
ABSTRACT
The nuclear receptor (FXR) plays essential roles in maintaining bile acid and lipid homeostasis by regulating diverse target genes. And its agonists were promising agents for treating various liver diseases. Nevertheless, the potential side effect of chronic FXR activation by specific agonists is not fully understood. In this study, we investigated the mechanism of FXR agonist WAY-362450 induced liver enlargement during treating liver diseases. We demonstrated that chronic ingestion of WAY-362450 induced liver hypertrophy instead of hyperplasia in mouse. Global transcriptional pattern was also examined in mouse livers after treatment with WAY-362450 by RNA-seq assay. Through GO and KEGG enrichment analyses, we demonstrated that the expression of Cyclin D1 (Ccnd1) among the cell cycle-regulating genes was notably increased in WAY-362450-treated mouse liver. Activation of FXR-induced Ccnd1 expression in hepatocyte in a time-dependent manner in vivo and in vitro. Through bioinformatics analysis and ChIP assay, we identified FXR as a direct transcriptional activator of Ccnd1 through binding to a potential enhancer, which was specifically active in livers. We also found active histone acetylation was essential for Ccnd1 induction by FXR. Thus, our study indicated that activation of FXR-induced harmless liver hypertrophy with spatiotemporal modulation of Ccnd1. With a better understanding of the mechanism of tissue-specific gene regulation by FXR, it is beneficial for development and appropriate application of its specific agonist in preventing hepatic diseases.
Introduction
The farnesoid X receptor (FXR or NR1H4) is a member of the nuclear receptor superfamily, which is constituted by ligand-activated transcription factors and involved in the modulation of various fundamental biological processes [Citation1,Citation2]. FXR could be activated by both endogenous ligands (including cholic acid (CA) and chenodeoxycholic acid (CDCA)) and synthetic exogenous agonist with high specificity (such as INT-747, GW4064, and WAY-362450) [Citation3–Citation5]. Studies demonstrated that active FXR played fundamental roles in maintaining bile acid and lipid homeostasis [Citation6,Citation7] through strictly regulating the expression of its target genes, such as small heterodimer partner (SHP; NR0B2), cholesterol 7α-hydroxylase (CYP7A1), sterol 12α-hydroxylase (CYP8B1), and bile salt export pump (BSEP; ABCB11) [Citation7–Citation9]. FXR regulates target gene expression through binding to FXR response element (FXRE) within the promoter and enhancer regions [Citation10].
FXR is abundantly expressed in liver, gastrointestinal tract, kidney, and adrenal gland. Unlike other organs, adult liver maintains a unique ability to regrowth/regenerate in response to various injuries primarily by hepatocyte duplication, which is tightly controlled by multiple pathways [Citation11,Citation12]. The essential role of FXR in supporting liver regeneration after partial hepatectomy has been highlighted [Citation13–Citation15]. Nevertheless, it was reported that feeding with excessive bile acid, including CA and CDCA, induces FXR-dependent liver growth by hepatocyte hyperplasia or by hypertrophy, respectively [Citation16]. Liver hypertrophy is also regarded as an adaptation of hepatocytes to increased metabolic demand against toxic drug induced-liver injury [Citation17,Citation18] and is also linked to liver regeneration after partial hepatectomy [Citation19].
Previous studies have characterized FXR as a potential target for the treatment of liver diseases and metabolic disorders [Citation20–Citation23]. In our previous work, we also showed that the FXR agonist WAY-362450 was effective in preventing alcoholic liver disease [Citation21] and intrahepatic cholestasis [Citation24]. However, we observed an unresolved phenomenon that liver size was obviously larger despite recovery from liver injury in WAY-362450 treated mice. Whether chronic ingestion of FXR agonist WAY-362450 induced liver enlargement by potential harmful hyperplasia or by hypertrophy was still unclear. In the present study, we identified that activation of FXR by WAY-362450 promoted liver growth by hypertrophy instead of hyperplasia with direct targeting Ccnd1 through binding to a tissue-specific enhancer.
Materials and methods
Animals and treatments
Wild-type (WT) C57BL/6 mice obtained from Shanghai SLAC laboratory animal Co., Ltd. (Shanghai, China) and FXR knockout (FXR-KO) mice from Jackson Laboratories (Bar Harbor, ME) were maintained in cages with a 12:12 h light-dark cycle. The animal experiments were approved by the Ethics Committee of the International Peace Maternity and Child Health Hospital. All experiments were performed in accordance with ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines (http://www.nc3rs.org.uk/arrive-guidelines) and relevant institutional regulations. All mice received humane care and had free access to water and food. Mouse models for estrogen-induced cholestasis were conducted as described in our previous reports [Citation25]. Briefly, female mice were subcutaneously injected with 5 mg/kg 17a-ethynylestradiol (Sigma-Aldrich Inc., St. Louis, MO) for four weeks. The synthetic agonist of FXR (WAY-362450, 30 mg/kg body weight) from Selleck Chemicals (Houston, TX) was administered to mice via gavage once a day for up to four weeks. At sacrifice, mice were anesthetized with sodium pentobarbital (75 mg/kg, ip). Liver, intestine and kidney tissues were harvested.
RNA isolation and reverse transcription-quantitative PCR (RT-qPCR)
Isolation of total RNA from tissues and cells was conducted using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instruction. RNA was reverse transcribed using the PrimeScript RT kit (Takara Biotech, Dalian, China). To detect gene expression, semi-quantitative PCR was performed using Quantitect SYBR Green PCR kit (Qiagen, Hilden, Germany) on the StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA). Primers for qPCR assays were included in supplementary material, Table S1. Relative mRNA expression of target genes was calculated using the 2−∆∆Ct method with normalization to β-Actin.
mRNA-seq assay and bioinformatics analysis
Library preparation and high throughput sequencing were conducted as described previously [Citation26]. Briefly, purified RNA was subjected to cDNA libraries construction using the KAPA Stranded RNA-Seq Library Preparation Kit for Illumina Platforms (KAPA biosystems) following the manufacturer’s protocol. After purification and quantification, the prepared libraries were subjected to high throughput sequencing on an Illumina HiSeq Xten platforms. Sequential quality control procedures were included. The data were analyzed on the free online platform of Majorbio I-Sanger Cloud Platform (http://www.i-sanger.com). Heatmap was plotted using the OmicShare tools (http://www.omicshare.com/tools). Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotations of DEGs were applied using the DAVID Bioinformatics Resources (https://david.ncifcrf.gov/). The RNA-seq raw data were deposited to the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra/SRP165945).
Public datasets
Released public data, including ChIP-seq and DNase-seq data on mouse tissues and HepG2 cells, were downloaded from ENCODE (https://www.encodeproject.org/) and GEO database. Accession numbers of these data were included in supplementary material, Table S2. These data were reanalyzed and visualized using IGV 2.4 [Citation27] (http://software.broadinstitute.org/software/igv/).
Western blotting assay
Western blotting assays were conducted as previously described [Citation24]. In brief, tissues and cells were lysed in a RIPA lysis buffer (Beyotime Institute of Biotechnology, Jiangsu, China) with protease inhibitors. Total proteins were separated by SDS-PAGE gels and transferred PVDF membranes (Roche Applied Science). The membranes were incubated with primary antibodies (listed in supplementary material, Table S3) following incubation with HRP-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA). The blots were visualized using an enhanced chemiluminescence (ECL) kit (Tiangen Biotech, Beijing, China) on the ImageQuant LAS 4000 mini (GE Healthcare, Piscataway, NJ).
Immunohistochemistry and immunofluorescence assay
Immunohistochemistry (IHC) and immunofluorescence (IF) assays in the liver tissues were performed as described previously [Citation24]. In brief, tissue sections were subjected to deparaffinization in xylene and antigen retrieval by boiling in EDTA buffer. Anti-Cyclin D1 antibody (Santa Cruz, Santa Cruz, CA), anti-phospho-histone-H3 (Ser10), a mouse IHC-specific anti-Ki-67 antibody (Cell Signaling Technology) and anti-pan-cadherin (Cell Signaling Technology, Danvers, MA) were applied as primary antibodies, which were incubated overnight at 4°C. For IHC, the tissue sections were incubated with the horseradish peroxidase polymer conjugate (Invitrogen, Carlsbad, CA) and developed with diaminobenzidine chromogen. For IF, Alexa Fluor 488-conjugated Goat anti-rabbit IgG (Invitrogen) was used as the secondary antibody, and the nucleus was stained with DAPI. Images were captured using a Leica fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
Chromatin immunoprecipitation (ChIP) assay
ChIP assays with mouse liver tissue using SimpleChIP Plus Enzymatic Chromatin IP Kit were performed according to the manufacturer’s protocol (Cell Signaling Technology, Danvers, MA). Briefly, liver tissues were subjected to cross-linking with 1% formaldehyde solution, nuclei preparation and treatment with micrococcal nuclease to digest genomic DNA to a length of approximately 150–900 bp. Furthermore, the digested cross-linked chromatin samples were incubated with the ChIP-grade FXR antibody (Santa Cruz Biotechnology), H3K4me1 and H3K27ac (Cell Signaling Technology) or normal rabbit IgG for immunoprecipitation. After elution and DNA purification, real-time PCR was used to assess target DNA content obtained from immunoprecipitation assay. The results of the ChIP assay in each sample were shown as a proportion of the total input.
Determination of nuclear DNA content in hepatocytes
Nuclear DNA content in mouse hepatocytes was quantified according to a previous report [Citation16]. Briefly, liver sections were deparaffinized, rehydrated and stained with 1 μg/ml Hoechst 33342 (Yeasen Biotech, Shanghai, China). Fluorescent images were acquired under 400x magnifications with a Leica fluorescence microscope (DM2500, Leica Microsystems, Wetzlar, Germany). The fluorescence within the hepatocyte nucleus was identified in images and quantified by CellProfiler Analyst 1.0 software [Citation28].
Cell culture and transfection
HEK293T, HepG2, and Hep3B cells were obtained from the cell bank of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). HEK293T and HepG2 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS, Gibco, Grand Island, NY) and antibiotics (100 U/ml penicillin and 0.1 mg/ml streptomycin) at 37°C in 5% CO2. Hep3B cells were maintained in Eagle’s Minimum Essential Medium (EMEM, Invitrogen) supplemented with 10% FBS, 0.11 mg/ml sodium pyruvate and antibiotics. Transient transfection using Lipofectamine 2000 reagent (Invitrogen) was applied according to the manufacturer’s protocol.
Plasmid construction and dual-luciferase assay
Full-length coding regions of FXR and RXRα cDNA were cloned from mouse liver cDNA library and placed into pcDNA3.1 and pcDNA3.0 vectors (Invitrogen, Carlsbad, CA), respectively. DNA fragments containing mouse Ccnd1 FXRE region (or FXRE region with deletion of putative IR1) were cloned into the pGL3-promoter reporter vector (Promega Corporation, Madison, WI), resulting in pGL3-Ccnd1-FXRE and pGL3-Ccnd1-FXRE-ΔIR1. One copy of the IR1-type FXR response element (FXRE) from mouse Nr0b2 gene was cloned into the pGL3-promoter vector (pGL3-Nr0b2-FXRE). The putative IR1-type FXRE (CGGTGATTCACCC) from mouse Ccnd1 gene was also cloned into the pGL3-promoter vector (pGL3-Ccnd1-IR1). All the constructs were confirmed by DNA sequencing.
For the dual-luciferase reporter assay, HEK293T cells transfected with pcDNA3.1-FXR, pcDNA3.0-RXRa, pRL-CMV (Promega) and corresponding pGL3-promoter constructs were treated with FXR agonist for next 24 h and subjected to Firefly and Renilla luciferase activity assays. The signals of dual luminescence were recorded in a SpectraMax M3 microplate reader. The relative activity of firefly luciferase was determined by normalizing to Renilla luciferase signals. All experiments were conducted in triplicate.
Mouse primary hepatocyte isolation and treatment
Mouse primary hepatocytes were isolated using an in situ perfusion collagenase IV method as described previously [Citation29]. Hepatocytes were cultured in 1:1 mixture of DMEM/Ham’s F-12 medium (Invitrogen, Carlsbad, CA) supplemented with 10% FBS. Primary hepatocytes were treated with 2 μM GW4064 (Sigma-aldrich) or 2 μM WAY-362450 with or without (+)-JQ1 (MedChem Express, Monmouth Junction, NJ) or C646 (MedChem Express) for several hours prior to gene expression analysis.
Cell growth and cell cycle assays
Cell growth and cell cycle assays were applied as previously described [Citation30]. Briefly, cells were treated with vehicle or FXR agonist for indicated time periods. For cell growth assay, cells were incubated with the Cell Counting Kit-8 (CCK-8) (Dojindo, Kamimashiki-gun Kumamoto, Japan) reagent for 1 h and subject to OD450 measurement with a SpectraMax M3 microplate reader (Molecular Devices, Sunnyvale, CA). For cell cycle assay, cells were fixed in ice-cold 70% ethanol before staining with propidium iodide/RNase solution (BD Biosciences, Rockville, MD) and subjected to DNA content determination under a flow cytometer (Beckman Coulter Inc., Miami, FL).
Statistical analysis
Data are shown as means ± SEM. Statistical analysis of numerical data was performed using the GraphPad Prism 5 (GraphPad Software, San Diego, CA) by two-tailed Student’s t-test and one-way ANOVA followed by Tukey’s post hoc test for parametric test or Mann–Whitney test for non-parametric test. Fisher’s exact test and chi-square test using SPSS 16.0 (SPSS Inc., Chicago, IL) were performed for the analysis of categorical data. Statistical significance was considered as p < 0.05.
Results
Chronic ingestion of WAY-362450 induced liver enlargement by hepatocyte hypertrophy in mouse
Previously, we found that administration of WAY-362450 protected against excessive estrogen-mediated cholestasis and hepatotoxicity by lowering bile acid level and induction of antioxidant response in female mouse [Citation24]. To our surprise, we also found liver size in WAY-362450-treated mouse was noticeably larger than that in vehicle- or estrogen (EE2) alone- treated mice, although liver injury was resolved (). As shown, the ratio of liver weight and body weight was increased in wild-type (p < 0.001) but not in FXR-deficient mouse upon WAY-362450 treatment ()). Moreover, WAY-362450 treatment led to cellular and nuclear size increment in hepatocytes. The number of Ki-67 positive hepatocytes was also increased ()). Subsequently, we also investigated the effect of WAY-362450 on normal quiescent liver. We demonstrated that liver weight, as well as liver/body weight ratio, was slightly increased in male mice treated with WAY-362450 for one-week ()). Analysis of liver sections revealed that the number of hepatocytes per microscopic field was noticeably reduced with cell size increment in WAY-362450-treated mice ()). There were slight increases in Ki67 (Mki67 gene) expression ()) and the number of Ki-67-positive hepatocytes ()). In addition, gavage with WAY-362450 increased nuclear DNA content hepatocytes (), p < 0.001) as demonstrated by quantitative analysis of Hoechst 33342-stained hepatocyte nuclei. However, the number of mitotic cells (as indicated by positive phospho-histone H3 (serine10) immunostaining [Citation31], )) was not obviously changed in WAY-362450-treated mouse. These results suggested that chronic ingestion of WAY-362450 induced FXR-dependent liver hypertrophy in mouse liver.
Figure 1. WAY-362450 promoted liver growth by induction of hepatocyte hypertrophy in mouse. (a-b) Wild type (WT) or FXR deficient (FXR-KO) mice (female; 8–10-week-old) treated with 17α-ethynylestradiol (EE2; 5 mg/kg, s.c.) were simultaneously gavaged with (EE2-WAY) or without (EE2-Veh) WAY-362450 for four weeks. (a) The ratios of liver/body weight in each group were shown; (b) Representative images were shown for staining with H&E (upper panels) or immunohistochemistry of Ki67 (lower panels) in WT liver tissues (Original magnification 200 x, bar: 100 μm). (C-H) WT mice (male; 8 weeks old) were administrated with either vehicle (Veh) or WAY-362450 (WAY) once daily for one-week. (c) The liver weight and ratios of liver/body weight in vehicle (Veh) or WAY-362450 (WAY)-treated mice were shown; (d) Plasma membrane of hepatocytes was immuno-stained by pan-cadherin (red), and the nucleus was stained by DAPI (blue). Original magnification 200 x, bar: 25 μm. The number of hepatocytes per microscope field. (e) The expression of Mki67 and Pcna in liver tissues was determined by RT-qPCR assay. (f) Representative images are shown for immunohistochemistry staining of Ki-67 in liver tissues from vehicle- or WAY-362,450-treated mice; Original magnification 200 x, bar: 100 μm for upper panel. The proportions of Ki67-positive hepatocytes in livers of vehicle or WAY-362,450 treated mice were shown in the right panel. (g) Nuclear DNA content in hepatocytes was semi-quantified in vehicle and WAY-362,450- treated mice; 1,500 nuclei were considered in hepatocytes. (h) The proportions of mitotic figure (as indicated by phospho-histone-H3 (Ser10) positive hepatocytes) in livers of vehicle or WAY-362450 treated mice were shown. (Data are represented as mean ± SEM; *p < 0.05, ***p < 0.001, ns: not significant (p > 0.05)).
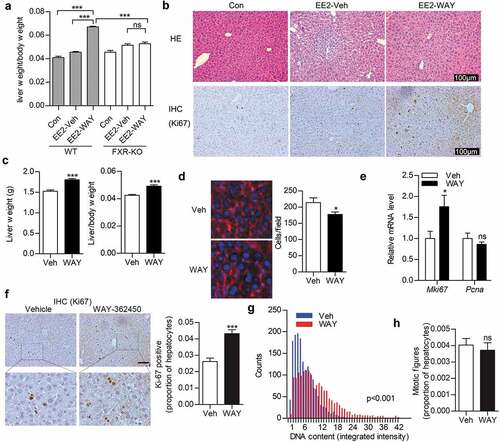
Activation of FXR by WAY-362450 markedly induced Ccnd1 expression in mouse liver
To explore the underlying mechanism for FXR-induced liver hypertrophy, we examined global gene expression pattern by RNA-seq in mouse livers after WAY-362450 treatment. We found that 346 genes were upregulated and 246 genes were downregulated among a total of 592 differentially expressed genes (DEGs; false discovery rate<0.05, |fold change|≥2) in WAY-362450-treated mouse compared with and vehicle-treated control (). By gene ontology ()) and KEGG pathway ()) enrichment analyses of DEGs, we found that the cell cycle pathway was also significantly enriched in WAY-362450-treated mouse liver. Moreover, we showed that Ccnd1 was the most significantly up-regulated and the first-ranked gene among the cell cycle-regulating genes ()). Subsequently, we applied RT-qPCR assay to verify the RNA-seq result. As shown, FXR was effectively activated by WAY-362450 as demonstrated by significantly upregulated well-established target genes (Nr0b2 and Abcb11) and downregulated Cyp7a1 ()). It was also validated that the mRNA level of Ccnd1 among cell cycle genes was significantly upregulated (p < 0.001) by WAY-362450 in the liver ()). We also tested the expression of Hhex (also known as HEX) and Foxm1 (also known as FOXM1b), which were previously reported as FXR-target genes in liver growth regulation [Citation14,Citation32]. Whereas Hhex was downregulated (p < 0.001), the expression of Foxm1 was not significantly changed (p > 0.05) in WAY-362450-treated mouse livers (supplementary material, Figure S1). Thus, activation of FXR by WAY-362450 could especially enhance Ccnd1 expression during induction of hepatocyte hypertrophy in mouse liver in vivo.
Figure 2. Activation of FXR by WAY-362450 markedly induced Ccnd1 expression in mouse liver. Male mice (8 weeks old) were administrated with either vehicle (Veh) or WAY-362450 (WAY) once daily for one-week. Gene expression pattern in mouse livers was analysis by RNA-seq assay. (a-b) Volcano plot (a) and heatmap (b) for identification of differentially expressed genes (DEGs, |fold change| ≥2, FDR < 0.05; n = 3 in each group) in mouse liver tissues upon WAY-362450 treatment. (c-d) Gene ontology (GO; C) and KEGG pathway (d) enrichment analysis of DEGs in mouse liver after WAY-362450 treatment was shown (p < 0.05). (e) The expression of cell cycle-regulating genes in the RNA-seq data was displayed as a heatmap. (f-g) Liver tissues from mice treated with vehicle or WAY-362450 were subjected to RT-qPCR assays for determination mRNA expression of the well-known FXR-target genes (Nr0b2, Abcb11 and Cyp7a1) and cell cycle genes (Ccna2, Ccnb1, Ccnb2, Ccnd1, Ccne1, Cdkn1a, Cdkn1b, Cdkn2c, Cdk1, Cdk2, Cdk4 and Cdk6). (n = 8; *p < 0.05, ***p < 0.001; ns, not significant).
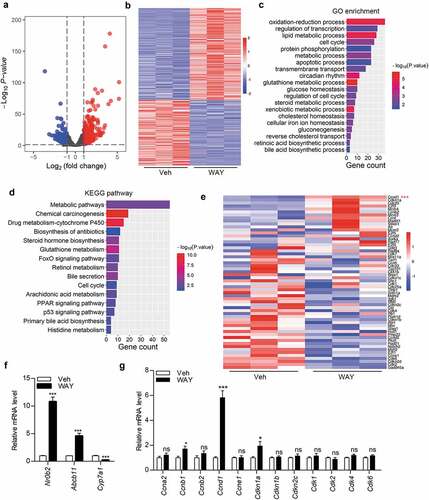
WAY-362450 enhanced hepatic Ccnd1 expression time-dependently in vivo
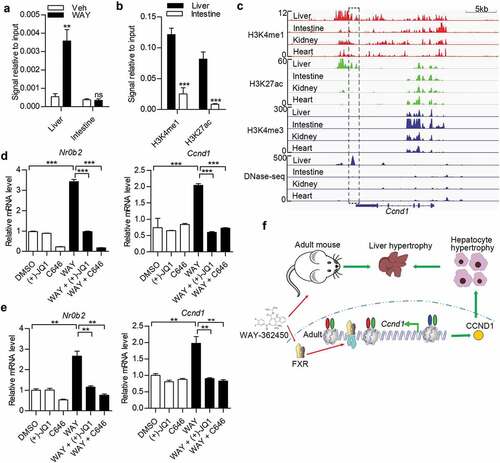
To validate the specificity of FXR agonist in Ccnd1 induction, we tested the mRNA levels of Nr0b2 and Ccnd1, both of which were not obviously changed, in FXR-deficient mice livers after treatment with WAY-362450 ()). Furthermore, wild-type mice were treated with WAY-362450 for a short period (4 h) or 1–3 days. As shown, activation of FXR increased Nr0b2 and Ccnd1 expression in mouse liver (). Administration of WAY-362450 for one-week () and four weeks ()) led to Cyclin D1 protein increment in mouse livers. Additionally, administration of WAY-362450 enhanced Nr0b2 while repressed Ccnd1 expression in both intestine and kidney tissues ()). These results further confirmed that the activation of FXR might regulate Ccnd1 expression in time-dependent and tissue-specific manners.
Figure 3. WAY-362450 enhanced Ccnd1 expression time-dependently in mouse livers. (a) FXR-deficient mice were treated with either vehicle (Veh) or WAY-362450 (WAY) for 24 h. The mRNA levels of Nr0b2 and Ccnd1 in mouse livers were determined by RT-qPCR (n = 6 each). (b) The mRNA levels of Nr0b2 and D-type cyclin family members (Ccnd1, Ccnd2, and Ccnd3) in mouse livers after WAY-362450 treatment for 4 h were determined by RT-qPCR (n = 6). (c) The hepatic mRNA levels of Nr0b2 and Ccnd1 in wild-type male mice treated with vehicle or WAY-362450 for 1–3 days were determined by RT-qPCR. (d-e) Male mice were treated with WAY-362450 for one-week. (d) Representative images are shown for immunohistochemistry staining of Cyclin D1 in liver tissues from vehicle- or WAY-362,450-treated mice. Original magnification 200 x, bar: 100 μm for upper panel. (e) The protein level of Cyclin D1 in mouse livers was determined by Western blot assay. Densitometric analysis of Cyclin D1 band density with normalization to β-Actin was shown in the lower panel. (f) The protein level of Cyclin D1 in female mouse livers treated with WAY-362450 for four weeks was determined by Western blot. (g) The expression levels of Nr0b2 and Ccnd1 in the intestine and kidney tissues from mice treated with vehicle or WAY-362450 for one-week were determined by RT-qPCR. (Mean ± SEM, ***p < 0.001; ns, not significant).
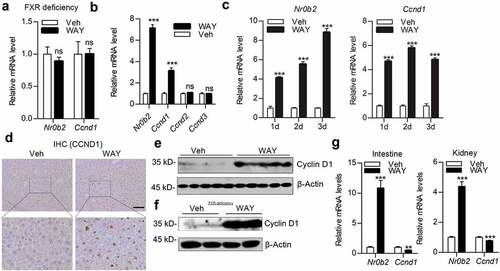
FXR promoted Ccnd1 expression time-dependently in hepatocyte in vitro
Subsequently, we analyzed the effect of FXR agonists on the expression of Ccnd1 in hepatocytes in vitro. Mouse primary hepatocytes (mPH) treated with WAY-362450 or GW4064 for 24 h showed abundantly increased Nr0b2 and Ccnd1 mRNA levels compared to vehicle-treated controls ()). Similarly, incubation with these agonists enhanced CCND1 expression in the HepG2 ()) and Hep3B ()) human hepatocellular carcinoma cells. In addition, incubation with the FXR antagonist, Z-guggulsterone, led to decreased expression of NR0B2 and CCND1 mRNA ()). We further treated HepG2 cells with FXR agonists for various time periods. As expected, activation of FXR-induced NR0B2 and CCND1 expression in a time-dependent manner in HepG2 cells ()). Activation of FXR continually increased NR0B2 mRNA expression for 72 h, whereas the mRNA level of CCND1 peaked at 24 h and decreased gradually ()). The protein level of CCND1 in HepG2 cells showed a similar expression pattern with WAY-362450 and GW4064 treatment ()). However, we did not observe the obvious difference in cell cycle progress in mPH (supplementary material, Figure S2A) or cell growth in HepG2 (supplementary material, Figure S2B) cells upon WAY-362450 or GW4064 treatment alone in vitro. These data indicated that Ccnd1 expression was tightly regulated by FXR in a time-dependent manner in hepatocytes.
Figure 4. FXR promoted Ccnd1 expression time-dependently in hepatocyte in vitro. (a-c) Mouse primary hepatocytes (A; mPH) and human hepatocellular carcinoma cell lines HepG2 (b) and Hep3B (c) were treated with DMSO (Veh), WAY-362450 (WAY, 2 μM) or GW4064 (GW, 2 μM) for 24 h. (d) HepG2 cells were treated with an FXR antagonist (z-guggulsterone, 20 μM) for 24 h. The mRNA expression levels of Nr0b2 (or NR0B2) and Ccnd1 (or CCND1) were determined by RT-qPCR. (e) HepG2 cells treated with DMSO, WAY-362450 (2 μM) or GW4064 (2 μM) were harvested at indicated time points and subjected to RT-qPCR analysis of NR0B2 and CCND1 mRNA levels. (f) Western blot analysis of CCND1 protein levels in 2 μM WAY-362450-treated HepG2 cells for 12, 24, 48 and 72 h. Densitometric analysis of Western blot result with normalization to β-Actin was shown in the right panel. (Mean ± SEM; *p < 0.05, *p < 0.01, ***p < 0.001).
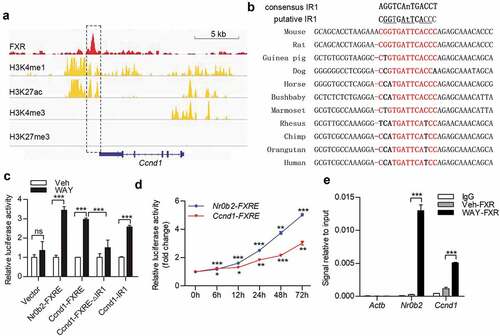
FXR regulated Ccnd1 transcription through direct binding to a downstream IR-1 element
We next explored whether FXR modulated Ccnd1 expression through transcriptional regulation. Through exploring FXR ChIP-Seq dataset in GEO database (accession: GSE73624), we found that FXR might associate with a downstream region of the mouse Ccnd1 gene in the liver ()). The region was also suggested as a potential distal (approximately 10 kb away from transcription start site) active enhancer of Ccnd1 gene in mouse liver, as indicated by positive histone markers of enhancer, H3K4me1/H3K27ac [Citation33] at the flanking region with an assessment of ENCODE epigenomic data (accession: ENCSR854HXZ) [Citation34,Citation35]. Through prediction of FXR-binding element by NUBIScan V2.0 (https://www.nubiscan.unibas.ch), we found that the region harbored a putative FXR responsive element (a canonical IR-1 consensus sequence), which was conservative in mammals ()). By dual-luciferase assay, we confirmed the positive transcriptional regulatory effect of the downstream enhancer and IR-1 element (). ChIP-qPCR assays verified that FXR was associated with the IR-1 region downstream the mouse Ccnd1 gene through direct binding ()). Thus, these data indicated that FXR regulated Ccnd1 expression through direct binding to IR-1 element within a downstream enhancer in mouse liver.
Figure 5. FXR regulates Ccnd1 transcription through direct binding to a downstream potential enhancer. (a) Public data, including FXR ChIP-seq (GEO: GSE73624), and H3K4me1, H3K4me3 and H3K27ac ChIP-seq (ENCODE: ENCSR854HXZ), on adult mouse livers were analyzed for potential FXR-binding sites and enhancer signature on mouse Ccnd1 gene. The results were exhibited on IGV 2.4 software. (b) Sequence alignment of the putative FXR responsive element (inverted repeat-1, IR-1) downstream Ccnd1 gene in mouse, rat, guinea pig, dog, horse, bushbaby, marmoset, rhesus, chimp, orangutan, and human. The consensus sequence in IR-1 elements was shown in red. (c) HEK293T cells were transfected with constructs containing FXR response element (FXRE)-approximal region (Ccnd1-FXRE) in Ccnd1, Ccnd1-FXRE region with IR1 deletion (Ccnd1-FXRE-ΔIR1), or Ccnd1 IR1 core element (Ccnd1-IR1). The construct containing the mouse Nr0b2 FXRE-containing region was used as a positive control (Nr0b2-FXRE). After treatment with vehicle (Veh) or WAY-362450 (WAY), the cells were subjected to dual-luciferase reporter assays. (d) HEK-293T Cells transfected with constructs containing Ccnd1-FXRE or the FXR response element (FXRE) from the mouse Nr0b2 gene (Nr0b2-FXRE) were treated with 2 μM WAY-362450 for the indicated time period. The activity of IR1 element of Ccnd1 was determined by dual-luciferase reporter assay. (e) A chromatin immunoprecipitation-quantitative PCR (ChIP-qPCR) assay was conducted using liver tissues from the vehicle or WAY-362450-treated mice (n = 3 each). ChIP signals are expressed as the ratio of immunoprecipitated DNA to the total input chromatin. A region of the Actb promoter was used as a negative control. The proximal region of the Nr0b2 promoter containing FXRE was used as the positive control. (Mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant).
Induction of Ccnd1 by FXR depended on active histone acetylation signature
We then explore the potential tissue-specific activity of FXR-binding region on Ccnd1 gene. As shown, we demonstrated that WAY-362450 treatment evidently increased the binding of FXR to the IR-1 region in the liver instead of the intestine ()). We also found that positive H3K4me1 and H3K27ac signals were present at the Ccnd1 enhancer region in the liver but not in the intestine ()). In accord with our findings, the H3K4me1/H3K27ac peaks and the structure accessibility of chromatin at the Ccnd1 downstream region specially present in mouse liver but absent in intestine, kidney, and heart tissues ()) with comparing ChIP and DNase-seq (DNase I hypersensitive sites sequencing) data for epigenome in ENCODE database [Citation34,Citation35]. The corresponding downstream region of CCND1 gene in HepG2 cells also harbored active enhancer markers (H3K4me1/H3K27ac positive) and showed high chromatin accessibility (supplementary material, Figure S3). Furthermore, the binding signals of transcriptional cofactors EP300 and BRD4, which were commonly associated with active enhancer [Citation33], were also observed in HepG2 cells (supplementary material, Figure S3). Subsequently, we tested whether the histone acetylation was involved in FXR-induced Ccnd1 expression. HepG2 cells and mPH were treated with for histone acetylation inhibitors, BET inhibitor ((+)-JQ1) [Citation36] and EP300/CBP inhibitor (C646) [Citation37]. We showed that the induction effect of WAY-362450 on CCND1/Ccnd1 and NR0B2/Nr0b2 expression was almost abrogated by these inhibitors (). Hence, these results indicated that FXR modulated Ccnd1 transcription through directly binding to an IR-1 element within a tissue-specific enhancer.
Figure 6. The signature of downstream enhancer of Ccnd1 was tissue-specific and developmental stage-dependent in the liver. (a) ChIP-qPCR analysis of FXR binding on Ccnd1 gene in vehicle or WAY-362450 treated liver and intestine tissues (n = 4). (b) The levels of H3K4me1 and H3K27ac at FXR-binding region of Ccnd1 gene in liver and intestine tissues (n = 4) were examined. (c) ChIP-seq data of histone modifications (H3K4me1, H3K4me3, H3K27ac, and H3K27me3) and chromatin accessibility (DNase-seq) on Ccnd1 gene from ENCODE were visualized using IGV 2.4 software. Data on liver, small intestine, kidney and heart tissues in adult mouse (8 weeks) were analyzed. (d-e) Mouse primary hepatocytes (d) and HepG3 cells (e) were treated with 2 μM WAY-362450 with or without 3 μM (+)-JQ1 and 10 μM C646 for 4 h. The mRNA levels of NR0B2 and CCND1 genes were determined by RT-qPCR assay. (Mean ± SEM, ***p < 0.001) (f) Schematic diagram for FXR agonist in the induction of liver and hepatocyte hypertrophy by regulating Ccnd1 expression through binding to a potential enhancer region (H3K4me1/H3K27ac positive and H3K4me3 negative). Red oval, H3K4me1; Green oval, H3K27ac; Blue oval, H3K4me3.
Discussion
In this study, we discovered that chronic activation of FXR by specific exogenous agonist WAY-362450 induced liver hypertrophy instead of hyperplasia. FXR regulated Ccnd1 expression by direct targeting in time-dependent and tissue-specific manners in mouse liver ()). In contrast with previous reports, activation of FXR by WAY-362450 did not obviously upregulate the expression of Hhex, which was previously identified as FXR-target gene during excessive bile acid-induced liver hypertrophy [Citation32]. Foxm1 was also reported as a target gene of FXR during liver regeneration [Citation16]. Although Foxm1 was slightly upregulated by not reached statistical significance, its expression is still very low in WAY-362450-treated mouse liver (TPM<0.5). The effect of FXR on target gene regulation may depend on the affinity of its agonist and microenvironment context.
Although FXR is highly expressed in various tissues, including liver, intestine, kidney, and adrenal gland, it is supposed that FXR plays distinct roles in different tissues. A study employing chromatin immunoprecipitation-sequencing (ChIP-seq) to evaluate genome-wide FXR/gene interaction revealed that hepatic and intestinal tissues share only a minor portion of total FXR-binding sites in mice [Citation38]. In particular, genes involved in the enterohepatic circulation of bile acid are regulated by FXR in a liver- and intestine-specific manner [Citation38–Citation41]. Nevertheless, the mechanism of tissue-specific gene regulation by FXR is rarely reported. Gene expression under strict spatiotemporal-regulation is essential to maintain specific cellular identity. Tissue-specific cis-regulatory elements were shown to play important roles in spatiotemporal gene regulation [Citation42]. Here, we showed FXR modulated Ccnd1 expression tissue-specifically in mouse liver. A possible explanation could be the activity of the downstream enhancer for Ccnd1 gene was tissue-specific, as indicated by H3K4me1/H3K27ac and chromatin accessibility signals were exclusively present in the liver. Indeed, we also showed that FXR agonist did not induce the expression of Ccnd1 by in both intestine and kidney tissues. A recent study in non-small cell lung cancer also demonstrated that FXR activated Cyclin D1 transcription through binding to an inverted repeat-0 element within CCND1 promoter [Citation43]. However, we did not observe the binding signal of FXR on Ccnd1 promoter in mouse liver. Previous reports showed that SHP (NR0B2) could repress CCND1 transcription in human hepatocellular carcinoma cell lines [Citation44–Citation46]. However, our data suggested that activation of FXR-induced CCND1 expression despite SHP induction both in mouse and human hepatocytes. Accumulated Nr0b2 would limit the level of Ccnd11 induction by FXR. Interestingly, we also found that FXR also induced the expression of a negative regulator in cell cycle progression Cdkn1a in mouse liver. Thus, FXR plays a comprehensive role in modulation of cell cycle protein expression by FXR depending on cellular context.
Aberrant bile acid signaling in the absence of hepatic FXR may lead to dysregulated proliferation and malignancy of hepatocytes [Citation46]. In addition, aged FXR-deficient mice spontaneously develop hepatocellular carcinoma and uncontrolled hepatocyte proliferation with dysregulated expression of cell cycle-related genes [Citation47,Citation48]. Thus, FXR may play a comprehensive role in the tight control of cell cycle events and hepatocyte growth. Here, we demonstrated that FXR agonists promoted liver hypertrophy. Although the well-characterized positive regulator of G1/S phase Cyclin D1 was substantially upregulated, the expression of other negative cell cycle-regulated genes (including Cdkn1a and Nr0b2) was also increased in WAY-362450-treated mice liver under both quiescent and excessive estrogen-induced chronic injury scenarios. Indeed, we demonstrated that a small portion of hepatocytes in WAY-362450-treated mice entered the cell cycle with increased nuclear DNA content. However, most of the hepatocytes were arrested before mitosis. These findings were in accordance with the hypertrophic phenotype of liver growth [Citation16]. Additionally, the findings that FXR agonist alone did not induce hepatocyte proliferation and cell cycle progression in vitro also excluded the evident mitogenic effect of FXR agonist. Thus, we proposed that FXR agonist WAY-362450 could stimulate liver growth through hepatocyte hypertrophy, not hyperplasia.
In addition to its role in metabolic regulation, FXR is also considered as a hepato-protective factor. Numerous studies have characterized FXR agonists, such as GW4064, WAY-362450 and INT-747, as promising agents for the treatment of liver diseases and metabolic disorders, including alcoholic liver disease, non-alcoholic fatty liver disease, liver fibrosis, and intra- or extrahepatic cholestatic disease [Citation20–Citation23], etc. Some agonists of FXR are undergoing a preclinical and clinical trial. However, the potential side effect of chronic FXR agonist ingestion is still not fully clarified, whereas a previous report suggested that chronic activation of FXR in liver and intestine could lead to partial neonatal lethality and growth restriction [Citation49]. In this study, we showed chronic ingestion of WAY-362450 led to liver enlargement. We also demonstrated that WAY-362450 induced benign hypertrophic effect (without obvious hepatocyte proliferation) rather than harmful pathological hyperplasia. We still suggest that correct management of dosage in the application of FXR agonist would reduce the risk of potential side effect.
In summary, our study demonstrated that activation of FXR by its specific agonist promoted liver hypertrophy and induced Ccnd1 expression through direct transcriptional regulation in mouse livers. Furthermore, we identified a novel FXR-binding element and a potential enhancer, the activity of which is proposed to be tissue-specific at Ccnd1 gene. These findings also extend our knowledge on the epigenetic mechanism of FXR-target gene interaction during physiological and pathological conditions. Moreover, it is beneficial for development and appropriate application of specific FXR agonist in preventing hepatic and metabolic diseases in human with extensive understanding of the effect of FXR activation at tissue-specific level.
Supplemental Material
Download MS Word (1.2 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Forman BM, Goode E, Chen J, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–693.
- Seol W, Choi HS, Moore DD. Isolation of proteins that interact specifically with the retinoid X receptor: two novel orphan receptors. Mol Endocrinol. 1995;9:72–85.
- Maloney PR, Parks DJ, Haffner CD, et al. Identification of a chemical tool for the orphan nuclear receptor FXR. J Med Chem. 2000;43:2971–2974.
- Pellicciari R, Fiorucci S, Camaioni E, et al. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45:3569–3572.
- Flatt B, Martin R, Wang TL, et al. Discovery of XL335 (WAY-362450), a highly potent, selective, and orally active agonist of the farnesoid X receptor (FXR). J Med Chem. 2009;52:904–907.
- Laffitte BA, Kast HR, Nguyen CM, et al. Identification of the DNA binding specificity and potential target genes for the farnesoid X-activated receptor. J Biol Chem. 2000;275:10638–10647.
- Sinal CJ, Tohkin M, Miyata M, et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744.
- Lu TT, Makishima M, Repa JJ, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6:507–515.
- Goodwin B, Jones SA, Price RR, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526.
- Massafra V, Pellicciari R, Gioiello A, et al. Progress and challenges of selective Farnesoid X Receptor modulation. Pharmacol Ther. 2018;191:162-177.
- Yanger K, Knigin D, Zong Y, et al. Adult hepatocytes are generated by self-duplication rather than stem cell differentiation. Cell Stem Cell. 2014;15:340–349.
- Borude P, Edwards G, Walesky C, et al. Hepatocyte-specific deletion of farnesoid X receptor delays but does not inhibit liver regeneration after partial hepatectomy in mice. Hepatology. 2012;56:2344–2352.
- Huang W, Ma K, Zhang J, et al. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science. 2006;312:233–236.
- Chen WD, Wang YD, Zhang L, et al. Farnesoid X receptor alleviates age-related proliferation defects in regenerating mouse livers by activating forkhead box m1b transcription. Hepatology. 2010;51:953–962.
- Merlen G, Ursic-Bedoya J, Jourdainne V, et al. Bile acids and their receptors during liver regeneration: “Dangerous protectors”. Mol Aspects Med. 2017;56:25–33.
- Milona A, Owen BM, van Mil S, et al. The normal mechanisms of pregnancy-induced liver growth are not maintained in mice lacking the bile acid sensor Fxr. Am J Physiol Gastrointest Liver Physiol. 2010;298:G151–158.
- Dai X, De Souza AT, Dai H, et al. PPARalpha siRNA-treated expression profiles uncover the causal sufficiency network for compound-induced liver hypertrophy. PLoS Comput Biol. 2007;3:e30.
- Hall AP, Elcombe CR, Foster JR, et al. Liver hypertrophy: a review of adaptive (adverse and non-adverse) changes–conclusions from the 3rd international ESTP expert workshop. Toxicol Pathol. 2012;40:971–994.
- Miyaoka Y, Ebato K, Kato H, et al. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr Biol. 2012;22:1166–1175.
- Verbeke L, Mannaerts I, Schierwagen R, et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep. 2016;6:33453.
- Wu W, Zhu B, Peng X, et al. Activation of farnesoid X receptor attenuates hepatic injury in a murine model of alcoholic liver disease. Biochem Biophys Res Commun. 2014;443:68–73.
- Zhang S, Wang J, Liu Q, et al. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol. 2009;51:380–388.
- Liu Y, Binz J, Numerick MJ, et al. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra- and extrahepatic cholestasis. J Clin Invest. 2003;112:1678–1687.
- Wu WB, Xu YY, Cheng WW, et al. Agonist of farnesoid X receptor protects against bile acid induced damage and oxidative stress in mouse placenta–a study on maternal cholestasis model. Placenta. 2015;36:545–551.
- Fiorucci S, Clerici C, Antonelli E, et al. Protective effects of 6-ethyl chenodeoxycholic acid, a farnesoid X receptor ligand, in estrogen-induced cholestasis. J Pharmacol Exp Ther. 2005;313:604–612.
- Zeng W, Liu Z, Liu X, et al. Distinct Transcriptional and Alternative Splicing Signatures of Decidual CD4(+) T Cells in Early Human Pregnancy. Front Immunol. 2017;8:682.
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192.
- Carpenter AE, Jones TR, Lamprecht MR, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100.
- Ryu D, Oh KJ, Jo HY, et al. TORC2 regulates hepatic insulin signaling via a mammalian phosphatidic acid phosphatase, LIPIN1. Cell Metab. 2009;9:240–251.
- Wu W, Wang Y, Xu Y, et al. Dysregulated activation of c-Src in gestational trophoblastic disease contributes to its aggressive progression. Placenta. 2014;35:824–830.
- Tetzlaff MT, Curry JL, Ivan D, et al. Immunodetection of phosphohistone H3 as a surrogate of mitotic figure count and clinical outcome in cutaneous melanoma. Mod Pathol. 2013;26:1153–1160.
- Xing X, Burgermeister E, Geisler F, et al. Hematopoietically expressed homeobox is a target gene of farnesoid X receptor in chenodeoxycholic acid-induced liver hypertrophy. Hepatology. 2009;49:979–988.
- Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15:272–286.
- Rosenbloom KR, Sloan CA, Malladi VS, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41:D56–63.
- Yue F, Cheng Y, Breschi A, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364.
- Booth CAG, Barkas N, Neo WH, et al. Ezh2 and Runx1 mutations collaborate to initiate lympho-myeloid leukemia in early thymic progenitors. Cancer Cell. 2018;33(274–291):e278.
- Lasko LM, Jakob CG, Edalji RP, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017;550:128–132.
- Thomas AM, Hart SN, Kong B, et al. Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology. 2010;51:1410–1419.
- Dawson PA, Hubbert M, Haywood J, et al. The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J Biol Chem. 2005;280:6960–6968.
- Kong B, Wang L, Chiang JY, et al. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology. 2012;56:1034–1043.
- Zhu Y, Li F, Guo GL. Tissue-specific function of farnesoid X receptor in liver and intestine. Pharmacol Res. 2011;63:259–265.
- Russo M, Natoli G, Ghisletti S. Housekeeping and tissue-specific cis-regulatory elements: recipes for specificity and recipes for activity. Transcription. 2018;9:177–181.
- You W, Chen B, Liu X, et al. Farnesoid X receptor, a novel proto-oncogene in non-small cell lung cancer, promotes tumor growth via directly transactivating CCND1. Sci Rep. 2017;7:591.
- Zhang Y, Xu P, Park K, et al. Orphan receptor small heterodimer partner suppresses tumorigenesis by modulating cyclin D1 expression and cellular proliferation. Hepatology. 2008;48:289–298.
- Ohno T, Shirakami Y, Shimizu M, et al. Synergistic growth inhibition of human hepatocellular carcinoma cells by acyclic retinoid and GW4064, a farnesoid X receptor ligand. Cancer Lett. 2012;323:215–222.
- Kong B, Zhu Y, Li G, et al. Mice with hepatocyte-specific FXR deficiency are resistant to spontaneous but susceptible to cholic acid-induced hepatocarcinogenesis. Am J Physiol Gastrointest Liver Physiol. 2016;310:G295–302.
- Kim I, Morimura K, Shah Y, et al. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis. 2007;28:940–946.
- Yang F, Huang X, Yi T, et al. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007;67:863–867.
- Cheng Q, Inaba Y, Lu P, et al. Chronic activation of FXR in transgenic mice caused perinatal toxicity and sensitized mice to cholesterol toxicity. Mol Endocrinol. 2015;29:571–582.