigma-Aldrich) was dissolved in ethanol and used at 400 nM in culture medium to initiate Cre induction.
Acknowledgments
We thank our lab members and Kathrin Bernt for discussion and critical comments.
Disclosure statement
Dr. Ernst owns Amgen stock and has consulted for Servier Oncology and Dr. Chen is an employee of Seattle Genetics.
Additional information
Funding
References
- Gu Y, Nakamura T, Alder H, et al. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell. 1992;71:701–708.
- Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992;71:691–700.
- Felix CA, Hosler MR, Winick NJ, et al. ALL-1 gene rearrangements in DNA topoisomerase II inhibitor-related leukemia in children. Blood. 1995;85:3250–3256.
- Milne TA, Kim J, Wang GG, et al. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol Cell. 2010;38:853–863.
- Meyer C, Kowarz E, Hofmann J, et al. New insights to the MLL recombinome of acute leukemias. Leukemia. 2009;23:1490–1499.
- Gan T, Jude CD, Zaffuto K, et al. Developmentally induced Mll1 loss reveals defects in postnatal haematopoiesis. Leukemia. 2010;24:1732–1741.
- Jude CD, Climer L, Xu D, et al. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell. 2007;1:324–337.
- Artinger EL, Mishra BP, Zaffuto KM, et al. An MLL-dependent network sustains hematopoiesis. Proc Natl Acad Sci U S A. 2013;110:12000–12005.
- Kataoka K, Kurokawa M. Ecotropic viral integration site 1, stem cell self-renewal and leukemogenesis. Cancer Sci. 2012;103:1371–1377.
- Lawrence HJ, Christensen J, Fong S, et al. Loss of expression of the Hoxa-9 homeobox gene impairs the proliferation and repopulating ability of hematopoietic stem cells. Blood. 2005;106:3988–3994.
- Chuikov S, Levi BP, Smith ML, et al. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol. 2010;12:999–1006.
- Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 2003;17:2298–2307.
- Kumar AR, Hudson WA, Chen W, et al. Hoxa9 influences the phenotype but not the incidence of Mll-AF9 fusion gene leukemia. Blood. 2004;103:1823–1828.
- Wang QF, Wu G, Mi S, et al. MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood. 2011;117:6895–6905.
- Arai S, Yoshimi A, Shimabe M, et al. Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood. 2011;117:6304–6314.
- Xu J, Li L, Xiong J, et al. MLL1 and MLL1 fusion proteins have distinct functions in regulating leukemic transcription program. Cell Discov. 2016;2:16008.
- Smith E, Lin C, Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011;25:661–672.
- Wang X, Chen CW, Armstrong SA. The role of DOT1L in the maintenance of leukemia gene expression. Curr Opin Genet Dev. 2016;36:68–72.
- Chen Y, Anastassiadis K, Kranz A, et al. MLL2, not MLL1, plays a major role in sustaining MLL-rearranged acute myeloid leukemia. Cancer Cell. 2017;31(755–770):e756.
- Thiel AT, Blessington P, Zou T, et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell. 2010;17:148–159.
- Cao F, Townsend EC, Karatas H, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell. 2014;53:247–261.
- Mishra BP, Zaffuto KM, Artinger EL, et al. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep. 2014;7:1239–1247.
- Matsuo Y, MacLeod RA, Uphoff CC, et al. Two acute monocytic leukemia (AML-M5a) cell lines (MOLM-13 and MOLM-14) with interclonal phenotypic heterogeneity showing MLL-AF9 fusion resulting from an occult chromosome insertion, ins(11;9)(q23;p22p23). Leukemia. 1997;11:1469–1477.
- Li BE, Gan T, Meyerson M, et al. Distinct pathways regulated by menin and by MLL1 in hematopoietic stem cells and developing B cells. Blood. 2013;122:2039–2046.
- Artinger EL, Ernst P. Cell context in the control of self-renewal and proliferation regulated by MLL1. Cell Cycle. 2013;12:2969–2972.
- Krivtsov AV, Figueroa ME, Sinha AU, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia. 2013;27:852–860.
- Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–1093.
- Slany RK. The molecular mechanics of mixed lineage leukemia. Oncogene. 2016;35(40):5215–5223. doi:10.1038/onc.2016.30.
- Garcia-Cuellar MP, Buttner C, Bartenhagen C, et al. Leukemogenic MLL-ENL fusions induce alternative chromatin states to drive a functionally dichotomous group of target genes. Cell Rep. 2016;15:310–322.