
ABSTRACT
Chromosomal instability (CIN) is defined as a high rate of whole chromosome loss or gain and is a hallmark of many aneuploid solid tumors. CIN positively correlates with poor patient prognosis and chemotherapeutic resistance. Despite this clinical importance, the role of CIN in tumor initiation, growth and/or progression remains poorly understood. To date, the only strategies developed to determine how CIN contributes to tumorigenesis have relied on transgenic mouse models that deliberately increase the rate of chromosomal mis-segregation. Here we develop a strain of transgenic mice that is designed to strategically decrease the rate of chromosome mis-segregation and suppress CIN. These animals modestly overexpress the kinesin-13 microtubule depolymerase Kif2b, a strategy proven successful in restoring faithful chromosome segregation to human cancer cells in culture. Using the LA2 K-Ras G12D-induced model for lung cancer, we show that Kif2b expression reduces the number of chromosome segregation defects but does not change the incidence of lung tumor lesions. However, pulmonary tumors were significantly larger in animals expressing Kif2b and those tumors exhibited elevated rates of Ki-67 positive cells relative to controls. Thus, in lung cancers driven by mutations in K-Ras, CIN has little impact on tumor initiation but suppresses tumor growth. These data support a model in which CIN imposes a burden on tumor cells, and that enhancement of mitotic fidelity results in accelerated tumor growth.
KEYWORDS:
Introduction
The frequent mis-segregation of whole chromosomes, a phenomenon called chromosomal instability (CIN), is a hallmark of most solid tumors [Citation1]. The consequence of CIN is aneuploidy, a state in which a cell’s chromosome count deviates from normal euploidy [Citation2,Citation3]. A growing body of evidence correlates CIN and aneuploidy with poor patient prognosis [Citation4], chemotherapy resistance [Citation5], and advanced stage cancer [Citation6]. Although CIN’s correlation with tumor aggressiveness is well established, its role in tumorigenesis is still poorly understood. Because of this connection, aneuploidy was hypothesized to act as a primary driver of tumor formation [Citation7]. Furthermore, because CIN ultimately leads to karyotypic heterogeneity, it is believed to provide tumors with superior adaptability that drives drug resistance and cancer progression [Citation8].
In principle, CIN and aneuploidy can be caused by defects in multiple events during mitosis in cancer cells. Defects in the spindle assembly checkpoint (SAC), cohesion, cell cycle regulation, centrosome number and importantly kinetochore-microtubule attachment dynamics have all been shown to drive CIN in cell culture [Citation9]. Many of these form the basis for the development of mouse models for CIN. However, in human cancer cells, the most common mitotic defect causing CIN is the persistence of errors in the attachment of spindle microtubules to chromosomes at kinetochores [Citation10,Citation11]. CIN cancer cell lines frequently exhibit hyperstable kinetochore-microtubule (kMT) attachments when compared to stable diploid control cells [Citation12]. This diminishes the cells ability to correct erroneous kMT attachments and manifests as a high rate of mis-segregation prone lagging chromatids at anaphase [Citation12,Citation13]. Importantly, when the mitotically active kinesin-13 protein Kif2b is overexpressed in cancer cells in culture, its potent MT depolymerase activity destabilizes kMT attachments and suppresses CIN to restore faithful chromosome segregation [Citation11,Citation13,Citation14]. Thus, overexpression of Kif2b provides an attractive strategy to suppress CIN in a physiologically relevant tumor environment.
Here, we develop a novel mouse model overexpressing Kif2b to study the effect of CIN suppression on tumorigenesis. This is the first mouse model designed to directly target the most common cause of CIN in cancer cells and to deliberately decrease the rate of chromosome mis-segregation. Using this strategy, we investigate the role of CIN in lung tumors driven by mutant K-Ras. We focused on lung cancer because there is an unmet clinical need to develop effective therapeutics for lung tumors that possess mutations in K-Ras [Citation15]. Moreover, lung adenoma is a common outcome in mouse models where CIN has been deliberately induced [Citation16] suggesting that changes in karyotype is an important step in the transformation of lung epithelial cells.
Materials and methods
Plasmids and targeting construct
Plasmid encoding 6xHis-tagged full-length human Kif2b was a gift of L. Wordeman (University of Washington, Seattle, WA). The pBTG (pBigT-IRES-GFP; DM#268) vector which contains a neomycin resistance marker and a STOP cassette (three consecutive polyadenylation signals for mRNA synthesis termination) both flanked by two LoxP sites (LSL), an internal ribosome entry site (IRES), and downstream enhanced green fluorescent protein (eGFP) was a gift from Douglas Melton (Addgene plasmid # 15037). The pRosa26Pam1 modified Roas26 targeting vector which contains both 5ʹ and 3ʹ Rosa26 targeting arms and the PGK-DTA negative selection marker was a gift from Philippe Soriano (Addgene plasmid # 21714) [Citation17]. 6xHis-tagged Kif2b cDNA was amplified by PCR using forward (5ʹ-TATATGCTAGCGCCACCATGGCCAGCCAGTTC-3ʹ) and reverse (5ʹ-CGCGCCTCGAGTTAATGGTGATGGTGGTGATGCTC-3ʹ) containing NheI/XhoI restriction sites and the Kozak consensus sequence. The NheI/XhoI cut fragment was inserted into the pBTG vector to generate the LSL-Kif2b-6xHis-IRES-eGFP intermediate construct. AscI and PacI digested intermediate construct was then inserted into the corresponding site of pRosa26Pam1 to generate the final Rosa26-LSL-Kif2b-6xHis-IRES-eGFP targeting vector. All constructs were confirmed by DNA sequencing (Dartmouth Molecular Biology Core Facility, Hanover NH).
Transgenic animals
Gene targeted transgenic mice conditionally overexpressing 6x-His-tagged human Kif2b protein were generated according to standard techniques [Citation18]. Briefly, the Rosa26-LSL-Kif2b-6xHis-IRES-eGFP targeting vector was linearized with MluI restriction enzyme and electroporated into 129/Sv ES cells. Targeted ES cells were grown under selection in G418 and colonies were isolated and expanded. G418 resistant ES cell clones were screened for recombination by a PCR genotyping strategy (see below). One positive ES clone was selected for injection into C57BL/6 (B6) blastocysts which were then implanted into a pseudopregnant B6D2.F1 female. Resulting chimeric mice were mated to wild type B6 mice to obtain Rosa26-LSL-Kif2b-6xHis-IRES-eGFP (R26-LSL-Kif2b) mice. To remove the STOP cassette sequence constitutionally, R26-LSL-Kif2b mice were crossed with homozygous B6.C-Tg(CMV-cre)1Cgn/J (CMV-Cre) (The Jackson Laboratory, Stock # 006054) and resulting R26-LSL-Kif2b; CMV-Cre (R26-Kif2b) mice were screened by PCR for STOP cassette excision. A forward primer (5ʹ-TCCCAAAGTCGCTCTGAGTT-3ʹ) and reverse primer (5ʹ-CCTCTTGGATGTCTCCGAAA-3ʹ) flanking the STOP cassette amplified ~0.5 kb and ~3.2 kb fragments in STOP cassette excised and unexcised conditions respectively. B6.129S-Krastm3Tyj/Nci (K-rasLA2) [Citation19] mice were obtained from the NCI Mouse Repository (Frederick, MD). The experiments presented here included male and female animals are approximately 50/50 ratio throughout, and gender was not used as an independent variable. All animal study protocols were approved by the Geisel School of Medicine at Dartmouth Institutional Animal Care and Use Committee (IACUC).
Mouse genotyping
Genomic DNA for genotyping was extracted from mouse tail or ES cells using the Purgene® Core Kit A (Qiagen) according to manufacturer’s instructions. All PCR reactions were performed with OneTaq® 2X Master Mix with GC Buffer (New England BioLabs Inc.). Mice with the R26LSL-Kif2b allele were genotyped using one forward (5ʹ-TCCCAAAGTCGCTCTGAGTT-3ʹ) and two reverse (5ʹ-GCCAGAGGCCACTTGTGTAG-3ʹ and 5ʹ-CTTTAAGCCTGCCCAGAAGA-3ʹ) primers spanning the recombination junction in the same PCR reaction. 242 bp and 538 bp amplicons were amplified for the wild-type and targeted Rosa26 locus respectively. Mice were genotyped for the R26Kif2b using one forward (5ʹ-TCCCAAAGTCGCTCTGAGTT-3ʹ) and one reverse primer (5ʹ-CCTCTTGGATGTCTCCGAAA-3ʹ). K-rasLA2 mice were genotyped according the established NCI PCR protocol (NCI Mouse Repository website).
MEF cell culture
Pairs of R26Kif2b/+ and heterozygous K-RasG12D/+ mice were crossed to obtain individual wild type, single-mutant, and double-mutant MEF cell lines. Pairs of R26LSL-Kif2b/+ mice were mated to generate inducible overexpression Kif2b MEF cell lines. All MEF lines were derived from individual E12.5 – E14.5 mouse embryos dissected from timed pregnant females. After removing head (retained for DNA extraction and genotyping) and internal organs, each dissected embryo was washed in sterile PBS. Embryo bodies were then suspended in 2 mL of 0.25% trypsin each and forced through a 5 mL syringe with an 18-gauge needle. The homogenate was incubated for 20 min at 37°C, passed through the syringe again, and then plated in a 100 cm tissue culture dish coated with 0.2% gelatin in PBS. MEFs were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% FBS (Hyclone), 50U/mL penicillin (Mediatech) and 50µg/mL streptomycin (Mediatech) at 37°C in a humidified atmosphere with 5% CO2 for no more than 4 passages.
Immunoblotting
MEF lysates were prepared by solubilizing cells directly in 1X Laemmli buffer and boiling for 10 min. For inducible human Kif2b expression, MEFs were incubated over-night with adenovirus (multiplicity of infection (MOI) of 500) containing a CMV-Cre expression vector (Viral Vector Core Facility, University of Iowa) prior to solubilizing. Mouse tissue lysates were prepared from either fresh or liquid nitrogen snap-frozen tissue samples. For lysis, 5 mg of tissue was suspended in ice cold 1X RIPA buffer with general protease inhibitor cocktail (Sigma Aldrich) and homogenized by bead-beating with 1.0 mm diameter silica beads (BioSpec Products Inc.). Homogenates were incubated on a rotator at 4°C and then centrifuged at 16,000 rcf at 4°C for 20 min. Total protein concentration was measured with Pierce™ BCA Protein Assay Kit (Life Technologies) and samples were diluted and boiled for 10 min in 4X Laemmli buffer (final concentration 1X). SDS-PAGE and Western-blot analysis was performed as previously described [Citation14].
MEF karyotype analysis
For karyotype analysis, MEF cells were first treated with 150 ng/mL nocodazole for 4 hours then washed twice with PBS. Cells were then fixed to glass slides as above. Metaphase spreads were probed using a pan-centromeric α-satellite FISH probe (Empire Genomics) and counter-stained with DAPI according to manufacturer’s instructions. Chromosomes were counted in a blinded fashion with the researcher unaware of the condition.
Surface lung tumor analysis
R26Kif2b/+ mice were crossed with KrasG12D/+ mice and F1 6 week old progeny were analyzed. Mice were sacrificed and whole lungs were removed with all lobes intact. Prior to dissection, lungs were re-inflated by tracheal instillation of 10% neutral buffered formalin (NBF, Polysciences, Inc.) immediately followed by closing off the trachea to maintain pressure. Inflated lungs were then submerged in 10% NBF prior to trimming and examination. Surface pulmonary lesions were quantified manually with a dissection microscope. Images of entire lung lobes were acquired with a 5.0 Megapixel color camera (SPOT Imaging Solutions) mounted on a Nikon SMZ 1000 dissection microscope (Nikon Instruments) with a 0.5x, Plan Apo objective. Tumor surface area measurements were performed using SPOT Basic 5.1 software (SPOT Imaging Solutions).
Histopathology, fluorescent immunohistochemistry and FFPE FISH
Mouse lungs were formalin fixed, paraffin-embedded (FFPE), sectioned and stained with hematoxylin and eosin (H&E). Entire H&E stained sections were imaged in bright a SCN400 slide scanner (Leica Microsystems) and analyzed with SlidePath Gateway Client software (Leica Microsystems). For immunohistochemistry, unstained sections were deparaffinized, rehydrated with graded ethanol, and subjected to heat-induced antigen retrieval in 0.01 M citrate buffer (pH 6.0). A fluorophore-conjugated secondary antibody was used for fluorescent detection. Sections were counter-stained with DAPI and mounted using ProLong® Gold antifade reagent (Molecular Probes). Antibodies used are listed below.
For FISH on FFPE lung tissue, unstained sections were deparaffinized, rehydrated with graded ethanol and then treated with 0.01 M citrate buffer (pH 6.0) with 0.05% Tween 20 for 2 hours at 80°C. Next, sections were digested in 0.025% pepsin in 0.01M HCl (pH 2.0) for 30 min at 37°C then dehydrated with graded ethanol. Pre-treated sections were then probed for a unique segment on chromosome 2 (D2Mit74) a using Mouse IDetect™ point FISH probe (Empire Genomics) and counter-stained with DAPI according to manufacturer’s instructions.
Antibodies
The following antibodies and concentrations were used for fluorescent immunohistochemistry (F-IHC) and immunoblotting (IB): Kif2b [Citation14] (IB at 1:1000), Ki-67 (AbCam; F-IHC at 1:1000), α-Tubulin DM1α (Sigma Aldrich; IB at 1:10,000). Secondary antibodies used were Alexa Fluor® 568 (F-IHC at 1:2000) and horseradish peroxidase (Bio-Rad; IB at 1:3000-5000).
Fluorescent microscopy
Images of FISH probed metaphase chromosome spreads and immunohistochemical stained sections were acquired with a cooled charge-coupled device camera (Andor Technology) mounted on a Nikon Eclipse Ti microscope (Nikon Instruments) with a 60x, 1.4 numerical aperture objective. Image series in the z-axis were obtained using 0.2-μm optical sections. Image deconvolution (3D adaptive blind) and contrast enhancement was performed using AutoQuant X3 (Media Cybernetics), Elements software (Nikon) and Adobe Photoshop. Manual image analysis was performed using Nikon Elements. All displayed fluorescent images are maximum intensity projections of deconvolved Z-series stacks.
Statistical analysis
All statistical analyses were performed as indicated in figure legends using Graphpad Prism 5 software.
Results
Generation of conditional overexpression Kif2b mice
To determine how suppression of CIN affects tumorigenesis and progression in vivo, we generated a strain of transgenic mice that conditionally overexpress human Kif2b. These mice were generated through homologous recombination of the human Kif2b cDNA into the mouse Rosa26 (R26) locus ()). The Rosa26 locus is a well characterized safe gene harbor which allows for moderate ubiquitous expression of transgenes in all mouse tissues driven by the endogenous promoter [Citation17]. To ensure conditional expression, a STOP cassette (triple SV40 polyadenylation sequence) was inserted between the endogenous Rosa26 promoter and the transgenic Kif2b cDNA sequence. A neomycin resistance gene was included for selection and these sequences were flanked by loxP sequences (LSL) to allow for Cre-recombinase-mediated excision of the STOP cassette ()).
Figure 1. Cre induced expression of transgenic human Kif2b in mice. (a). pBigT-IRES-GFP (pBTG) & pROSA26PAm1: Design of construct for targeting the Cre inducible human Kif2b cDNA to the endogenous mouse Rosa26 locus (See Methods for details). Rosa26 WT locus (R26+), Targeted locus (R26LSL-Kif2b) & + Cre Recombinase (R26Kif2b): Schematic of the Rosa26 alleles used in this study for conditional expression of human Kif2b. White arrow heads indicate LoxP sites. Numbered arrows represent genotyping primer hybridization sites on the endogenous (black) and recombined (red) Rosa26 locus. Kif2b, 6xHis-tagged human Kif2b cDNA; PGK-Neo, neomycin selection cassette; 3x STOP, transcriptional and translational stop cassette; IRES, internal ribosomal entry site; PGK-DTA, diphtheria toxin gene negative selection cassette. (b). Representative genotyping PCR using genomic DNA from mice with the indicated genotypes and primers 1, 2, & 3 (see A). Amplicons of 242 bp and 538 bp are produced from the R26+ and R26LSL-Kif2b alleles respectively. (c). PCR demonstrating Cre mediated recombination using genomic DNA derived from control and Ad-Cre infected MEFs carrying the R26LSL-Kif2b allele. Primers 1 & 4 (from A) were used. Amplicons of ~3200 bp and 500 bp are produced from the R26LSL-Kif2b & R26Kif2b alleles respectively. (d). Immunoblots of control MEFs and MEFs infected with Ad-Cre using an anti-Kif2b antibody. Anti-Tubulin (DM1α) antibody was used as a loading control. 2 independent lines of MEFs carrying the R26LSL-Kif2b allele are shown.
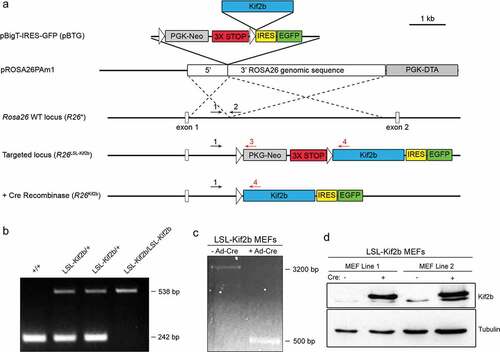
The linear DNA targeting construct containing the human Kif2b cDNA was introduced into mouse ES cells and neomycin resistant clones with the appropriate homologous recombination were selected by a PCR strategy (Supplementary Fig. S1A,B). A positive clone was expanded, injected into a mouse blastocyst and implanted into a pseudo-pregnant female using standard techniques (see materials and methods). The resulting chimeric animals were bred to wild type C57BL/6 mice and an independent transgenic strain was obtained. To confirm transmission of the recombined Rosa26 locus, F1 heterozygous mice were crossed and the resulting progeny were genotyped by PCR. The F2 progeny displayed the expected frequency of genotypes that were wild type for Rosa26 (R26+/+), heterozygous for the Kif2b transgene (R26LSL-Kif2b/+) and homozygous for the Kif2b transgene (R26LSL-Kif2b/LSL-Kif2b) ()).
To verify the conditional expression of transgenic Kif2b we isolated primary murine embryonic fibroblasts (MEFs) from R26LSL-Kif2b/+ embryos. We infected MEFs with adenovirus carrying Cre-recombinase driven by the CMV promoter. PCR using primers flanking the STOP cassette confirmed recombination and excision upon infection with this adenovirus that expresses Cre-recombinase ()). Immunoblot analysis of whole cell extract derived from MEFs infected with the andenovirus carrying Cre-recombinase (Ad-Cre) verifies robust expression of transgenic Kif2b protein ()). We are not able to define the level of transgenic Kif2b protein expression compared to endogenous Kif2b protein expression because this antibody has not been validated against the murine protein. These results demonstrate that we have generated a transgenic mouse strain with human Kif2b integrated into the endogenous Rosa26 locus, and that the promoter from the endogenous Rosa26 gene drives transgenic Kif2b expression upon STOP cassette excision by Cre recombinase.
Kif2b overexpression suppresses chromosomal instability in murine cells
Next we tested if expression of human Kif2b altered the frequency of chromosome mis-segregation in MEFs. Previous studies have shown that constitutive Ras signaling is sufficient to induce chromosome mis-segregation in both cancer cells and MEFs [Citation1,Citation20]. Thus, we generated MEFs from mice expressing mutant oncogenic K-Ras (G12D) with and without elevated levels of Kif2b. We bred mice carrying the LoxP flanked STOP Kif2b transgene to B6.C-Tg(CMV-cre)1Cgn/J (CMV-Cre) mice. CMV-Cre mice constitutively and ubiquitously express transgenic Cre recombinase driven by the CMV promoter from a randomly integrated construct on the X chromosome ()). The resulting progeny efficiently removed the STOP cassette (Supplementary Fig. S2A) and expressed transgenic Kif2b protein in all tissues tested (Supplementary Fig. S2B). The expression of transgenic Kif2b from the Rosa26 locus was sustained in the absence of exogenous Cre after the STOP cassette was excised. Mice were fertile and maintained as heterozygotes (R26Kif2b/+) or homozygotes (R26Kif2b/Kif2b) for the STOP excised allele. We crossed a R26Kif2b/+ mouse with a K-RasLA2 mouse ()) and derived individual fibroblasts from E14.5 embryos. K-RasLA2 (From here on referred to as K-RasG12D) mice conditionally express a latent oncogenic K-RasG12D allele that activates with stochastic intra-chromosomal homologous recombination [Citation19]. PCR analysis confirmed the expected genotypes of the resulting MEF lines () & Supplementary Fig. S3A). Furthermore, MEFs containing transgenic Kif2b expressed the protein at increased levels (Supplementary Fig. S3B).
Figure 2. Expression of human Kif2b reduces the frequency of aneuploidy in murine cells. (a). Breeding scheme to generate MEF cell lines and mice expressing Cre inducible exogenous human Kif2b and the latent K-RasLA2 mutant G12D allele (K-RasG12D). (b). Representative image of metaphase chromosome spreads from MEFs with the indicated genotypes after 4 hours of nocodazole treatment. A pan centromeric FISH probe was used to visualize centromeres (red) and DAPI was used to stain DNA (blue). Scale bar, 10 μm. (c). Percentage of aneuploid metaphases (chromosome count ≠ 40) from MEF cell lines each derived from embryos of the indicated genotype. n ≥ 60 metaphases per genotype; ***, P < 0.001, Fisher’s exact test. (d). Frequency of karyotypes containing the indicated number of chromosomes in MEFs of the genotypes from C.
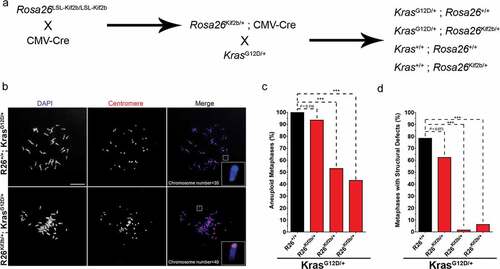
We determined the effect of Kif2b expression on chromosome mis-segregation by examining metaphase spreads from fibroblasts derived from wild type, transgenic R26Kif2b/+, transgenic K-RasG12D/+ and double transgenic R26Kif2b/+; K-RasG12D/+ embryos. Importantly, these MEFs were passaged under normoxic conditions in serum-containing media. These conditions are known to induce MEF populations to rapidly become chromosomally unstable and accumulate aneuploidies before ultimately senescing [Citation20,Citation21]. A FISH probe specific for all mouse centromeric regions was used to probe the metaphase spreads to assist in the quantification of chromosomes per karyotype ()). The combination of DAPI staining and centromeric FISH probe allowed us to quantify both whole chromosome ()) and structural aneuploidies (), inset). Compared to passage and litter matched wild type MEFs, karyotypes examined from the R26+/+; K-RasG12D/+ MEFs were significantly more aneuploid (66.7% vs. 100.0%, p < 0.001) (,d)) indicating that G12D mutant K-Ras acts synergistically with normoxia and serum shock to further drive increased chromosome mis-segregation. Conversely, expression of exogenous Kif2b in fibroblasts derived from double transgenic R26Kif2b/+; K-RasG12D/+ embryos reduced the frequency of aneuploidy (62.5%) to levels comparable to those observed in WT MEFs (,d)). The same trend was observed for MEFs derived from fibroblast lines derived from multiple embryos, although there was variability in the effect size and statistical significance among the different lines. Notably, expression of exogenous Kif2b, independently of mutant K-Ras, in R26Kif2b/+ MEFs did reduce the frequency of aneuploidy caused by normoxia and serum shock compared to wild type MEFs (61.3% vs. 66.7%, respectively) (,d)), though this trend did not reach significance. Taken together, these results show that stable expression of human Kif2b is tolerated in mice and that it reduces the frequency of chromosome mis-segregation induced in cells in response to expression of mutant oncogenic Ras.
Increased Kif2b expression synergizes with oncogenic K-ras to enhance tumor growth
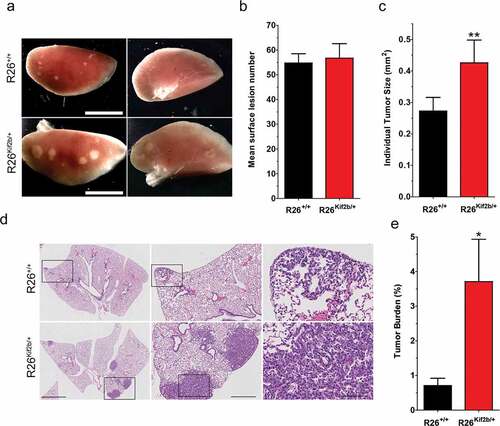
Studies in transgenic animals engineered to elevate the rate of chromosome mis-segregation have shown that CIN can either enhance or suppress tumorigenesis and/or progression depending on the tissue context [Citation16,Citation22,Citation23]. To assess the impact of suppression of CIN on tumor initiation and progression we measured the tumor burden on mice carrying the human Kif2b transgene (R26Kif2b/+) and the stochastically activated oncogenic allele of K-Ras (K-RasLA−2). We measured lung tumor burden at 6 weeks of age as previous data showed that adult mice harboring the latent K-RasLA−2 develop multiple lung adenomas with complete penetrance at this age [Citation19].
Quantification of visible surface lung lesions revealed no significant difference in the number of lung tumors in mice expressing human Kif2b compared to control animals (mean of 56.7 vs 54.8, tumors per mice; p = 0.78) (,b)). In contrast, the lung lesion surface area was 59.3% larger in animals expressing human Kif2b. Tumors from control R26+/+; K-RasG12D/+ mice had a mean size of 0.27 mm2 while tumors in R26Kif2b/+; K-RasG12D/+ animals had a mean size of 0.43 mm2 (p < 0.05) (,c)). Analysis of single histological sections of each lobe of the lung per mouse revealed a similar outcome. Analysis of H&E stained lung FFPE sections showed that the tumor burden increased from 0.71% in control mice to 3.71% (p < 0.05) in Kif2b overexpressing mice (,e)). Although the tumors were consistently larger in R26Kif2b/+; K-RasG12D/+ mice relative to control animals, none of the tumors had progressed beyond the adenoma stage. Additionally, none of the wild type (n = 13) or R26Kif2b/+ (n = 12) mice inspected harbored any visible lung lesions at 6 weeks of age indicating that expression of trangenic Kif2b alone is not sufficient to drive tumorigenesis in the lung.
Figure 3. Expression of exogenous Kif2b accelerates mutant K-Ras driven adenoma growth in mice. (a). Representative images of the anterior (left) and posterior (right) surfaces of left lung lobes isolated from control (R26+/+) or Kif2b expressing (R26Kif2b/+) 6 week old K-RasG12D/+ mice. Scale bars, 5mm. (b). The mean number of visible surface lung lesions per K-RasG12D/+ mouse. Bars, mean ± SEM; n ≥ 10 mice per genotype. (c). The mean surface area (mm2) of lung lesions from K-RasG12D/+ mice. Bars, mean ± SEM; n ≥ 100 lesions from 7 mice per genotype; **, P < 0.01, Mann-Whitney test. (d). Representative hematoxylin and eosin (H&E) stained lung tissue sections from K-RasG12D/+ mice. Black boxes indicate magnified region shown in panel directly to the right. Scale bars (left to right), 2 mm, 0.5 mm, and 0.1 mm. (e). Mean tumor burden percentage (tumor tissue area: total lung tissue area) in H&E stained lung sections from K-RasG12D/+ mice. Bars, mean ± SEM; n = 4 mice per genotype; *, P < 0.05, Mann-Whitney test.
Next, we measured parameters of lung tumor cell turnover by quantification of proliferative and apoptotic cells in the tumors from the different strains of mice. In line with the differences in overall tumor size, we observed a significant (~2-fold) increase in proliferating tumor cells in the adenomas from R26Kif2b/+; K-RasG12D/+ mice compared to control R26+/+; K-RasG12D/+ mice (8.8% vs 4.6% respectively, p < 0.05) based upon staining for the proliferation marker Ki-67 (,b)). TUNEL analysis revealed very few apoptotic cells in tumors from both control and human Kif2b expressing mice and tumors in mice expressing human Kif2b displayed fewer apoptotic cells relative to control mice, but this trend did not reach statistical significance ()).
Figure 4. Suppression of chromosomal instability enhances cell proliferation in lung adenomas. (a). Representative fluorescent immunohistochemistry images of lung adenoma tissue sections stained for Ki67 (red) and DNA (blue) from 6 week old control (R26+/+) and Kif2b expressing (R26Kif2b/+) K-RasG12D/+ mice. Bar, 5 μm. (b). Mean percentage of Ki67 (Ki67+) positive cells per tumor section from K-RasG12D/+ mice of indicated Rosa26 genotype. Bars, mean ± SEM; n indicates number of tumor sections scored from ≥ 3 mice per genotype (> 300 cells scored per tumor section); *, P < 0.05, Student’s t-test. (c). Quantification of apoptotic cells per lung tumor section from K-RasG12D/+ mice of indicated Rosa26 genotype as scored via TUNEL. Bars, mean ± SEM n indicates number of tumor sections scored from ≥ 4 mice per genotype (> 300 cells scored per tumor section). (d). Representative fluorescent images of lung adenoma tissue sections probed for chromosome 2 (red) and stained with DAPI to visualize DNA (blue) from 6 week old control (R26+/+) and Kif2b expressing (R26Kif2b/+) K-RasG12D/+ mice. Bar, 5 μm. (e). Percentage of lung tumor cells aneuploid for chromosome 2 (copy number ≠ 2) in control (R26+/+) and Kif2b expressing (R26Kif2b/+) tissue sections from K-RasG12D/+ mice. Copy number was judged by a FISH point probe for the D2Mit74 (98 cM) on mouse chromosome 2. Bars, mean; n > 1400 nuclei from 6 tumor sections (from 2 mice) per genotype; *, P < 0.05, χ2 test. (f). Conceptual model demonstrating enhanced proliferation of adenoma cells upon reduction of chromosome segregation error rate. Single diploid cells are depicted containing 3 pairs of chromosomes (blue, black, and white). Stochastic activation of the mutant K-Ras G12D allele (lightning bolt) transforms control (left, white fill) and Kif2b expressing (right, gray fill) cells resulting in hyperplasia and adenoma formation. Chromosome mis-segregation events result in aneuploidy (yellow fill) which can burden cells with delayed proliferation (hashed lines), senescence, or apoptosis. In control mice, high rates of mis-segregation result in small adenomas with a high percentage of aneuploid cells (bottom left). However, in Kif2b expressing mice, decreased karyotypic heterogeneity reduces the growth burden imposed by aneuploidy and results in large adenomas (bottom right).
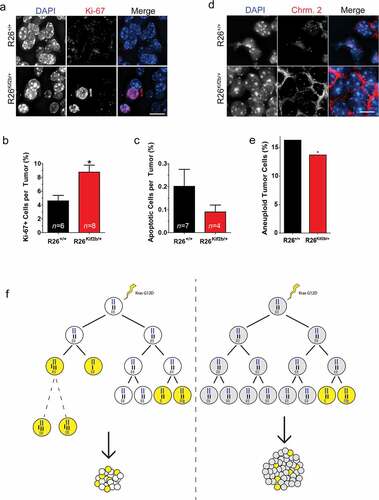
Finally, we measured the impact of human Kif2b expression on the chromosome constitution of tumor cells in vivo using tissue FISH. Using a probe specific to chromosome 2 we quantified the copy number of this chromosome in tumor cells in the adenomas from R26Kif2b/+; K-RasG12D/+ mice compared to control R26+/+; K-RasG12D/+ mice (,e)). We observed a significant decrease in the fraction of aneuploid cells for chromosome 2 in the adenomas of R26Kif2b/+; K-RasG12D/+ mice relative to control animals. Assuming an equivalent effect on all chromosomes, these data indicate that that human Kif2b expression reduces the frequency of aneuploidy in lung adenoma by approximately 24-fold. Taken together, these data demonstrate that expression of human Kif2b effectively suppresses CIN in mouse lung tumors consistent with previous data from human cell culture experiments. The consequence of this suppression of CIN is that although the initiation of K-Ras-induced tumors is unchanged, the growth rate of the tumors accelerates without progressing to malignant adenocarcinomas.
Discussion
Here we introduce a novel mouse model engineered to improve mitotic fidelity by directly targeting the machinery responsible for the most common cause of CIN in human cancer cells. We demonstrate that transgenic human Kif2b can be constitutively expressed at moderate levels in all mouse tissues with no apparent effects on development and fertility. Similar to previous observations in human cell culture experiments, increased expression of Kif2b increases the fidelity of chromosome segregation in murine fibroblasts (under normoxic and serum shock conditions) and reduces the frequency of aneuploidy in mouse lung tumors. Strikingly, there is a significant increase in lung tumor size in animals where Kif2b expression suppresses CIN.
These data fit a model where the ongoing karyotypic change driven by CIN is burdensome to tumor cells and retards their growth ()). This concept that CIN is burdensome is speculative based upon only the data shown here, but is well supported by accumulating data published elsewhere. Suppression of CIN alleviates that burden and supports more robust tumor cell growth leading to larger overall tumor size without changing the rate of tumor initiation. Multiple lines of evidence show that in otherwise diploid cells, chromosome mis-segregation causes apoptosis, senescence, and slowed proliferation unless other permissive mutations are acquired [Citation24–Citation26]. It is unlikely that cells in these early adenomas have acquired those additional mutations making their growth rate sensitive to karyotype changes. Furthermore, because these tumors are in the early stage of progression, they are relatively small and do not encounter the same selection pressures that larger, more malignant tumors face as resources become limited. In the absence of those selective pressures, the genetic landscape created by CIN does not provide a platform for an evolutionary advantage. Therefore, chromosome mis-segregation results in the accumulation of aneuploid cells of lowered viability which do not contribute to the overall growth of the tumor. Expression of human Kif2b reduces the frequency of a cell becoming aneuploid to alleviate that burden. This provides the tumor with more actively dividing cells which increase its overall size ()). This is supported by our data showing that Kif2b reduces the fraction of aneuploid cells in MEFs (under normoxic and serum shock conditions) and lung tumors, although we can’t formally rule out that expression of Kif2b also influences other cellular properties such as the invasiveness of tumor cells [Citation27].
These data add to the growing list of mouse models that have been generated to study the relationship of CIN and cancer in vivo. All previous models have been engineered to increase the rate of chromosome mis-segregation by disrupting the mitotic machinery that contributes to faithful chromosome segregation [Citation28,Citation29]. In contrast, the model developed here is designed to suppress CIN in vivo. The pattern emerging from these mouse models is complex. Many models show that elevating chromosome mis-segregation rates induces spontaneous tumor formation in some tissues, but the incidence is often relatively low, occurs late in life, and dependent on tissue context [Citation23,Citation30–Citation32]. Elevating chromosome mis-segregation rates has also been shown to exacerbate carcinogen-induced tumorigenesis and reduce tumor latency suggesting that CIN can cooperate with additional mutations to drive malignancy [Citation33,Citation34]. In contrast, some studies have revealed a tumor suppressive role for CIN [Citation22,Citation23,Citation35,Citation36]. For example, mice heterozygous for CENP-E develop spontaneous lung and spleen tumors and exhibit an increase in aneuploidy. However, in p19/ARF knockout mice that predominately develop lymphomas and sarcomas, an increase of overall tumor-free survival is observed in combination with CENP-E heterozygosity [Citation23]. Similar results have been observed in CIN prone mice that are haploinsufficient for Aurora B [Citation36] or Cdh1 [Citation35]. Additionally, a recent study has demonstrated that enhancing microtubule assembly rates suppresses chromosomal instability and accelerates tumor growth in a xenograft model [Citation37]. To explain these findings, it has been proposed that a high rate of chromosome mis-segregation might be tumor suppressive due to proliferation defects as karyotype diversity exceeds a tolerable threshold [Citation22,Citation23,Citation38,Citation39]. This is supported by the observation of reduced viability in aneuploid strains of budding yeast [Citation26] and mammalian cells [Citation24,Citation25], and is consistent with the data shown here where CIN suppresses lung tumor growth induced by mutant K-Ras.
Taken together, it will be important to discern how the specific molecular design of each mouse model impacts on the rates of tumor initiation vs growth and progression. With respect to tumors of the lung, it is possible that the induction of CIN makes a more significant impact on tumor initiation as evidenced by the increase in lung adenomas in many models that elevate CIN, and that the suppression of CIN makes a more significant impact on tumor growth and/or progression and shown here. The new mouse model described here will aid in the detailed comparison of the effects of induction versus suppression of CIN. Just as certain cancer subtypes are more responsive to some chemotherapeutic agents, the success of targeting CIN therapeutically will depend on multiple factors including the tissue of origin, and whether CIN makes the larger contribution to tumor initiation and/or growth in that tissue.
Supplemental Material
Download Zip (1.5 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
The supplementary material for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837.
- Geigl JB, Obenauf AC, Schwarzbraun T, et al. Defining “chromosomal instability”. Trends Genet. 2008;24:64–69.
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627.
- Bakhoum SF, Danilova OV, Kaur P, et al. Chromosomal instability substantiates poor prognosis in patients with diffuse large B-cell lymphoma. Clin Cancer Res. 2011;17:7704–7711.
- Lee AJX, Endesfelder D, Rowan AJ, et al. Chromosomal instability confers intrinsic multi-drug resistance. Cancer Res. 2011;71:1858–1870.
- Walther A, Houlston R, Tomlinson I. Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut. 2008;57:941–950.
- Boveri T. Concerning the origin of malignant tumours by theodor boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008;121:1–84.
- Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313.
- Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010;20:R285–R295.
- Thompson SL, Compton DA. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol. 2008;180:665.
- Godek KM, Kabeche L, Compton DA. Regulation of kinetochore-microtubule attachments through homeostatic control during mitosis. Nat Rev Mol Cell Biol. 2015;16:57–64.
- Bakhoum SF, Genovese G, Compton DA. Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr Biol. 2009;19:1937–1942.
- Bakhoum SF, Thompson SL, Manning AL, et al. Genome stability is ensured by temporal control of kinetochore–microtubule dynamics. Nat Cell Biol. 2008;11:27–35.
- Hood EA, Kettenbach AN, Gerber SA, et al. Plk1 regulates the kinesin-13 protein Kif2b to promote faithful chromosome segregation. Mol Biol Cell. 2012;23:2264–2274.
- Zhang J, Park D, Shin DM, et al. Targeting KRAS-mutant non-small cell lung cancer: challenges and opportunities. Acta Biochim Biophys Sin (Shanghai). 2016 Jan;48(1):11–16. Epub 2015 Nov 17. doi:10.1093/abbs/gmv118.
- Ricke RM, van Ree JH, van Deursen JM. Whole chromosome instability and cancer: a complex relationship. Trends Genet. 2008;24:457–466.
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71.
- Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet. 2005;6:507–512.
- Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116.
- Woo RA, Poon R. Activated oncogenes promote and cooperate with chromosomal instability for neoplastic transformation. Genes Dev. 2004;18:1317–1330.
- Busuttil RA, Rubio M, Dollé MET, et al. Oxygen accelerates the accumulation of mutations during the senescence and immortalization of murine cells in culture. Aging Cell. 2003;2:287–294.
- Silk AD, Zasadil LM, Holland AJ, et al. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci. 2013;110:E4134–E4141.
- Weaver BA, Silk AD, Montagna C, et al. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36.
- Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol. 2010;188:369.
- Williams BR, Prabhu VR, Hunter KE, et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–709.
- Torres EM, Sokolsky T, Tucker CM, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–924.
- Zaganjor E, Osborne JK, Weil LM, et al. Ras regulates kinesin 13 family members to control cell migration pathways in transformed human bronchial epithelial cells. Oncogene. 2014;33:5457–5466.
- Schvartzman J-M, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer. 2010;10:102–115.
- Simon JE, Bakker B, Foijer F. CINcere modelling: what have mouse models for chromosome instability taught us? In: Ghadimi BM, Ried T, editors. Chromosomal instability in cancer cells. Recent Results Cancer Res. 2015;200:39–60. doi:10.1007/978-3-319-20291-4_2.
- Baker DJ, Jin F, Jeganathan KB, et al. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell. 2009;16:475–486.
- Diaz-Rodríguez E, Sotillo R, Schvartzman J-M, et al. Hec1 overexpression hyperactivates the mitotic checkpoint and induces tumor formation in vivo. Proc Natl Acad Sci. 2008;105:16719–16724.
- Sotillo R, Hernando E, Díaz-Rodríguez E, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23.
- Iwanaga Y, Chi Y-H, Miyazato A, et al. Heterozygous deletion of mitotic arrest–deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res. 2007;67:160–166.
- Babu JR, Jeganathan KB, Baker DJ, et al. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol. 2003;160:341–353.
- García-Higuera I, Manchado E, Dubus P, et al. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol. 2008;10:802–811.
- Fernández-Miranda G, Trakala M, Martín J, et al. Genetic disruption of aurora B uncovers an essential role for aurora C during early mammalian development. Development. 2011;138:2661–2672.
- Ertych N, Stolz A, Stenzinger A, et al. Increased microtubule assembly rates influence chromosomal instability in colorectal cancer cells. Nat Cell Biol. 2014;16:779–791.
- Godek KM, Venere M, Wu Q, et al. Chromosomal instability affects the tumorigenicity of glioblastoma tumor-initiating cells. Cancer Discov. 2016;6:532–545.
- Janssen A, Kops GJPL, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci. 2009;106:19108–19113.