
ABSTRACT
Myocardial ischemia-reperfusion (I/R) injury is caused by endothelial dysfunction and enhanced oxidative stress. The overexpression of JAZF1, a zinc finger protein, has been reported to promote cell proliferation and suppress myogenic differentiation in type 2 diabetes. However, the involvement of JAZF1 in myocardial I/R injury remains to be unclear. The current study aims to investigate the role by which JAZF1 influences cardiac microvascular endothelial cells (CMECs) in a rat model of myocardial I/R injury. A total of 50 rats were established as a myocardial I/R model to isolate CMECs, with alterations in JAZF1 expression. After that, the gain- or loss-function of JAZF1 on the proliferation, apoptosis and tube formation ability of CMECs were evaluated by a series of in vitro experiments. Results indicated that JAZF1 was down-regulated in CMECs of rats with myocardial I/R injury. After treatment with JAZF1, the levels of VEGF, Bcl-2, PDGF and p-Akt/Akt were all increased; however, the expression of Bax, caspase-3, caspase-9, p-Bad/Bad, c-caspase-3/caspase-3, c-caspase-9/caspase-9, and p-FKHR/FKHR exhibited decreased levels; CMEC proliferation and angiogenesis were increased, while cell apoptosis was attenuated. CMECs transfected with JAZF1 shRNA exhibited the contrary tendencies. The key findings of this study suggest that the over-expression of JAZF1 alleviates myocardial I/R injury by enhancing proliferation and angiogenesis of CMECs and in turn inhibiting apoptosis of CMECs via the activation of the Akt signaling pathway.
Introduction
Early reperfusion is the solitary way to inhibit further tissue damage after the occurrence of myocardial ischemia (MI); however, it induces reversible and irreversible organ injuries, which are referred to as ischemia-reperfusion (I/R) injuries [Citation1]. Several critical factors have been identified by experimental studies as potential mediators of the detrimental effects often observed in myocardial I/R, including oxidative stress, inflammation, intracellular Ca2+ overload, rapid restoration of physiological pH at the time of reperfusion, mitochondrial permeability transition pore (mPTP), as well as late myocardial reperfusion injury [Citation2–Citation5]. Currently, therapeutic strategies, such as ischemic preconditioning (IPC) or postconditioning (Ipost), remote ischemic preconditioning, therapeutic hyperoxemia and hypothermia, pharmacologic agents for preventing myocardial I/R are used to treat the condition [Citation6]. Despite revascularization strategies including surgical revascularization or percutaneous catheter intervention achieving great progresses; angiogenic therapies have drawn to the attention of numerous researchers [Citation7]. Additionally, as the most common cells in a normal heart, cardiac microvascular endothelial cells (CMECs) are comprised of an elementary component of the myocardial microcirculation, and can secretes cytokines associated with cardiac growth, contractile performance, and rhythmicity under normal conditions [Citation8]. Additionally, a string of studies has emphasized the roles of CMECs in the preservation of cardiomyocytes against I/R [Citation9].
With the development of thrombolysis, percutaneous transluminal coronary angioplasty and coronary artery bypass grafting have resulted in myocardial I/R injury becoming the primary blockade in the search for the optimal therapeutic effect from ischemic heart disease during reperfusion treatment [Citation10]. Ischemic heart disease is widely thought to be related to various genetic and hereditary factors in the incidence of the condition [Citation11]. Akt, also known as protein kinase B, is a serine protein kinase that acts as a regulator of heart growth, myocardial angiogenesis and cardiomyocyte apoptosis [Citation12]. Furthermore, previous evidence dictates that activated Akt substantially reduces infarction size and can dramatically enhance post-ischemic myocardial functioning [Citation13]. The juxtaposed with another zinc finger protein 1 (JAZF1), also called TAK1-interacting protein 27 (TIP27) or zinc finger protein 802 (ZNF802) gene, encodes for a nuclear protein with a molecular mass of 27-kDa, that is expressed in various human tissues [Citation14,Citation15]. Previous literature has asserted that up-regulated JAZF1 can in turn result in higher blood pressure, abnormal electrocardiogram readings as well as impaired mitochondria [Citation16]. In addition, JAZF1 has been highlighted due to its auxo-action on visfatin expression through the peroxisome proliferator-activated receptor alpha (PPARα) and PPARβ/δ signaling pathways [Citation17]. Moreover, multiple studies have verified that Akt function is mediated by its upstream pathways during and post-transcription, while JAZF1 exerts its effect through transcriptional activation as well [Citation18,Citation19]. Furthermore, a study demonstrated that the phosphorylation of Akt was increased in the muscle, liver and adipose tissues of TIP27-Tg mice, indicating an Akt-dependent signaling pathway for TIP27-induced changes in insulin action [Citation20]. Based on the aforementioned content, a bold assertion was made during the conception of this current study, suggesting the existence of a potential mechanism between the Akt signaling pathway and JAZF1, a mechanism to which has not been previously explored present. Therefore, the current study was performed to validate the role of JAZF1 on CMEC proliferation, angiogenesis, and apoptosis in a rat model of I/R injury with involvement of the Akt signaling pathway.
Results
I/R rats show severe pathological changes in myocardial tissues
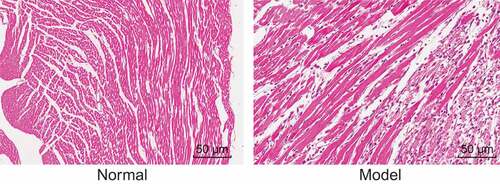
Initially, hematoxylin-eosin (HE) staining was employed in order to observe the pathological changes in myocardial tissues of normal and I/R rats. As shown in , cells in the normal myocardial tissues in the normal group were observed to have a neat arrangement and clear boundaries with complete nucleus and homogeneous cytoplasm, while cells in the myocardial tissues with I/R in the model group were largely disordered, as well as exhibiting necrocytosis, inflammatory cell infiltration and karyopyknosis. Moreover, tissue space was observed to have widened in addition to a great amount of fibrous tissue, and cross-striations were blurry and broken.
Figure 1. I/R rats show severe pathological changes in myocardial tissues (× 400). I/R, ischemia-reperfusion.
CMECs were successfully cultured and identified
When cell confluence reached 85–90% after 7–10 d of primary culture of CMECs, cells exhibited a typical “paving-stone” shape upon observation under an inverted phase microscope (). Cells in subculture were found to be more likely to adhere to the wall within 24 h of culturing compared to the primary cells. In the early stage of growth, cells were observed to be spindle, triangular or polygonal in shape. After 3–4 days, cellular morphology exhibited a “paving-stone” shape, when cell confluence reached 90% (). In addition, immunohistochemistry was applied in order to identify cells using factor VIII, which is the specific label for microvascular endothelial cells (MVECs). Microscopic observation revealed that the cytoplasm of most cells was tan-colored and uniformly distributed, while the nucleus was blue-colored (). These results demonstrated that the cells in primary culture were CMECs, as well as indicating that the positive rate of cells exceeded 95%. Therefore, purified CMECs could be provided for the subsequent experiments.
Figure 2. CMECs were successfully cultured and identified. (a), primary CMECs of rats (7 d after adherence to wall) (× 100); (b), sub-cultured CMECs (× 100); (c), cell identification showing that factor VIII was tan-colored in cytoplasm (× 400). CMECs, cardiac microvascular endothelial cells.

Successful transfection of JAZF1 plasmids into CMECs
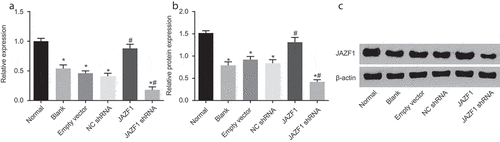
Subsequently, reverse transcription quantitative polymerase chain reaction (RT-qPCR) () and Western blot analysis () were performed to verify the successful transfection of JAZF1 plasmids. Following CMECs transfection, when compared with the normal group, the mRNA and protein levels of JAZF1 were observed to be significantly decreased in the blank, empty vector, negative control (NC) short hairpin RNA (shRNA) and JAZF1 shRNA groups (p < 0.05). However, no significant differences were observed among the blank, empty vector, and NC shRNA groups (p > 0.05). Compared with the empty vector and NC shRNA groups, the mRNA and protein levels of JAZF1 in the JAZF1 group were significantly elevated, whereas opposite trends were observed in the JAZF1 shRNA group (all p < 0.05). On the whole, JAZF1 plasmids were successfully transfected into CMECs.
Figure 3. JAZF1 plasmids were successfully transfected into CMECs. (a), relative mRNA levels of JAZF1 in response to the treatment of JAZF1 or JAZF1 shRNA; (b), relative protein levels of JAZF1 in response to the treatment of JAZF1 or JAZF1 shRNA; (c), the gray value of JAZF1 protein band in response to the treatment of JAZF1 or JAZF1 shRNA; CMECs, cardiac microvascular endothelial cells; NC, negative control; shRNA, short hairpin RNA; JAZF1, the juxtaposed with another zinc finger protein 1; *, p < 0.05 vs. the normal group; #, p < 0.05 vs. the blank, empty vector and NC shRNA group.
Over-expression of JAZF1 elevates levels of vascular endothelial growth factor (VEGF) and B-cell lymphoma-2 (Bcl-2) while reducing that of Bcl-2 associated X protein (Bax), Bcl-2/Bcl-XL-associated death promoter (Bad), caspase-3 and caspase-9 as well as p-Bad/Bad, p-FKHR/FKHR, c-caspase3/t-caspase3, and c-caspase9/t-caspase9
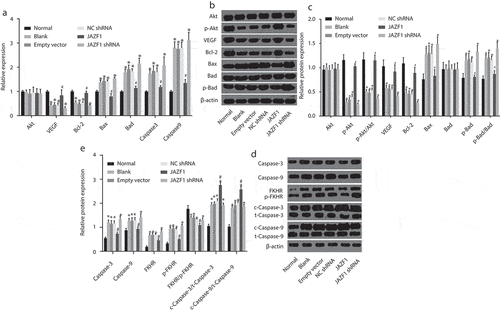
The effect of JAZF1 on mRNA and protein levels of related genes and proteins were investigated by means of RT-qPCR and Western blot analysis (), the results of which revealed that all groups presented with similar mRNA levels of Akt (p > 0.05). Compared with the normal group, the mRNA and protein levels of VEGF and Bcl-2 as well as p-Akt/Akt were significantly lower while the mRNA levels of Bad, the mRNA and protein levels of Bax, caspase-3 and caspase-9 as well as p-Bad/Bad, p-FKHR/FKHR, c-caspase3/t-caspase3, and c-caspase9/t-caspase9 were higher in the blank, empty vector, NC shRNA and JAZF1 shRNA groups (all p < 0.05). Relative to the blank, empty vector and NC shRNA groups, increased mRNA and protein levels of VEGF and Bcl-2 as well as p-Akt/Akt, c-caspase3/t-caspase3 and c-caspase9/t-caspase9 and declined mRNA levels of Bad, mRNA and protein levels of Bax, caspase-3, and caspase-9 as well as p-Bad/Bad and p-FKHR/FKHR were detected in the JAZF1 group (all p < 0.05). However, no significant differences were detected among the blank, empty vector and NC shRNA groups (p > 0.05). These results suggested that over-expression of JAZF1 elevates the mRNA levels of VEGF and Bcl-2 while reducing that of Bax, Bad, caspase-3 and caspase-9. In addition, over-expressed JAZF1 up-regulates the protein levels of VEGF and Bcl-2 while down-regulating Bax, caspase-3, caspase-9, p-Bad/Bad, p-FKHR/FKHR, c-caspase3/t-caspase3 and c-caspase9/t-caspase9.
Figure 4. Over-expressed JAZF1 elevates levels of VEGF and Bcl-2 while reducing that of Bax, Bad, caspase-3 and caspase-9 as well as p-Bad/Bad, p-FKHR/FKHR, c-caspase3/t-caspase3 and c-caspase9/t-caspase9. (a). mRNA expression levels of related genes in cells before and after transfection; (b) & (d), protein bands of related proteins in cells of each group; (c) & (e), protein expression levels of related proteins in cells before and after transfection; NC, negative control; Akt, protein kinase (b); VEGF, vascular endothelial growth factor; Bcl-2, B-cell lymphoma-2; Bax, Bcl-2 associated X protein; Bad, Bcl-2/Bcl-XL-associated death promoter; shRNA, short hairpin RNA; JAZF1, the juxtaposed with another zinc finger protein 1; *, p < 0.05 vs. the normal group; #, p < 0.05 vs. the blank and NC groups.
Over-expression of JAZF1 promotes CMEC proliferation while silencing of JAZF1 inhibits proliferation of CMECs
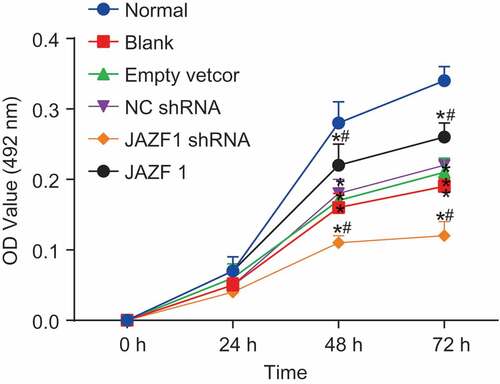
Additionally, the current study performed 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) assay so as to ascertain whether JAZF1 could affect the viability of CMECs. As shown in , MTT assay results demonstrated that cell growth in the blank, empty vector and NC shRNA groups was significantly down-regulated compared with that in the normal group (p < 0.05). In comparison to the blank, empty vector and NC shRNA groups, cells in the JAZF1 group exhibited faster growth rates which was indicated by higher optical density (OD) values noted at 48 h and 72 h (p < 0.05). However, cell growth in the JAZF1 shRNA group was found to be inhibited with no notable increases in regard to the OD values between the 48–72 h time points. Cell viability was not significantly different among the blank, empty vector, and NC shRNA groups (all p > 0.05). These findings suggested that over-expression of JAZF1 promotes CMEC proliferation while silencing of JAZF1 results in inhibition.
Figure 5. Over-expression of JAZF1 promotes while silencing of JAZF1 inhibits cell viability. *, p < 0.05 vs. the normal group; #, p < 0.05 vs. the blank, empty vector and NC shRNA groups; NC, negative control; shRNA, short hairpin RNA.
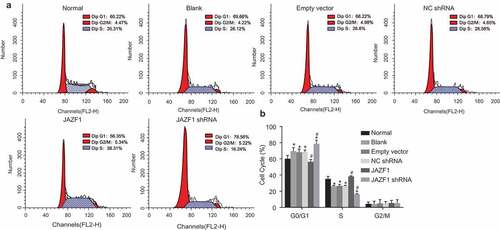
Over-expression of JAZF1 arrests CMECs at S stage and inhibits apoptosis of CMECs while silencing of JAZF1 induces apoptosis
Flow cytometry with propidium iodide (PI) staining was employed to explore the effects of JAZF1 on cell cycle distribution of CMECs (). The results highlighted that in comparison with the normal group (G0/G1: 60.22 ± 4.11%; S: 35.31 ± 3.08%), the number of cells at the G0/G1 stage was significantly higher among the blank, empty vector and NC shRNA groups (G0/G1: blank group: 69.66 ± 4.32%; empty vector: 68.22 ± 3.98%; NC shRNA group: 68.79 ± 2.13%), however, the number of cells at the S stage was significantly lower (S: blank group: 26.12 ± 1.10%; empty vector: 26.80 ± 1.78%; NC shRNA group: 26.56 ± 0.77%) (all p < 0.05), suggesting that proliferation of CMECs was inhibited. Compared with the blank, empty vector, and NC shRNA groups, the JAZF1 group had a fewer number of cells at the G0/G1 stage (56.35 ± 3.12%) and more cells at the S stage (38.31 ± 0.83%), while the JAZF1 shRNA group displayed a greater amount of cells at the G0/G1 stage (78.56 ± 4.54%) and lesser cells at the S stage (16.24 ± 0.75%) (all p < 0.05). There were no significant differences regarding cell cycle distribution of CMECs among the blank, empty vector and NC shRNA groups, while the number of cells at G2/M stage was also not significantly different among each group (all p > 0.05). Taken together, these results suggest that over-expression of JAZF1 arrests CMECs at the S stage while down-regulation brings about opposite results.
Figure 6. More cells are arrested at the S phase after transfected with JAZF1, and few cells are arrested at the S phase after transfected with silenced JAZF1. (a), cell cycle mapping in each group; (b), the percentage of cells in each phase of cell cycle in each group; *, p < 0.05 vs. the normal group; #, p < 0.05 vs. the blank, empty vector and NC shRNA groups; NC, negative control; shRNA, short hairpin RNA; CMECs, cardiac microvascular endothelial cells; JAZF1, the juxtaposed with another zinc finger protein 1.
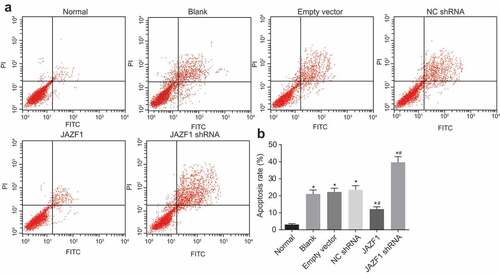
In addition, Annexin V/PI double staining in combination with flow cytometry was employed to explore the effects of JAZF1 on apoptosis of CMECs (). According to the results, the normal group presented with the lowest apoptosis rate of CMECs (3.11 ± 0.55%), while the rates in the blank group, the empty vector group, and the NC shRNA group were 21.07 ± 2.34%, 22.23 ± 2.21% and 23.51 ± 2.42%, respectively, which were all significantly higher than that in the normal group. Compared with the blank group and NC shRNA group, the JAZF1 group presented with decreased rate of apoptosis (12.16 ± 1.35%), while that was increased in the JAZF1 shRNA group (39.64 ± 3.37%) (all p < 0.05). These results indicated that over-expression of JAZF1 inhibited CMEC apoptosis while down-regulation of JAZF1 lead to induction of apoptosis.
Figure 7. Rate of cell apoptosis of CMECs is inhibited after transfection with JAZF1, and the rate is promoted after transfection with silenced JAZF1. (a), cell apoptosis mapping measured by flow cytometry; (b), apoptosis rates in six groups; *, p < 0.05 vs. the normal group; #, p < 0.05 vs. the blank, empty vector and NC shRNA group; NC, negative control; shRNA, short hairpin RNA; CMECs, cardiac microvascular endothelial cells; JAZF1, the juxtaposed with another zinc finger protein 1.
Over-expression of JAZF1 promotes angiogenesis in CMECs
In addition, we assessed the number and length of newly-formed blood vessels in each group in order to determine whether JAZF1 could affect angiogenesis in CMECs. Compared with the number of newly-formed blood vessels in the normal group (17.33 ± 2.08), significantly reduced number of newly-formed blood vessels were noted in the blank (9.33 ± 1.15), empty vector (9.67 ± 1.15) and NC shRNA group (11.00 ± 1.00) (all p < 0.05). However, as illustrated in , a far greater amount of blood vessels were formed in the JAZF1 group (23.33 ± 2.52), while low blood vessels formation was observed in the JAZF1 shRNA group (4.67 ± 1.15) in comparison with the blank, empty vector and NC shRNA groups (all p < 0.05).
Figure 8. Over-expressed JAZF1 promotes angiogenesis and silencing of JAZF1 inhibits angiogenesis in CMECs. (a), formation of blood vessel observed under the inverted microscope (× 100); (b), the number of blood vessel formed in each group; (c), the length of blood vessel formed in each group; *, p < 0.05 vs. the normal group; #, p < 0.05 vs. the blank, empty vector and NC shRNA group; NC, negative control; shRNA, short hairpin RNA; CMECs, cardiac microvascular endothelial cells; JAZF1, the juxtaposed with another zinc finger protein 1.
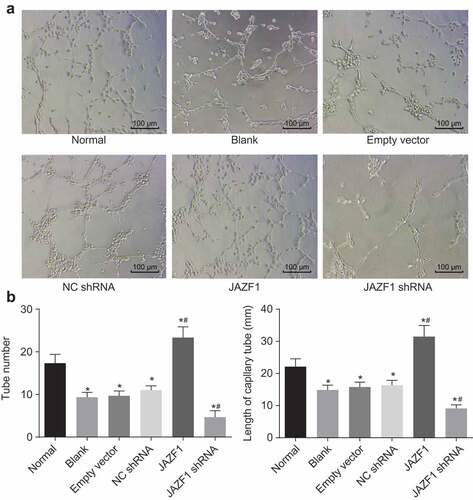
Compared with the length of blood vessel in the normal group (22.14 mm), significantly decreased length of blood vessel was noted in the blank (14.89 mm), empty vector (15.78 mm) and NC shRNA group (16.34 mm) (all p < 0.05). However, as shown in , the length of blood vessels was significantly increased in the JAZF1 group (31.47 mm), while that in the JAZF1 shRNA group was decreased (9.11 mm) in comparison with the blank, empty vector and NC shRNA groups (all p < 0.05).
From the above results, it was concluded that over-expression of JAZF1 promoted angiogenesis in CMECs while silencing JAZF1 leads to inhibited angiogenesis.
Over-expression of JAZF1 increases the expression of PDGF
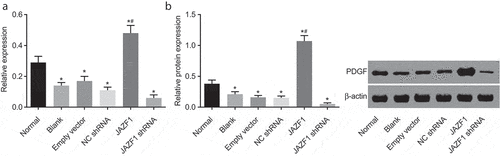
The effect of JAZF1 on PDGF was investigated using RT-qPCR () and Western blot analysis (). The crucial role of PDGF in heart has been widely reported [Citation21,Citation22], and thus we also try to test the expression of PDGF after different transfections. After transfection with JAZF1, decreased mRNA and protein levels of PDGF in CMECs were observed among the blank, empty vector, NC shRNA and JAZF1 shRNA groups, while that in the JAZF1 group were increased (all p< 0.05). No significant differences were detected among the blank, empty vector and NC shRNA groups (p > 0.05). In comparison with the empty vector and NC shRNA groups, the mRNA and protein levels of PDGF were found to be elevated in the JAZF1 group but decreased in the JAZF1 shRNA group (all p< 0.05). Taken together, over-expression of JAZF1 increases the expression of PDGF while JAZF1 down-regulation brought about the opposite results.
Figure 9. Over-expressed JAZF1 increases the expression of PDGF while silencing brings about the opposite results. (a), relative mRNA level of PDGF in CMECs after transfection using RT-qPCR; (b), relative protein level of PDGF in CMECs after transfection detected by western blot analysis; *, p < 0.05 vs. the normal group; #, p < 0.05 vs. the blank, empty vector and NC shRNA group; NC, negative control; shRNA, short hairpin RNA; CMECs, cardiac microvascular endothelial cells; JAZF1, the juxtaposed with another zinc finger protein 1; PDGF, platelet-derived growth factor.
Discussion
Ischemia-reperfusion (I/R) injury, ultimately results in heart failure, and causes grave morbidity and mortality across the world [Citation23]. JAZF1 exerts functions over insulin sensitivity, lipid metabolism, gluconeogenesis as well as inflammation [Citation24]. A previous study revealed that JAZF1 plays an important role in regulating hepatic glucose metabolism and insulin sensitivity by activation of the Akt signaling pathway in the process of glucose homeostasis [Citation20]. In addition, TIP27 was suggested to regulate insulin action via Akt activation, resulting in the suppression of the gluconeogenetic and glycogenolytic genes and ultimately HGP [Citation20]. Furthermore, Lee et al. have supported that Akt phosphorylates zinc finger protein at Thr385, and the phosphorylated zinc finger protein is released from the p53 promoter, resulting in transcriptional activation of p53 [Citation25]. Interestingly, Sussman et al. revealed that activation of Akt exerts protective effects on cardiomyocytes against I/R injury by means of reducing cell apoptosis [Citation26]. However, there is no biological explanation elucidating the true function of JAZF1 on I/R via the Akt signaling pathway. Hence, the current study attempted to investigate the underlying mechanism by which JAZF1 affects I/R via the Akt signaling pathway in rat models. Our findings evidenced that JAZF1 exerts cardioprotective effects through the proliferation of CMECs and angiogenesis and suppressing apoptosis of CMECs through the Akt signaling pathway in rats with I/R.
Another significant finding of the current study was in the form of the observation that CMEC proliferation is enhanced following over-expression of JAZF1, based on the fact that cellular activity was improved in the JAZF1 group and undermined in the JAZF1 shRNA group embodied by lower OD values. A previous study suggested that, JAZF1 functions as a transcriptional repressor of testicular orphan nuclear receptor 4 (TR4), which regulates a number of biological procedures, including human metabolism and growth [Citation16,Citation27]. Furthermore, reduced expression of Bax, Bad, caspase-3, caspase-9, p-Bad/Bad, p-FKHR/FKHR c-caspase3/t-caspase3 and c-caspase9/t-caspase9 and increased expression of p-Akt/Akt, VEGF, Bcl-2 and PDGF were found in cells treated with JAZF1. Our results were largely in line with another recently published review in which the phosphorylation of Akt enhanced cell survival by reducing levels of pro-apoptotic molecules in the Bcl-2 family and caspases as well as increasing levels of anti-apoptotic molecules in the Bcl-2 family [Citation28]. Several reports have further suggested that caspase-3 and −9 are induced after the hindering of wortmannin in the PI3K/Akt pathway, indicating that the PI3K/Akt pathway could potentially regulate caspases [Citation29]. Moreover, inhibition of Akt also induced caspase-3-dependent cell apoptosis in Jurkat T-lymphocytes [Citation30]. The majority of Akt could be activated by α-platelet–derived growth factor receptor (α-PDGFR) signaling, PDGF and its tyrosine kinase receptor including α-PDGFR and β-PDGFR [Citation31]. Strikingly, JAZF1 was previously demonstrated to be a transcriptional repressor of TAK1 [Citation32]. In myogenic differentiation, TAK1 is a crucial signal transducer downstream of TRAF6, while knockdown of TRAF6 compromised the Akt pathways and elicited differentiation defects in mouse myoblasts, while the deliberate activation of Akt pathway could alleviate the aforementioned differentiation defect [Citation33]. Moreover, inhibition of TAK1 accelerates cell apoptosis of AKT-transformed cells in anchorage-independent cell growth accompanying by the down-regulation of Mcl-1 and Bcl-2 expression [Citation34]. Various research literature findings have also verified that Akt is capable of enhancing cell proliferation via the phosphorylation of several proteins that can regulate cell cycle in either a direct or indirect manner [Citation35]. In particular, a study reported that Akt is capable of modulating endothelial cell proliferation by phosphorylating various target proteins, especially angiogenesis related to VEGF [Citation12]. The latter function was also consistent with a key finding of our study, highlighted by the up-regulated levels of JAZF1 and its contribution to the formation of tubes.
As illustrated in , a greater amount of tubes were formed in the JAZF1 group, where the length of blood vessels was increased significantly as well, while opposite trends were observed in the JAZF1 shRNA group. The loss of JAZF1 expression may bring about growth retardation in the infant period of mice and muscle mass reduction in the mature period [Citation36]. A prior study also found that prolonged activation of Akt signaling pathway by stimuli like VEGF in the cardiovascular system may contribute to the formation of tubes of endothelial cells [Citation12,Citation18]. Regarding the current study, we found it reasonable to infer that increased levels of VEGF caused by JAZF1 could activate the Akt signaling pathway so as to promote functioning.
During the current study, an additional crucial finding was characterized by over-expressed JAZF1 and its inhibition of VEC apoptosis. Our findings revealed that over-expression of JAZF1 exerted an influential role on cell cycle distribution of CMECs, which arrested fewer CMECs at the G0/G1 stage and more CMECs at the S stage. In addition, flow cytometry results indicated that the apoptosis rate in the JAZF1 group was much lower than that of other groups, with the exception of the normal group. The homology encoded by JAZF1 is involved in cancer-associated process and expression of JAZF1 in tumor-suppression procedures [Citation37]. In addition, it has been previously demonstrated that the activation of Akt plays a vital role in cell apoptosis through Bax and Bad, both of which belong to Bcl-2 family. For instance, the regulation of Bad phosphorylation can create conditions for inhibition of cell apoptosis inhibition [Citation26]. In line with the aforementioned statement, JAZF1 causes inhibition of both the levels of Bax and Bad in the current study. Similarly, recent findings have indicated that increased expression and phosphorylation of Akt may result in reduced cell apoptosis and enhanced cardiac function [Citation38]. The serine/threonine-specific protein kinase Akt is activated by several myocardial protective ligand-receptor systems such as insulin, insulin-like growth factor-1, and gp130 signaling pathways [Citation39]. One study further reported that cardiac stretching could induce Akt signaling [Citation40]. Activated Akt could advance cell survival by phosphorylating and inactivating glycogen synthase kinase-3β (GSK-3β), the pro-apoptotic Bcl-2 family member BAD, and caspase-9; therefore, it can be inferred that activated Akt can substantially reduce the infarction size of the heart and dramatically improve post-ischemic myocardial functioning [Citation39]. Taken together, the aforementioned findings demonstrate that over-expression of JAZF1 exerts protective effects against I/R in rats through the activation of the Akt signaling pathway. The observation results are also in accord with the research assertions that activated Akt exhibits anti-apoptotic and pro-proliferative effects in numerous types of cells [Citation41].
In conclusion, the current study suggests that JAZF1 promotes CMEC proliferation and angiogenesis while suppressing apoptosis via the Akt signaling pathway in I/R. These findings may open novel avenues for future therapies of I/R. However, the limited sample size and inability to recreate exact human situations serve as limitations to the current study. Therefore, a relative larger sample shall be employed for further study to verify our conclusion in order to raise the quality of life of patients suffering from I/R injury.
Methods and materials
Ethics statement
All animal experiments were approved by the Ethics committee of Yantai Yuhuangding Hospital. In addition, best efforts were made to minimize the suffering of the included animals.
Animal grouping and model establishment
A total of 60 healthy male specific pathogen-free (SPF) Wistar rats (aged between 8 ~ 9 weeks and weighing 200 ± 10 g) were purchased from the Beijing Vital River Laboratory Animal Technology Co., Ltd. (license number: SCXK 2013–0012, Beijing, China). After a week of adaptive feeding, the rats were randomly assigned into normal group (n = 10) and model group (n = 50). Rats in the model group were maintained in a state of anesthesia using an anesthesia machine for small animals (oxygen pressure: 0.1 MPa, oxygen flow: 300 mL/min, concentration of anesthetic drugs: 2%). The 3rd and 4th intercostal space on the left side was opened using a hemostat, and the heart was obtained by pressing down on the thoracic cavity gently. The root of the left anterior descending branch of the coronary artery was ligated using NO.0 sutures, after which lidocaine carbonate was injected onto the surface of heart. The heart was promptly placed back in the thoracic cavity with the expulsion of air. In addition, penicillin (0.1–0.2 mL) was injected as soon as the overlying skin was sutured to avoid microbial interference.
HE staining
Within 24 h of model establishment, all rats were euthanized, and the cardiac tissues were extracted and immediately fixed in 10% formalin. After 24 h, the tissues were dehydrated conventionally using gradient ethanol of 70%, 80%, 90%, 95% and 100% (1 min each time), cleared two times with 500 mL of xylene (X820585, HZ Biotech Co., Ltd., Hefei, Anhui, China) (5 min each time), immersed with wax and embedded with paraffin. The embedded tissue blocks were sectioned into 4 μm slices, with some sections prepared for immunohistochemistry. Next, the sections were dewaxed to the liquid state, stained with hematoxylin (C0007, Banmanbio Co., Ltd., Shanghai, China) at room temperature for 10 min, and quickly washed under running water for 30 s to 60 s. Differentiation was conducted using 1% hydrochloric acid ethanol for 1 min. Following differentiation, the sections were washed under running water for 1 min, stained with eosin at room temperature for 5–10 min, dehydrated using gradient ethanol (1 min each time), cleared two times in xylene (1 min each time), and then mounted with neutral balsam. Subsequently, morphological changes of cardiac tissues were observed and imaged using an optical microscope (XSP-36, Boshida Optical Instrument Co., Ltd., Shenzhen, Guangdong, China).
Culture and identification of CMECs
Within 24 h of model establishment, all rats were euthanized and the infarction sections of the cardiac tissues were extracted. CMECs were isolated and then cultured using the tissue explants adherent method. Next, the rats were anesthetized with an intra-peritoneal injection of pentobarbital sodium at 300 mg/kg and 500 U heparin. CMECs of rats were isolated from the infarcted section of the heart, and periadventitial fat and connective tissues around the vessels were cleared with forceps and iris scissors under a dissecting microscope. Matrigel (BD Biosciences, San Jose, CA, USA) was added into a 24-well plate at the density of 250 µL each well and polymerized at 37℃ for 30 min. The aorta was sliced into 8–10 small pieces, which were then placed with the intima side facing down on the Matrigel in the wells (4–5 pieces in each well). Next, in order to keep the explants moist, a small amount of culture media was added without submerging them. The explants were placed in a humidified incubator with 5% CO2 in air at 37℃, and cells migrated from the aortic segments. The aortic pieces were removed after 5–7 d, and 2 mL of culture medium was added to the wells. When the cells reached confluence, the medium was discarded, and the cells were cultured at 37℃ for 60 ~ 90 min in 2 mL/10 cm2 (100 U) dispase (BD Biosciences, San Jose, CA, USA) to detach the cultured cells from the Matrigel. After that, the culture medium was subsequently added to terminate the detachment by dilution. Following termination, the cells were harvested and centrifuged at 1000 rpm for 5 min with the supernatant decanted, and small samples of the pellet were transferred to a six-well plate. When the cells had grown to confluence, the medium was removed, and the cell pellets obtained were rinsed two times with Ca2+-free phosphate buffered solution (PBS) containing 0.1 mM ethylenediaminetetraacetic acid (EDTA). Cells were passaged after removing PBS. The culture medium (100 mL) was composed of 80 mL of Dulbecco’s modified Eagle’s medium (DMEM; BRL 41965; Gibco, Carlsbad, CA, USA), 10 mL of fetal calf serum (FCS; BRL 10270; Gibco, Carlsbad, CA, USA), 7.5 mg of endothelial cell growth supplement (ECGS; E-2759; Sigma-Aldrich Chemical Company, St Louis, MO, USA), 200 µL of heparin (final concentration of 10 U/mL), 2 mL of penicillin/streptomycin (final concentration of 100 U/mL, BRL 15070; Gibco, Carlsbad, CA, USA), 1 mL of L-glutamine (100 ×, BRL 25030–024; Gibco, Carlsbad, CA, USA) and 1 mL of Minimal Essential Amino acids (100 ×, BRL 11140–035; Gibco, Carlsbad, CA, USA). Addition of ECGS to the culture medium was intended to promote endothelial cell growth, while heparin was employed for the prevention of vascular smooth muscle cells growth. The cells at passage 2 were selected for immunocytochemistry staining. Factor VIII revealed positive antigen, indicating that the cultured cells were CMECs. The CMECs at passage 3–4 were selected for further experimentation. Myocardial tissues were sliced into blocks (1 mm3) using ophthalmic scissors and cultured with 4–5 mL of Dulbecco modified Eagle medium (DMEM) high glucose culture medium (100 mg/mL, 100 μ/mL penicillin) containing 20% fetal bovine serum (FBS), and then placed in a humidified incubator with 5% CO2 in air at 37°C, until cells were adherent to the wall. After 2–3 h, when tissue blocks were in a dry adherent state, the incubator was flipped over to fully immerse the blocks in the medium. A further culture was conducted for 48 h. After 7–10 d of primary culture, subculture was conducted when the cells settled at the bottom of culture bottle in monolayer. The cells at passage 2 were then cryopreserved, and cells at passage 3–4 were all adopted for culture experiments. The antigen was labeled with the primary antibody, specific rabbit-anti-rat antibody to factor VIII, and the corresponding secondary antibody, goat-anti-rabbit antibody to IgG was combined with the primary antibody for color development. Subsequently, an inverted phase contrast microscope was applied for observation purposes.
Cell grouping and transfection
CMECs were grouped into the following six groups: the normal group (normal CMECs, liposome and 20% FBS); the blank group (CMECs obtained from rats in the model group, liposome and Opti-MEM), the empty vector group (CMECs obtained from rats in the model group, NC blank plasmid of PIRES2-GFP, liposome and Opti-MEM), the NC shRNA group (CMECs obtained from rats in the model group, NC blank plasmid of pGenesil-HK, liposome and Opti-MEM), the JAZF1 group (CMECs obtained from rats in the model group, PIRES2-JAZF1 recombinant plasmid, liposome, and Opti-MEM) and the JAZF1 shRNA group (pGenesil-JAZFl-shRNA recombinant plasmid, liposome and Opti-MEM). Three parallel wells were set for each group. Transfection was conducted in accordance with the instructions of the LipofectamineTM 2000 with Opti-MEM mixed with the combination of plasmid and liposome at the ratio of 1: 2.5. Twenty-four hours later, mRNA was extracted accordingly.
Liposome was adopted for transfection according to instructions of Lipofectamine 2000. Firstly, cells at the logarithmic phase of growth were collected and counted. After cell concentration was adjusted to 5 × 105–1.25 × 106 cells/mL in serum-free DMEM, the cells were seeded into a six-well plate (1 mL in total). Secondly, solution A was obtained as follows: 5 µg of plasmid DNA was prepared, and PBS was added to the blank group, and PIRES2-GFP empty plasmid was added in the NC group. PIRES2-JAZF1 recombinant plasmid and pGenesil-JAZFl-shRNA recombinant plasmid + OPTI-MEM (100 µL in total) were used to transfect cells in each group, respectively, following the above-mentioned grouping rules and then allowed to stand for 5 min. Solution B was obtained by mixing 2.5 µL of Lipofectamine 2000 and 97.5 µL of OPTI-MEM. Thirdly, solution B was added to solution A, and the mixture was placed on a clean bench at room temperature for 20 min. Then after, the mixture was added to a six-well plate which was then gently shaken for uniform mixing, followed by incubation for approximately 4–6 h. Finally, the culture solution was replaced with DMEM containing 10% FBS (Gibco, Carlsbad, CA, USA), and cells were cultured at 37°C with 5% CO2 in air. Next, mRNAs were extracted 24 h after transfection and proteins were extracted 48 h after transfection.
RT-qPCR
Total RNA content was extracted using TRIZOL, and the RNA concentration was measured by an ultraviolet spectrophotometer. Reverse transcription was performed using a PrimeScript reverse transcription kit (RR014A, Takara Biotechnology Ltd., Beijing, China). The total reaction system was 10 μL, and reaction conditions were as follows: reverse transcription at 42°C for 15 min and inactivation of reverse transcriptase at 85°C for 2 min. An appropriate amount of complementary DNA (cDNA) was used as template for PCR. The primer sequences () were designed by Primer 5.0 and synthetized by GeneScript Co., Ltd. (Nanjing, Jiangsu, China). Next, RT-qPCR was performed according to instructions of the PCR kit (KR011A1, Beijing Tiangen Biotech Co., Ltd., Beijing, China) with reaction conditions as follows: pre-denaturation at 95°C for 5 min, followed by 30 cycles of denaturation at 95°C for 40 s, 57°C for 40 s and 72°C for 40 s, annealing/extension at 72°C for 10 min and 4°C for 5 min. The reaction system comprised of 10 µL of SYBR Premix Ex TaqTM II, 0.4 µL of PCR Forward Primer (10 µM), 0.4 µL of PCR Reverse Primer (10 µM), 2 µL of DNA template and 7.2 µL of sterile purified water. Expression of target genes, including JAZF1, vascular endothelial growth factor (VEGF), B-cell lymphoma-2 (Bcl-2), Bcl-2 associated X protein (Bax), Bcl-2/Bcl-XL-associated death promoter (Bad), caspase-3 and caspase-9 was calculated using the 2−ΔΔCt method [Citation42] as follows: ΔCt = Ct target gene – Ct reference gene; ΔΔCt = ΔCt experiment group – ΔCt control group. Subsequently, the expression of the target genes in each group was compared and recorded [Citation43,Citation44].
Table 1. Primer sequences for RT-qPCR
Western blot analysis
CMECs were rinsed three times with pre-cooled PBS and lysed with protein lysis buffer on ice for 10 min in order to obtain the protein content. Protein concentration of each sample was determined using a bicinchonininc acid (BCA) kit. After carrying out sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) at 100 V, the proteins were transferred onto nitrocellulose membranes (30 mA, 120 min). The membranes were then blocked with 5% bovine serum albumin (BSA)/Tris-buffered saline with Tween 20 (TBST) for 60 min and then incubated overnight at 4°C with several primary antibodies, namely, rabbit-anti-rat antibodies to β-actin (ab8227, dilution ratio of 1: 1000), JAZF1 (ab199791, dilution ratio of 1: 1000), Akt (ab8805, dilution ratio of 1: 500), p-Akt (ab38449, dilution ratio of 1: 1000), VEGF (ab32152, dilution ratio of 1: 1000), Bax (ab32503, dilution ratio of 1: 1000), Bcl-2 (ab59348, dilution ratio of 1: 1000), caspase-3 (ab13847, dilution ratio of 1: 500), caspase-9 (ab2013, dilution ratio of 1: 1000), c-caspase-3 (ab2302, dilution ratio of 1: 1000), c-caspase-9 (WL01838, dilution ratio of 1: 1000), Bad (ab32445, dilution ratio of 1: 2000), FKHR (ab39670, dilution ratio of 1: 500) and p-FKHR (ab131339, dilution ratio of 1: 500). Next, the membranes were hybridized with the secondary antibody, horseradish peroxidase (HRP) labeled goat-anti-rabbit antibody to IgG diluted at 1: 1000 by TBST at room temperature for 120 min. All aforementioned antibodies were provided by Abcam, Inc. (Cambridge, MA, USA), except c-caspase-9 which was provided by Shenyang Wanlei Biotech Co., Ltd. (Shenyang, Liaoning, China). Following incubation, the membranes were rinsed three times with TBST. Enhanced Chemiluminescence (ECL) kit was used for luminous reaction, and the images were acquired after compression, fixing and observation. The quantity-One software was adopted to detect the gray values of protein bands. The relative protein expression of target genes was calculated using the following formula: gray value target band/gray value internal control.
MTT assay
CMECs at the logarithmic phase of growth were seeded in a 96-well plate at a density of 1 × 104 cells/well, and eight parallel wells were set for each group. When cells reached 70% confluence, each well was added with 10 μL of MTT solution (5 mg/mL, ST316, Beyotime Biotechnology, Shanghai, China) and cultured at 37°C for 4 h. The supernatant was then removed. After PBS rinsing, each well was added with 100 μL of dimethyl sulfoxide (DMSO) (D5879, Sigma-Aldrich Chemical Company, St Louis, MO, USA), and the plate was placed on a shaking table for 10 min. An OD value of 492 nm in each group was measured using a microplate reader (MK3, Thermo Pittsburgh, PA, USA) at numerous time points (0 h, 24 h, 48 h, and 72 h) after incubation, respectively. The cell proliferation rate was calculated as follows: (OD experiment well – OD blank well)/OD blank well. The experiment was performed three times in order to obtain the mean value.
Flow cytometry
The current study employed PI staining (Roche Co., Ltd., Shanghai, China) in order to detect cell cycle composition in CMECs. Forty-eight hour after transfection, CMECs were collected, fixed with 70% iced ethanol and incubated at 4°C overnight, followed by centrifugation and removal of the supernatant. Next, the cells were rinsed two times with PBS containing 1% FBS and re-suspend with 400 μL of binding buffer. Subsequently, 50 μL of RNase (Sigma-Aldrich Chemical Company, St Louis, MO, USA) was added for incubation at 37°C for 30 min, after which 50 μL of PI (50 mg/L, Sigma-Aldrich Chemical Company, St Louis, MO, USA) was added to stain cells in dark conditions for 30 min at room temperature. Following staining, a flow cytometer was used to detect cell cycle composition.
In addition, the Annexin V/PI double staining method was used to determine cell apoptosis in CMECs. After transfection for 48 h, the cells were treated by 0.25% EDTA-free trypsin, collected in a flow tube and centrifuged. After the supernatant was discarded, the cells were then rinsed three times with pre-cold PBS and centrifuged, with the supernatant discarded. Annexin-V-FITC/PI staining solution was obtained by mixing Annexin-V-FITC, PI and N- 2- Hydroxyethylpiperazine-N’-2-Ethane-sulfonic acid (HEPES) buffer at a ratio of 1: 2: 50 according to the instructions of Annexin-V-FITC cell apoptosis detection kit (Roche Co., Ltd., Shanghai, China). Following this, the cells were cultured in the mixture at room temperature for 15 min. Another 1 mL of HEPES buffer was added (PB180325, Procell Life Science & Technology Co., Ltd., Wuhan, Hubei, China) and mixed uniformly with the cells. Subsequently, cell apoptosis was analyzed by means of FITC detection and PI fluorescence through activation of band pass at 525 nm and 620 nm by a wavelength of 488 nm. The experiment was repeated three times to obtain the mean value.
Matrigel angiogenesis assay
Matrigel (Shanghai Shanran Biotech Co., Ltd., Shanghai, China) was placed at 4°C overnight and allowed to melt to a yellow jelly. Next, 70 μL of Matrigel (concentration of 0.5 mmol/L) was added to a pre-cooled 96-well plate using a pre-cooled fin-pipette instantly. The culture plate was incubated at 37°C for 30 min in order to allow solidification. After the cells were detached into the cell suspension, the cell suspension (density of 1 × 105 cells/mL) was inoculated in the culture well paved with gel. Each well was added with the corresponding cell culture medium and then incubated in an incubator for 18 h. A total of three visual fields were randomly selected from each well for micrography at low magnification. Subsequently, the average length and number of blood vessels formed in each visual field were calculated and recorded.
Statistical analysis
Statistical analyses were performed using the SPSS 21.0 software (IBM Corp. Armonk, N.Y., USA). Each experiment was repeated three times to obtain the mean value. Measurement data were expressed as mean ± standard deviation (SD). Comparisons between two groups were conducted by t-test, while comparisons among multiple groups were assessed by one-way analysis of variance (ANOVA). A value of p < 0.05 was considered to be statistically significant.
Acknowledgments
We would like to give our sincere appreciation to the reviewers for their helpful comments on this article.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Koeppen M, Harter PN, Bonney S, et al. Adora2b signaling on bone marrow derived cells dampens myocardial ischemia-reperfusion injury. Anesthesiology. 2012;116:1245–1257.
- Neri M, Fineschi V, Di Paolo M, et al. Cardiac oxidative stress and inflammatory cytokines response after myocardial infarction. Curr Vasc Pharmacol. 2015;13:26–36.
- Cao J, Xie H, Sun Y, et al. Sevoflurane post-conditioning reduces rat myocardial ischemia reperfusion injury through an increase in NOS and a decrease in phopshorylated NHE1 levels. Int J Mol Med. 2015;36:1529–1537.
- Lincoff AM, Roe M, Aylward P, et al. Inhibition of delta-protein kinase C by delcasertib as an adjunct to primary percutaneous coronary intervention for acute anterior ST-segment elevation myocardial infarction: results of the PROTECTION AMI Randomized Controlled Trial. Eur Heart J. 2014;35:2516–2523.
- Lim SY, Davidson SM, Hausenloy DJ, et al. Preconditioning and postconditioning: the essential role of the mitochondrial permeability transition pore. Cardiovasc Res. 2007;75:530–535.
- Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100.
- Gao J, Shen M, Guo X, et al. Proteomic mechanism of myocardial angiogenesis augmented by remote ischemic training of skeletal muscle in rabbit. Cardiovasc Ther. 2011;29:199–210.
- Cui H, Li X, Li N, et al. Induction of autophagy by Tongxinluo through the MEK/ERK pathway protects human cardiac microvascular endothelial cells from hypoxia/reoxygenation injury. J Cardiovasc Pharmacol. 2014;64:180–190.
- Leucker TM, Bienengraeber M, Muravyeva M, et al. Endothelial-cardiomyocyte crosstalk enhances pharmacological cardioprotection. J Mol Cell Cardiol. 2011;51:803–811.
- Liu H, Shang J, Chu F, et al. Protective effects of Shen-Yuan-Dan, a traditional Chinese medicine, against myocardial ischemia/reperfusion injury in vivo and in vitro. Evid Based Complement Alternat Med. 2013;2013:956397.
- Lavu M, Gundewar S, Lefer DJ. Gene therapy for ischemic heart disease. J Mol Cell Cardiol. 2011;50:742–750.
- Chaanine AH, Hajjar RJ. AKT signalling in the failing heart. Eur J Heart Fail. 2011;13:825–829.
- Ku HC, Chen WP, Su MJ. DPP4 deficiency preserves cardiac function via GLP-1 signaling in rats subjected to myocardial ischemia/reperfusion. Naunyn Schmiedebergs Arch Pharmacol. 2011;384:197–207.
- Wang KS, Zuo L, Owusu D. et al. Prostate cancer related JAZF1 gene is associated with schizophrenia. J Schizophr Res. 2014;1:pii:1002.
- Yang M, Dai J, Jia Y, et al. Overexpression of juxtaposed with another zinc finger gene 1 reduces proinflammatory cytokine release via inhibition of stress-activated protein kinases and nuclear factor-kappaB. Febs J. 2014;281:3193–3205.
- Yang H, He J, Xu XL, et al. Molecular characterization and tissue expression profile analysis of the porcine JAZF1 gene. Genet Mol Res. 2015;14:542–551.
- Ming GF, Li X, Yin JY, et al. JAZF1 regulates visfatin expression in adipocytes via PPARalpha and PPARbeta/delta signaling. Metabolism. 2014;63:1012–1021.
- Abeyrathna P, Su Y. The critical role of Akt in cardiovascular function. Vascul Pharmacol. 2015;74:38–48.
- Rasheed MA, Kantoush N, Abd El-Ghaffar N, et al. Expression of JAZF1, ABCC8, KCNJ11and Notch2 genes and vitamin D receptor polymorphisms in type 2 diabetes, and their association with microvascular complications. Ther Adv Endocrinol Metab. 2017;8:97–108.
- Yuan L, Luo X, Zeng M, et al. Transcription factor TIP27 regulates glucose homeostasis and insulin sensitivity in a PI3-kinase/Akt-dependent manner in mice. Int J Obes (Lond). 2015;39:949–958.
- Raica M, Cimpean AM. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals (Basel). 2010;3:572–599.
- Bloomekatz J, Singh R, Prall OW, et al. Platelet-derived growth factor (PDGF) signaling directs cardiomyocyte movement toward the midline during heart tube assembly. Elife. 2017;6.
- Lin L, Yang Z, Zheng G, et al. Analyses of changes in myocardial long non-coding RNA and mRNA profiles after severe hemorrhagic shock and resuscitation via RNA sequencing in a rat model. BMC Mol Biol. 2018;19:11.
- Meng F, Lin Y, Yang M, et al. JAZF1 inhibits adipose tissue macrophages and adipose tissue inflammation in diet-induced diabetic mice. Biomed Res Int. 2018;2018:4507659.
- Lee WP, Lan KH, Li CP, et al. Akt phosphorylates myc-associated zinc finger protein (MAZ), releases P-MAZ from the p53 promoter, and activates p53 transcription. Cancer Lett. 2016;375:9–19.
- Sussman MA, Volkers M, Fischer K, et al. Myocardial AKT: the omnipresent nexus. Physiol Rev. 2011;91:1023–1070.
- Stevens VL, Ahn J, Sun J, et al. HNF1B and JAZF1 genes, diabetes, and prostate cancer risk. Prostate. 2010;70:601–607.
- Jubair S, Li J, Dehlin HM, et al. Substance P induces cardioprotection in ischemia-reperfusion via activation of AKT. Am J Physiol Heart Circ Physiol. 2015;309:H676–H684.
- Liu C, Yang J, Fu W, et al. Coactivation of the PI3K/Akt and ERK signaling pathways in PCB153-induced NF-kappaB activation and caspase inhibition. Toxicol Appl Pharmacol. 2014;277:270–278.
- Uriarte SM, Joshi-Barve S, Song Z, et al. Akt inhibition upregulates FasL, downregulates c-FLIPs and induces caspase-8-dependent cell death in Jurkat T lymphocytes. Cell Death Differ. 2005;12:233–242.
- Dolloff NG, Russell MR, Loizos N, et al. Human bone marrow activates the Akt pathway in metastatic prostate cells through transactivation of the alpha-platelet-derived growth factor receptor. Cancer Res. 2007;67:555–562.
- Ming GF, Xiao D, Gong WJ, et al. JAZF1 can regulate the expression of lipid metabolic genes and inhibit lipid accumulation in adipocytes. Biochem Biophys Res Commun. 2014;445:673–680.
- Xiao F, Wang H, Fu X, et al. TRAF6 promotes myogenic differentiation via the TAK1/p38 mitogen-activated protein kinase and Akt pathways. PLoS One. 2012;7:e34081.
- Qu Y, Zhang L, Ma A, et al. c-MYC overexpression overrides TAK1 dependency in efficient tumorigenicity of AKT-transformed cells. Cancer Lett. 2013;336:290–298.
- New DC, Wu K, Kwok AW, et al. G protein-coupled receptor-induced Akt activity in cellular proliferation and apoptosis. FEBS J. 2007;274:6025–6036.
- Langberg KA, Ma L, Sharma NK, et al. Single nucleotide polymorphisms in JAZF1 and BCL11A gene are nominally associated with type 2 diabetes in African-American families from the GENNID study. J Hum Genet. 2012;57:57–61.
- Koontz JI, Soreng AL, Nucci M, et al. Frequent fusion of the JAZF1 and JJAZ1 genes in endometrial stromal tumors. Proc Natl Acad Sci U S A. 2001;98:6348–6353.
- Tao L, Bei Y, Zhang H, et al. Exercise for the heart: signaling pathways. Oncotarget. 2015;6:20773–20784.
- Kim CH, Hao J, Ahn HY, et al. Activation of Akt/protein kinase B mediates the protective effects of mechanical stretching against myocardial ischemia-reperfusion injury. J Vet Sci. 2012;13:235–244.
- Skurk C, Izumiya Y, Maatz H, et al. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J Biol Chem. 2005;280:20814–20823.
- Li L, Zhao D, Jin Z, et al. Phosphodiesterase 5a inhibition with adenoviral short hairpin RNA benefits infarcted heart partially through activation of akt signaling pathway and reduction of inflammatory cytokines. PLoS One. 2015;10:e0145766.
- Ayuk SM, Abrahamse H, Houreld NN. The role of photobiomodulation on gene expression of cell adhesion molecules in diabetic wounded fibroblasts in vitro. J Photochem Photobiol B. 2016;161:368–374.
- Yao Y, Suo AL, Li ZF, et al. MicroRNA profiling of human gastric cancer. Mol Med Rep. 2009;2:963–970.
- Fang Y, Chen H, Hu Y, et al. Burkholderia pseudomallei-derived miR-3473 enhances NF-kappaB via targeting TRAF3 and is associated with different inflammatory responses compared to Burkholderia thailandensis in murine macrophages. BMC Microbiol. 2016;16:283.