
ABSTRACT
Autophagy plays a key role in cellular homeostasis since it allows optimal cellular functioning and provides energy under conditions of stress. Initial is revealed that alterations of macroautophagy disturb adipogenic differentiation in cultured cells, and in mice, leading to a drastic reduction of adipose tissue depots. Nevertheless, more recent studies indicate that autophagy participates in the control of adipose tissue biology in a more complex manner. The protein TP53INP2 activates the formation of autophagosomes by binding to ATG8 proteins such as LC3 or GATE16, and its genetic elimination reduces but does not cancel this activity. TP53INP2 deficiency increases adipogenic differentiation and induces a gain in adiposity in the mouse. At the cellular level, TP53INP2 promotes the sequestration of the regulatory protein GSK3β in multivesicular bodies (MVBs) by a process that involves autophagic activity and the participation of the endosomal sorting complexes required for transport (ESCRT) machinery. Through this mechanism, TP53INP2 stabilizes and activates β-catenin, which in turn triggers the inhibition of adipogenesis. In summary, autophagic pathways provide a whole set of mechanisms that may regulate in an opposite way the biology of adipose tissue, and consequently, have a variable impact on the whole body adiposity. This concept may be extensible to other cell types.
KEYWORDS:
Obesity and overweight have reached epidemic proportions globally and are now among the major causes underlying the development of chronic diseases such as type 2 diabetes (T2D), cardiovascular disease, hypertension, and cancer, among others [Citation1]. According to the World Health Organization, more than half of adults and one in three children were considered obese and overweight between 2014 and 2015 [Citation2]. In developed countries, a lifestyle linked to a high-caloric food intake and a sedentary lifestyle enhances the risk of obesity and associated pathologies. Surprisingly, the obesity epidemic is not restricted to industrialized societies. The incidence of obesity is now also increasing in developing countries. Given this context, identification of drug targets that permit efficacious treatment of this condition and a full understanding of its underlying causes are major research goals in the field.
The main feature of obesity is the accumulation of excessive and/or dysfunctional white adipose tissue (WAT) as a result of an imbalance between food intake and energy expenditure. Genetic susceptibility, in combination with environmental factors, also makes a significant contribution to the ethiopathogeny of obesity. Greater knowledge of the mechanisms involved in the regulation of adipose tissue is crucial to understand the development of obesity and its associated complications.
Autophagy in adipose tissue during obesity
Autophagy is a tightly regulated degradation process that is essential for eukaryotic cell homeostasis [Citation3]. There are various types of autophagy, namely non-selective macroautophagy (herein referred to as autophagy), selective autophagy, chaperone-mediated autophagy, and microautophagy [Citation4,Citation5]. Autophagy occurs when a double-membrane structure, named phagophore, engulfs cytosolic material, forming an organelle named an autophagosome. These autophagosomes fuse with lysosomes to form autolysosomes [Citation6].
Autophagy is a process that degrades defective cellular organelles and thus represents a renewal process. Autophagy is also responsible for the conversion of macromolecules into simpler compounds, and it is therefore crucial under starvation conditions [Citation7]. In addition, this degradation process plays more specialized functions, having been reported to participate in the degradation of lipids (lipophagy) in hepatocytes, and in the production and release of inflammatory adipokines in adipocytes [Citation8].
A key role of adipose tissue is the promotion of lipid storage under conditions of excess energy acquisition, and the activation of lipolysis and the concomitant release of fatty acids under circumstances of energy deficit. The tight regulation of triglyceride synthesis and degradation participates in the control of energy balance, as well as in glucose homeostasis. Thus, upon hypertrophy, adipocytes activate the production and release of inflammatory factors that become systemic under conditions of obesity or insulin resistance. All these functions differ depending on the type of adipose depots. Moreover, adipogenic capacity and the expansion of WAT are key inter-related aspects for the physiology of this tissue. In this regard, the composition of the extracellular matrix, the formation of new blood vessels [Citation9] and the secretion of adipokines are factors that also regulate the degree of WAT expansion necessary to prevent obesity-associated complications. In this respect, the differentiation capacity of visceral adipose depots is lower than that of subcutaneous ones [Citation10]. An increase in the abundance of subcutaneous fat therefore promotes beneficial effects on glucose homeostasis and insulin sensitivity [Citation11], whereas the accumulation of visceral adipose tissue (vWAT) favors the risk of T2D and other complications [Citation12].
The induction of hypertrophy in adipose cells under culture conditions or during obesity alters the autophagic process of this cell population. Thus, vWAT of mice on a high-fat diet (HFD) display less accumulation of a membrane-bound form of microtubule-associated 1A/1B light chain 3B (LC3-II) protein after treatment with chloroquine, and thus show a reduced autophagic flux in this tissue [Citation8]. Accordingly, an LC3-II turnover assay performed in vWAT explants from obese mice also shows that autophagic clearance is impaired. However, rapamycin treatment leads to accelerated autophagosome formation [Citation13]. Furthermore, hypertrophic 3T3-L1 adipocytes, as well as isolated visceral adipocytes from ob/ob mice, also show a diminished autophagic flux after treatment with a lysosomal activity inhibitor [Citation8,Citation14]. However, Jansen and coworkers demonstrated an enhancement in LC3-II protein in the absent of lysosomal inhibitor treatment, but they did not analyze autophagic flux [Citation15]. All of these results suggest that uniform criteria are required for the evaluation of the autophagic state of adipose tissue and adipose cells in obesity, and this should involve necessarily the measurement of autophagy flux.
In addition to altered autophagy in the adipose tissue of obese mice, others tissues such as the hypothalamus and kidney also show a decrease in autophagy [Citation16,Citation17], thereby suggesting the involvement of systemic factors.
In keeping with those data obtained in animal models of obesity, studies performed in WAT and in adipose cells derived from obese human subjects also reveal aberrant autophagy [Citation15,Citation18,Citation19]. Thus, subcutaneous white adipose tissue (sWAT) from obese subjects display an increase in the LC3-II protein levels [Citation15], suggesting that there is an enhancement of autophagic clearance and accumulation of autophagosomes. However, autophagic flux experiments would be necessary in order to clarify these results.
Furthermore, both the sWAT and vWAT of obese subjects show enhanced expression of autophagic genes such as Autophagy-regulated gene 5 (Atg5) and LC3 [Citation18,Citation19]. In addition, many studies have reported an increased abundance of autophagy proteins in sWAT and vWAT of obese subjects [Citation15,Citation18–Citation21]. This observation is in agreement with a higher abundance of autophagosomes in the adipocytes of these individuals [Citation19]. However, the accumulation of autophagosomes could indicate either a blockage in autophagy or an increased autophagosome formation. Studies performed on obese human subjects reveal that differences in autophagy markers caused by obesity are present in both subcutaneous and visceral adipose depots [Citation18,Citation19]. There have been conflicting observations regarding the impact of obesity on autophagic flux in adipose tissue and in adipocytes in humans. In this regard, studies performed in adipose explants obtained from the subcutaneous or visceral depots of obese subjects reported enhanced autophagic flux [Citation18]. Thus, adipocytes obtained from sWAT show enhanced autophagic flux in non-healthy individuals and in diabetic obese subjects compared to controls [Citation20]. In contrast, Soussi and coworkers described reduced autophagic flux in subcutaneous adipocytes of healthy obese subjects [Citation14]. These results highlight that the alterations in glucose homeostasis are a key aspect in the autophagy regulation of adipose tissue in obesity.
The discrepant findings documented may be explained by differences in the distribution of adipose depots in obese subjects in different studies and/or by variations in the autophagy pattern displayed by the distinct cells found in adipose tissue, such as adipocytes, macrophages or other immune cells, as well as preadipocytes, and adipose-derived stem cells. Given the key role of autophagy in cellular homeostasis, it is likely that this process is altered but in distinct manners, in the different cell populations present in WAT during obesity and overweight. Further studies are required to elucidate the complexities involved.
In keeping with the idea that different cell types present in adipose depots may regulate autophagy in different ways, it has been reported that adipose tissue macrophages (ATM) display an increase in LC3-II protein levels [Citation22] together with an enhanced autophagic flux [Citation22]. In contrast, peritoneal macrophages obtained from mice on a HFD show a decrease in the levels of LC3-II protein [Citation23], thereby suggesting the existence of local modulatory factors.
Regulatory factors of autophagy in adipose tissue
As mentioned above, the expression of some autophagy factors is altered in adipose tissue during obesity. Human obesity has been reported to induce the expression of autophagy genes Atg5, LC3A or LC3B in adipose depots [Citation18]. Furthermore, non-healthy obese subjects show a greater induction in autophagy genes compared to healthy obese subjects [Citation18] in sWAT as well as in vWAT depots [Citation18]. Although the regulation of autophagy genes does not provide a definitive idea on whether autophagy is enhanced or reduced in a specific condition, analysis of the factors involved in adipose tissue is certainly relevant.
Regarding the transcriptional regulation of Atg genes, E2F1 transcription factor modulates these genes in human adipose tissue during obesity [Citation24]. E2F1 promotes cell cycle progression and activates autophagy in non-adipose cells. The vWAT of obese patients shows an upregulation of E2F1 [Citation25], and its gene expression correlates with Atg5 and LC3 [Citation24]. Interestingly, E2F1 induction occurs in parallel to an inflammatory activation in adipose tissue, thereby suggesting that E2F1 activates autophagy in order to protect the tissue against obesity-associated inflammation. Furthermore, E2F1-deficient adipocyte-like cells show less activation of autophagy in response to inflammatory cytokines [Citation24]. In this regard, López-Vicario and coworkers reported that the inhibition of soluble epoxide hydrolase (sEH) reduces macrophage infiltration in adipose tissue of mice on a HFD [Citation26]. In addition, the blockage of sEH leads to a decrease in the expression of autophagy proteins in obese mice [Citation26].
The forkhead family of transcription factors (FoXO) also regulates the activity of Atg genes. In particular, FoxO1 and FoxO3 activate the autophagy program in non-adipose cells [Citation27]. In the adipogenic context, FoxO factors inhibit adipogenesis and contribute to obesity-associated inflammation [Citation28,Citation29]. In this regard, Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) also regulates the inflammatory process associated with obesity in adipose tissue [Citation30], and it could be a relevant factor mediating autophagy in the immune cells of adipose tissue. Other key factors that participate in autophagy include the lysosomal proteases cathepsin L (CTSL) and cathepsin B (CTSB) [Citation31]. Thus, there is a decrease in the expression and activity of mature CTSL in vWAT of mice on a HFD and ob/ob mice [Citation13]. The impaired lysosomal function in vWAT of obese mice decreases autophagic clearance, thus causing autophagosome accumulation [Citation13]. In this regard, Death-associated protein kinase 2 (DAPK2) participates in the regulation of lysosome-mediated autophagy in obesity [Citation14]. Thus, adipocytes isolated from obese subjects show a marked decrease in DAPK2 protein levels, together with a reduction in autophagy flux [Citation14]. Thereby, lysosomal proteases are synthesized in the endoplasmic reticulum, processed post-translationally, and then shuttled to the endosomal-lysosomal system [Citation32]. It is therefore feasible that an alteration in the endosomal machinery plays a relevant role in the alterations of adipose tissue in obesity.
Impact of genetic cancellation of autophagy on adipose tissue biology
Studies in cell cultures of both 3T3-L1 and mouse embryonic fibroblasts (MEFs) indicate that the inhibition of autophagy induced by the repression of Autophagy-related gene 7 or 5 (Atg7 or Atg5), or by pharmacological inhibitors, prevents correct adipogenesis [Citation33–Citation35]. Under these conditions, there is a decrease in the adipogenic regulatory proteins, namely CCAAT/enhancer-binding protein beta and alpha (C/EBPβ and C/EBPα), and Peroxisome proliferator-activated receptor gamma (PPARγ) [Citation35]. The molecular mechanisms by which the inhibition of autophagy causes a decrease in the expression of the master proteins of adipogenesis remain unknown.
In parallel to these observations in cell cultures, two groups simultaneously reported the phenotype of Atg7 knockout mice selectively for adipose tissue by using Cre-recombinase directed by the Adipocyte fatty acid-binding protein (aP2) promoter [Citation33,Citation35]. Atg7-deficient mice showed a lower WAT mass, together with a decrease in body weight. Under these conditions, the Atg7 KO mice showed reduced circulating levels of glucose, insulin, triglycerides, cholesterol and free fatty acids. At the cellular level, WAT contained a greater number of adipocytes, and in addition, fat showed enhanced expression of uncoupling protein 1 (UCP-1), and a greater expression of the proliferator-activated receptor coactivator 1α (PGC-1α) factor, a higher content of mitochondria, and greater β-oxidation of fatty acids. The absence of Atg7 in WAT leads to the appearance of beige adipose tissue, probably as a consequence of changes in the differentiation of precursor cells. Atg7-deficient mice also showed alterations in brown adipose tissue (BAT) metabolism such as a greater oxidation of fatty acids, increased expression of PGC-1α and UCP-1, and a reduced size of lipid droplets. The mechanism by which the inhibition of autophagy leads to a differentiation of the beige adipocytes is unknown. However, the disappearance of beige adipose tissue in response to increased temperature of housing conditions depends on the autophagic degradation of mitochondria [Citation36]. This observation could partly explain the phenotype of the Atg7 KO mouse.
Since macrophages, endothelial cells and brain, in addition to adipocytes, express aP2, it would be of interest to examine whether part of the phenotype of Atg7 KO mice results from alterations in tissues or cell types other than adipose cells. In this regard, a link between the central nervous system and other tissues, such as liver or BAT, regulates lipophagy [Citation37], an observation that points to the existence of more complex mechanisms of autophagy in WAT.
Studies using Atg7-deficient mice have provided a clear view of the role of autophagy in adipose tissue biology. However, other reports are more difficult to incorporate into a coherent model. Thus, the global overexpression of Atg5 activates autophagy and also promotes a lower adipose tissue mass in both subcutaneous and visceral deposits, as well as an increased sensitivity to insulin [Citation38]. In addition, the ablation of Atg3 or Atg16L1 in mature adipocytes promotes insulin resistance in the absence of changes in body weight or in the percentage of fat mass [Citation39]. Atg3 ablated mice display an increase in the crown-like adipocytes present in vWAT, thereby suggesting the implication of inflammation in the development of insulin resistance [Citation39].
Furthermore, data on p62 or Sequestosome-1 protein (SQSTM1/p62) are not coherent with the Atg7 model. Among other cellular functions, p62 participates in autophagy as an adapter protein that binds poly-ubiquitinated proteins for their degradation. The deficiency of p62 in MEFs accelerates adipogenic differentiation, and the global p62 KO mouse displays an increase in food intake, a reduction in energy expenditure, and an increase in adipose tissue mass, in parallel to a decrease in glucose tolerance [Citation40]. The phenotype of the global p62 KO mice could be a result of changes in autophagy or other cellular actions of p62. It is likely that p62 has distinct metabolic actions in different tissues. In this regard, the p62 brain-specific KO mouse shows hypothalamic resistance to leptin, hyperphagia and obesity [Citation41].
All of these findings highlight the complexity of the protein framework of autophagy in adipose tissue. Further research should seek to clarify the role of this degradation process in adipose tissue physiology.
TP53INP2 is a positive modulator of autophagy
Tumor protein p53 inducible nuclear protein 2 (TP53INP2) acts as a nuclear coactivator in mammals and Drosophila melanogaster [Citation42,Citation43]. In the context of autophagy, TP53INP2 regulates the basal autophagy machinery in these two models [Citation43,Citation44]. Thus, TP53INP2 gain-of function increases the number of autophagic vacuoles, and this effect is enhanced in the present of amino acid starvation or rapamycin treatment in human cells [Citation44]. In the fat body of Drosophila melanogaster, TP53INP2 also participates in the activation of autophagy [Citation44]. The mechanism by which TP53INP2 modulates autophagy is mediated by the interaction of this molecule with members of the ATG8 family of proteins. Thus, under autophagy activation, TP53INP2 moves to the cytosol, where it interacts with LC3 and GATE16 to promote autophagosome formation and protein degradation [Citation44] (). In the cytosol, TP53INP3 also interacts directly with ATG7, thus contributing to autophagosome biogenesis [Citation45]. The mechanism of TP53INP2 translocation to the cytosol appears to depend on its interaction with nuclear LC3. In this regard, Liu and coworkers have documented that TP53INP2, together with deacetylase SIRT1, promotes the translocation of nuclear LC3 to the cytosol [Citation46].
Figure 1. Modulation of TP53INP2 during autophagy. In basal conditions, TP53INP2 and LC3 remain mainly nuclear. Under autophagy activation by amino acid starvation or rapamycin treatment, TP53INP2 moves to the cytosol together with LC3. In the cytosol, TP53INP2 interacts with LC3, GATE16 and ATG7 to promote autophagosome formation and maturation. Under most conditions, TP53INP2 leaves the autophagosome before its fusion with lysosomes.
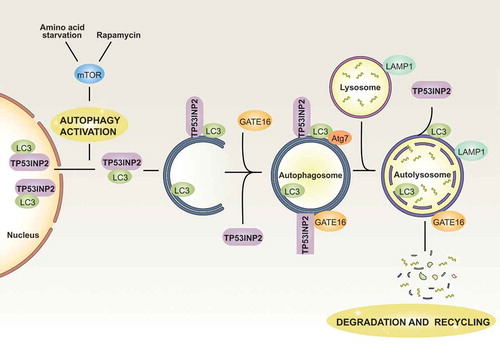
TP53INP2 also participates in the regulation of autophagy in active metabolic tissues. In this regard, transgenic mice overexpressing TP53INP2 specifically in skeletal muscle display an upregulation of basal autophagy to control the skeletal muscle mass [Citation47]. Furthermore, in obesity or T2D, the expression of TP53INP2 decreases in skeletal muscle in order to prevent the activation of autophagy. The TP53INP2-mediated action serves to mitigate the accelerated protein loss that occurs in these pathological contexts [Citation47].
TP53INP2 modulates adiposity in mice and humans
Obesity and overweight are major public health problems because they contribute to the development of a range of diseases [Citation48]. In this regard, the maintenance of WAT homeostasis is a key factor to avoid the problems associated with obesity. sWAT is the major storage site of lipids [Citation49]. Therefore, unhealthy obese subjects have fewer adipocytes (but hypertrophic) than healthy obese subjects [Citation49] and show deregulated adipogenesis [Citation50,Citation51]. Thus knowledge of the expandability of sWAT and adipogenesis may provide relevant insight into obesity.
TP53INP2 plays a role in obesity and T2D. Thus, there is a downregulation of TP53INP2 in skeletal muscle of T2D and obese non-diabetic patients [Citation47]. Moreover, the expression of TP53INP2 also decreases in sWAT of obese non-diabetic patients [Citation52], thereby suggesting that this protein modulates adiposity in humans. In relation with this, TP53INP2-deficient mice show a significant increase in body weight associated with a high accumulation of fat depots. In addition, the sWAT of these animals shows an early hyperplasic phase followed by a hypertrophic phenotype [Citation52]. The increase in the capacity to generate new adipose cells, together with the high accumulation of triglycerides induced by TP53INP2 deficiency, occurs in the absence of changes in glucose tolerance or insulin sensitivity. These observations thus point to the participation of the inflammatory process that occurs in adipose tissue in obesity. The induction of a pro-inflammatory response in this tissue promotes the development of insulin resistance. However, there are some mouse models in which an enhanced fat mass is not accompanied by insulin resistance [Citation53,Citation54]. In keeping with this, TP53INP2-deficient mice show a decrease in the expression of some pro-inflammatory genes, together with an increase in adipogenesis. In this regard, the activation of the pro-inflammatory program in human adipose tissue reduces the differentiation capacity of adipose cells [Citation55]. All of these results suggest that TP53INP2 participates in the inflammatory response and in the expansion of sWAT in healthy obese subjects in order to regulate glucose metabolism.
Moreover, the TP53INP2 homologous protein, TP53INP1, also participates in the metabolic syndrome. Thus, TP53INP1-deficient mice show an increase in body weight, together with a greater adipose depots compared to control littermates [Citation56].
TP53INP2 facilitates crosstalk between autophagy and the endosomal pathway
Autophagosomes are able to fuse with endosomal organelles, such as early endosomes and late endosomes or MVBs, in order to generate a hybrid organelle called amphisome [Citation57], which also fuses with lysosomes [Citation58]. In this regard, there is crosstalk between autophagy and the endosomal pathway [Citation59], and a functional endocytic pathway is a prerequisite for efficient autophagic flux [Citation60]. Thus, early endosomes are necessary components for correct autophagosome maturation [Citation61] and for MVB formation [Citation57]. Various multi-subunit protein complexes drive the maturation of the endosomal compartment. Therefore, the biogenesis of MVBs involves the formation of intraluminal vesicles (ILVs), a process that requires the ESCRT complex, among others [Citation62]. The ESCRT machinery is formed by four protein complexes, ESCRT-0, I, II and III, which are activated sequentially. First, ESCRT-0 binds PI3P in the endosomal membrane in order to promote the recruitment of ESCRT-I and ESCRT-II. This process facilitates vesicle budding from the endosomal membrane to the lumen and recruits ESCRT-III to the complex in order to cleave the vesicle buds, thus forming the ILVs that contain the cargo. Finally, vacuolar sorting protein-4 (VPS4), an ATPase complex, removes ESCRT machinery from the endosome [Citation63].
TP53INP2 modulates the maturation of MVBs, thereby regulating the number and cellular distribution of these endosomes. Thus, TP53INP2-deficient cells show a decrease in lysobisphosphatidic acid (LBPA)-positive puncta [Citation52], and a perinuclear distribution of these LBPA+ endosomes. These findings thus point to an alteration in late endosomal function [Citation52]. In addition, TP53INP2 overexpression induces the formation of LBPA+ puncta, but this effect is absent in both ESCRT-0- and ESCRT-III-deficient cells. These observations thus suggest that TP53INP2 modulates MVBs through an ESCRT-dependent mechanism. Disruption of the ESCRT machinery also impairs amphisome formation [Citation64,Citation65]. Therefore, in the presence of the active form of Rab5 (Rab5 Q79L), TP53INP2 moves from the nucleus, thus promoting the recruitment of LC3-II to these Rab5-labeled endosomes to facilitate amphisome formation [Citation52]. In all, TP53INP2 coordinates the crosstalk between autophagy and the endosomal pathway to regulate adipogenesis [Citation52].
TP53INP2 promotes the WNT signaling pathway through autophagy
The canonical WNT signaling pathway plays an important role in the regulation of adipogenesis [Citation66]. In the presence of WNTs, cytoplasmic β-CATENIN translocates to the nucleus where it binds to TCF binding sites to block the transcription of adipogenic genes [Citation67]. Various WNT proteins, including WNT6, WNT10a and WNT10b, are strongly expressed in pre-adipocytes and show a decrease in expression during the differentiation process [Citation68]. Furthermore, transgenic mice expressing WNT10 in adipose tissue are lean and resistant to diet-induced obesity [Citation69,Citation70]. The levels of β-CATENIN are regulated by a cytosolic degradation complex that consists of Glycogen Synthase Kinase 3 (GSK3β), Casein Kinase 1 (CK1), Adenomatous Polyposis Coli (APC) and AXIN. This complex drives β-CATENIN phosphorylation and its subsequent ubiquitination and proteasomal degradation. The activity of GSK3β is a key point in the regulation of WNT signaling. In this regard, dysregulation of GSK3β is associated with a range of pathological conditions, including diabetes mellitus, obesity, and inflammation [Citation71].
The endosomal pathway plays an important role in the regulation of WNT signaling. Thus, the internalization of GSK3β into MVBs is a prerequisite for the stabilization of cytosolic β-CATENIN [Citation72]. In the presence of WNT3, the molecules GSK3β and AXIN are recruited to the WNT receptor complex, where they phosphorylate LDL Receptor-Related Protein 6 (LRP6). After that, the complex is internalized into acidic vesicles, which correspond to MVBs. A similar effect occurs in the presence of an active mutant of LRP6. This observation thus reinforces the GSK3β sequestering hypothesis [Citation72]. Furthermore, the ESCRT machinery actively participates in this process. Thus, hepatocyte growth factor-regulated tyrosine kinase substrate (HRS)-deficient cells do not show an increase in β-catenin upon WNT3a treatment. In turn, a VPS4-dominant negative, which inhibits MVB formation, blocks the effects of WNT3a and an LRP6-active mutant [Citation72].
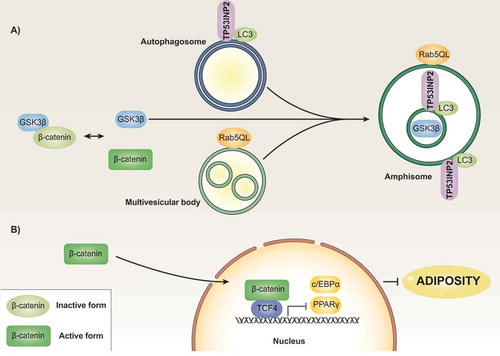
TP53INP2 increases β-CATENIN levels by a mechanism that involves the endosomal pathway. Thus, in the presence of an active Rab5 mutant, TP53INP2 promotes the internalization of GSK3β into MVBs. In addition, TP53INP2 recruits LC3-II to these vesicles to promote amphisome formation (). However, the action of TP53INP2 does not occur when there is an autophagy blockage. Thus, in Atg-deficient cell models such as Atg5 or Atg3 KO MEFs, TP53INP2 is unable to promote the internalization of GSK3β into MVBs and the subsequent upregulation of β-catenin protein levels [Citation52]. Furthermore, ESCRT complexes, in particular ESCRT-0 and III, are necessary for TP53INP2-induced GSK3β sequester. Thus, TP53INP2 is unable to perform its action in HRS- and VPS4-deficient cells. All of these results, together with the observation of TP53INP2 downregulation in sWAT of obese non-diabetic patients, suggest that TP53INP2 abolishes adiposity by modulating the WNT signaling pathway in an autophagy-dependent manner.
Figure 2. Model of TP53INP2 action in adipose precursor cells. Regulatory protein β-CATENIN is found in inactive and active states. The formation of a complex with GSK3β, among other proteins, leads to β-CATENIN phosphorylation followed by further proteasomal degradation. The presence of a constitutively active mutant form of Rab5 (Rab5QL) causes the formation of enlarged MVBs, which occurs in the absence of changes in the localization of GSK3β. Under these conditions, TP53INP2 promotes the fusion of autophagosomes with MVBs in such a way that GSK3β is internalized into the MVB/amphisome compartment in an ESCRT-dependent manner (panel A). This process leads to activation of β-CATENIN, which blocks the transcription of genes involved in the induction of the adipogenic program (panel B).
Conclusions and future perspectives
Data reviewed in prior sections permit us to draw a number of tentative conclusions and suggestions for future studies. Autophagy, or perhaps better autophagic pathways, provides a whole set of mechanisms that may regulate in an opposite way the biology of adipose tissue, depending on the pathway that gets modulated, and also depending on the developmental time. This concept is relevant to understand the variety of signals emanating from autophagy in adipose cells or precursor cells, and also because it may be extensible to other cell types present in adipose tissue. Future research should focus in the dissection of the different autophagy pathways present in adipose cells and in precursor cells, and also the delineation of those pathways that are relevant in the biology of adipose precursor cells or of mature adipocytes. An interesting angle to this study is to analyze whether subcutaneous and visceral adipose cells or precursor cells show any differences in terms of the organization of autophagy, and its regulation.
In addition, comparative analysis of alterations in autophagy in different animal models of obesity focused in visceral adipose tissue reveals that in most cases autophagy flux is enhanced but other studies show the reverse, and this suggests the variable contribution of different cell types in different models of obesity to the global analysis of autophagy. In this connection, future research should analyzed in detail the differences in response to obesity of different cell types present in adipose tissue namely adipose cells, precursor cells, and resident immune cells.
Acknowledgments
We gratefully acknowledge institutional funding from the MINECO through the Centres of Excellence Severo Ochoa Award, and from the CERCA Programme of the Catalan Government.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Zylke JW, Bauchner H. The unrelenting challenge of obesity. Jama. 2016;315:2277–2278.
- Abbasi A, Juszczyk D, van Jaarsveld CHM, et al. Body mass index and incident type 1 and type 2 diabetes in children and young adults: a retrospective cohort study. J Endocr Soc. 2017;1:524–537.
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662.
- Reggiori F, Komatsu M, Finley K, et al. Autophagy: more than a nonselective pathway. Int J Cell Biol. 2012;2012:219625.
- Watanabe T, Kuma A, Mizushima N. Physiological role of autophagy in metabolism and its regulation mechanism. Nihon Rinsho. 2011;69(Suppl 1):775–781.
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845–1846.
- Galluzzi, L., Baehrecke, E.H., Ballabio, A., et al. Molecular definitions of autophagy and related processes. Embo J. 2017;36:1811–1836.
- Yoshizaki, T., Kusunoki, C., Kondo, M., et al. Autophagy regulates inflammation in adipocytes. Biochem Biophys Res Commun. 2012;417:352–357.
- Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094–2101.
- Lee MJ, Wu Y, Fried SK. Adipose tissue heterogeneity: implication of depot differences in adipose tissue for obesity complications. Mol Aspects Med. 2013;34:1–11.
- Tran TT, Kahn CR. Transplantation of adipose tissue and stem cells: role in metabolism and disease. Nat Rev Endocrinol. 2010;6:195–213.
- Fox, C.S., Massaro, J.M., Hoffmann, U., et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation. 2007;116:39–48.
- Mizunoe, Y., Sudo, Y., Okita, N., et al. Involvement of lysosomal dysfunction in autophagosome accumulation and early pathologies in adipose tissue of obese mice. Autophagy. 2017;13:642–653.
- Soussi, H., Reggio, S., Alili, R., et al. DAPK2 downregulation associates with attenuated adipocyte autophagic clearance in human obesity. Diabetes. 2015;64:3452–3463.
- Jansen, H.J., Van Essen, P., Koenen, T., et al. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology. 2012;153:5866–5874.
- Meng Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J Biol Chem. 2011;286:32324–32332.
- Yamahara, K., Kume, S., Koya, D., et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J Am Soc Nephrol. 2013;24:1769–1781.
- Kovsan, J., Blüher, M., Tarnovscki, T., et al. Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab. 2011;96:E268–277.
- Kosacka, J., Kern, M., Klöting, N., et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol Cell Endocrinol. 2015;409:21–32.
- Ost, A., Svensson, K., Ruishalme, I., et al. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med. 2010;16:235–246.
- Nunez, C.E., Rodrigues, V.S., Gomes, F.S., et al. Defective regulation of adipose tissue autophagy in obesity. Int J Obes (Lond). 2013;37:1473–1480.
- Grijalva A, Xu X, Ferrante AW Jr. autophagy is dispensable for macrophage-mediated lipid homeostasis in adipose tissue. Diabetes. 2016;65:967–980.
- Liu, K., Zhao, E., Ilyas, G., et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11:271–284.
- Haim, Y., Blüher, M., Slutsky, N., et al. Elevated autophagy gene expression in adipose tissue of obese humans: A potential non-cell-cycle-dependent function of E2F1. Autophagy. 2015;11:2074–2088.
- Maixner, N., Bechor, S., Vershinin, Z., et al. Transcriptional dysregulation of adipose tissue autophagy in obesity. Physiology (Bethesda). 2016;31:270–282.
- Lopez-Vicario, C., Alcaraz-Quiles, J., García-Alonso, V., et al. Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: role for omega-3 epoxides. Proc Natl Acad Sci U S A. 2015;112:536–541.
- Sengupta A, Molkentin JD, Yutzey KE. FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem. 2009;284:28319–28331.
- Gross DN, van Den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27:2320–2336.
- Ito Y, Daitoku H, Fukamizu A. Foxo1 increases pro-inflammatory gene expression by inducing C/EBPbeta in TNF-alpha-treated adipocytes. Biochem Biophys Res Commun. 2009;378:290–295.
- Shoelson SE, Lee J, Yuan M. Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. Int J Obes Relat Metab Disord. 2003;27(Suppl 3):S49–52.
- Kaminskyy V, Zhivotovsky B. Proteases in autophagy. Biochim Biophys Acta. 2012;1824:44–50.
- Reiser J, Adair B, Reinheckel T. Specialized roles for cysteine cathepsins in health and disease. J Clin Invest. 2010;120:3421–3431.
- Zhang, Y., Goldman, S., Baerga, R., et al. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A. 2009;106:19860–19865.
- Baerga R, Zhang Y, Chen PH, et al. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy. 2009;5:1118–1130.
- Singh, R., Xiang, Y., Wang, Y., et al. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009;119:3329–3339.
- Altshuler-Keylin, S., Shinoda, K., Hasegawa, Y., et al. Beige adipocyte maintenance is regulated by autophagy-induced mitochondrial clearance. Cell Metab. 2016;24:402–419.
- Martinez-Lopez, N., Garcia-Macia, M., Sahu, S., et al. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 2016;23:113–127.
- Pyo, J.O., Yoo, S.M., Ahn, H.H., et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013;4:2300.
- Cai, J., Pires, K.M., Ferhat, M., et al. Autophagy ablation in adipocytes induces insulin resistance and reveals roles for lipid peroxide and Nrf2 signaling in adipose-liver crosstalk. Cell Rep. 2018;25(1708–1717):e1705.
- Rodriguez, A., Durán, A., Selloum, M., et al. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006;3:211–222.
- Harada, H., Warabi, E., Matsuki, T., et al. Deficiency of p62/Sequestosome 1 causes hyperphagia due to leptin resistance in the brain. J Neurosci. 2013;33:14767–14777.
- Baumgartner, B.G., Orpinell, M., Duran, J., et al. Identification of a novel modulator of thyroid hormone receptor-mediated action. PloS One. 2007;2:e1183.
- Francis VA, Zorzano A, Teleman AA. dDOR is an EcR coactivator that forms a feed-forward loop connecting insulin and ecdysone signaling. Curr Biol. 2010;20:1799–1808.
- Mauvezin, C., Orpinell, M., Francis, V.A., et al. The nuclear cofactor DOR regulates autophagy in mammalian and Drosophila cells. EMBO Rep. 2010;11:37–44.
- You, Z., Xu, Y., Wan, W., et al. TP53INP2 contributes to autophagosome formation by promoting LC3-ATG7 interaction. Autophagy. 2019.
- Liu X, Klionsky DJ. TP53INP2/DOR protein chaperones deacetylated nuclear LC3 to the cytoplasm to promote macroautophagy. Autophagy. 2015;11:1441–1442.
- Sala, D., Ivanova, S., Plana, N., et al. Autophagy-regulating TP53INP2 mediates muscle wasting and is repressed in diabetes. J Clin Invest. 2014;124:1914–1927.
- Jensen, M.D., Ryan, D.H., Apovian, C.M., et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American college of cardiology/American heart association task force on practice guidelines and the obesity society. J Am Coll Cardiol. 2014;63:2985–3023.
- Spalding, K.L., Bernard, S., Näslund, E., et al. Impact of fat mass and distribution on lipid turnover in human adipose tissue. Nat Commun. 2017;8:15253.
- Patel P, Abate N. Body fat distribution and insulin resistance. Nutrients. 2013;5:2019–2027.
- Spalding, K.L., Arner, E., Westermark, P.O., et al. Dynamics of fat cell turnover in humans. Nature. 2008;453:783–787.
- Romero, M., Sabaté-Pérez, A., Francis, V.A., et al. TP53INP2 regulates adiposity by activating beta-catenin through autophagy-dependent sequestration of GSK3beta. Nat Cell Biol. 2018;20:443–454.
- Maris, M., Overbergh, L., Gysemans, C., et al. Deletion of C/EBP homologous protein (Chop) in C57Bl/6 mice dissociates obesity from insulin resistance. Diabetologia. 2012;55:1167–1178.
- Perreault M, Marette A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med. 2001;7:1138–1143.
- Gustafson, B., Gogg, S., Hedjazifar, S., et al. Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am J Physiol Endocrinol Metab. 2009;297:E999–E1003.
- Seillier, M., Pouyet, L., N’guessan, P., et al. Defects in mitophagy promote redox-driven metabolic syndrome in the absence of TP53INP1. EMBO Mol Med. 2015;7:802–818.
- Morvan, J., Köchl, R., Watson, R., et al. In vitro reconstitution of fusion between immature autophagosomes and endosomes. Autophagy. 2009;5:676–689.
- Lamb CA, Dooley HC, Tooze SA. Endocytosis and autophagy: shared machinery for degradation. BioEssays. 2013;35:34–45.
- Liou W, Geuze HJ, Geelen MJ, et al. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J Cell Biol. 1997;136:61–70.
- Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011;13:495–504.
- Razi M, Chan EY, Tooze SA. Early endosomes and endosomal coatomer are required for autophagy. J Cell Biol. 2009;185:305–321.
- Henne WM, Buchkovich NJ, Emr SD. The ESCRT pathway. Dev Cell. 2011;21:77–91.
- Wollert T, Hurley JH. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature. 2010;464:864–869.
- Rusten, T.E., Vaccari, T., Lindmo, K., et al. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr Biol. 2007;17:1817–1825.
- Juhasz, G., Hill, J.H., Yan, Y.,et al. The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J Cell Biol. 2008;181:655–666.
- Christodoulides C, Lagathu C, Sethi JK, et al. Adipogenesis and WNT signalling. Trends Endocrinol Metab. 2009;20:16–24.
- Ross, S.E., Hemati, N., Longo, K.A., et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950–953.
- Cawthorn, W.P., Bree, A.J., Yao, Y, et al. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a beta-catenin-dependent mechanism. Bone. 2012;50:477–489.
- Longo, K.A., Wright, W.S., Kang, S., et al. Wnt10b inhibits development of white and brown adipose tissues. J Biol Chem. 2004;279:35503–35509.
- Wright, W.S., Longo, K.A., Dolinsky, V.W.,et al. Wnt10b inhibits obesity in ob/ob and agouti mice. Diabetes. 2007;56:295–303.
- Wada A. GSK-3 inhibitors and insulin receptor signaling in health, disease, and therapeutics. Front Biosci (Landmark Ed). 2009;14:1558–1570.
- Taelman, V.F., Dobrowolski, R., Plouhinec, J.L., et al. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143:1136–1148.