
ABSTRACT
Recent studies have shown that genomic instability in tumor cells leads to activation of inflammatory signaling through the cGAS/STING pathway. In this review, we describe multiple ways by which genomic instability leads to cGAS/STING-mediated inflammatory signaling, as well as the consequences for tumor development and the tumor microenvironment. Also, we elaborate on how tumor cells have apparently evolved to escape the immune surveillance mechanisms that are triggered by cGAS/STING signaling. Finally, we describe how cGAS/STING-mediated inflammatory signaling can be therapeutically targeted to improve therapy responses.
Genomic instability in cancer
Cells are equipped with a tightly regulated “DNA damage response” (DDR) to protect their genome from lesions that arise from endogenous and exogenous sources. In this way, various different DNA lesions are continuously being detected and repaired to maintain genomic stability. Conversely, alterations in the ability of cells to repair their DNA can lead to genomic instability, which occurs frequently in cancer. Depending on the underlying cause, genomic instability is characterized by accumulation of mutations, complex genomic rearrangements, and the progressive loss or gain of genomic regions or whole chromosomes.
Genomic instability has been recognized as a hallmark of cancer [Citation1], and various underlying mechanisms have been identified. For instance, germline mutations in DNA repair genes can drive the accumulation of genomic aberrancies and ensuing tumorigenesis. Prototypical examples are mutations in the breast and ovarian cancer susceptibility genes BRCA1 and BRCA2, which result in defective DNA repair of DNA double-strand breaks (DSBs) through homologous recombination (HR) [Citation2]. Alternatively, germline mutations in mismatch repair (MMR) genes, collectively known as Lynch syndrome, lead to cancer predisposition, which mainly involves endometrial and non-polyposis colorectal cancer [Citation3–Citation5]. These cancers are characterized by microsatellite instability (MSI), which involves an increased number of somatic mutations at repetitive genomic loci [Citation6]. Of note, HR or MRR is not the only DNA repair pathways in which defects are associated with an increased risk to develop cancer. Notably, besides germ-line mutations, also somatic alterations were shown to underlie cancer-associated DNA repair deficiencies [Citation7]. Interestingly, telomere dysfunction has also been described as an underlying mechanism of genomic instability in cancer cells. Cells that accumulate unprotected chromosome ends may bypass senescence, can lead to the formation of clones with high levels genomic instability [Citation8]. Cells that survive a telomere crisis gain various genomic alterations, involving chromothripsis and kataegis [Citation9,Citation10].
Another important cause of genomic instability in cancer is oncogene-induced replication stress [Citation11,Citation12]. Overexpression of specific oncogenes, including CCND1 (encoding Cyclin D1), CCNE1 (encoding Cyclin E1) or MYC (encoding c-MYC), leads to deregulation of the cell cycle and was shown to induce replication stress via different mechanisms [Citation11–Citation14]. A common theme in, this context involves elevated CDK activity, notably CDK2, which consequently leads to increased firing of replication origins [Citation15]. As a result, oncogene overexpression leads to depletion of the nucleotide pool activity, which limits replication fork progression and triggers genomic instability [Citation16,Citation17]. Indeed, Cyclin E1 or Cdc25A overexpression was shown to induce reversal and slowing of replication forks [Citation18]. In parallel, the elevated levels of origin firing combined with high transcriptional activity lead to frequent collisions between the replication machinery and the transcriptional apparatus [Citation19].
Single-stranded DNA (ssDNA) stretches that are exposed upon replication fork stalling and the DNA breaks that form upon collapse of stalled replication forks will trigger activation of the ATR and ATM kinases within the DNA damage response (DDR). Under physiological conditions, DDR activation leads to p53-mediated apoptosis or senescence to clear pre-cancerous cells [Citation20,Citation21]. The DNA lesion that arises as a consequence of oncogene-induced replication stress or defective DNA repair result in genetic pressure on tumor suppressor genes involved in DNA damage-induced cell cycle checkpoint activation [Citation22,Citation23]. Indeed, loss of p53 is one of the mechanisms by which transformed cells with high levels of replication stress and DNA damage escape cell cycle checkpoint activation and apoptosis to continue proliferation [Citation24]. In line with this notion, TP53 mutations are frequently observed in cancers (~42% of all human cancers), especially in those that are characterized by high levels of genomic instability, such as high-grade serous ovarian cancer (96% with TP53 alteration) or triple negative breast cancer (80% with TP53 alteration) [Citation25–Citation28]. Although p53-dependent cell cycle checkpoint control is frequently inactivated in genomically instable cancers, other levels of cell cycle control are typically retained. In fact, genomically instable cancers increasingly depend for their survival on the remaining cell cycle checkpoint components, including Chk1 and Wee1 [Citation29,Citation30].
Although residual cell cycle checkpoint control in genomically instable cancer cells can delay entry into mitosis in situations of DNA damage, we increasingly realize that these checkpoints do not fully prevent damaged cells from entering mitosis. Notably, cancer-associated genomic instability frequently involves DNA lesions that originate during DNA replication and remain unresolved at mitotic entry [Citation31–Citation34]. As a consequence, such DNA lesions interfere with normal chromosome segregation and lead to breakage-fusion-bridge cycles, ultimately resulting in structural genomic aberrations [Citation35].
Aberrant chromosome segregation is not only observed in situations of defective genome maintenance. Defects in spindle assembly checkpoint (SAC) functioning, or improper attachment of microtubules to the kinetochore leads to mis-segregation of entire chromosomes during mitosis. The resulting chromosomal instability (CIN) involves lagging chromosomes and numerical aneuploidies. Importantly, numerical chromosomal defects were shown to induce structural chromosomal abnormalities and vice versa, in good agreement with these phenotypes frequently co-occurring in cancers (, left panel) [Citation36–Citation38].
Figure 1. Genomic instability and cGAS/STING signaling in response to cytoplasmic DNA.
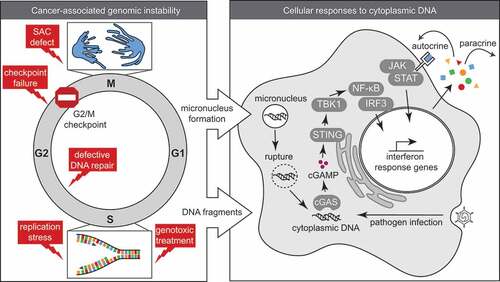
Micronuclei formation as a source of cytoplasmic DNA
The presence of unresolved mitotic DNA damage or chromosome mis-segregation often results in the formation of micronuclei upon mitotic exit (). Micronuclei contain complete chromosomes or chromosome fragments, which are surrounded by a nuclear envelope. However, multiple “non-core” envelope proteins, including nuclear pore complex (NPC) components, cannot be assembled on lagging chromosomes and therefore prevent the formation of a proper nuclear envelope [Citation39]. As a consequence, multiple “nuclear” processes do not function properly in micronuclei. [Citation40]. Among these disturbed processes, micronuclei show defects in nucleo-cytoplasmic transport, which impairs the recruitment of the MCM components of the replicative DNA helicase as well as DNA repair proteins [Citation37]. Therefore, DNA damage accumulates in micronuclei during S- and G2-phase of the cell cycle and leaves genomic regions under-replicated. Furthermore, chromatids in micronuclei that contain centromeric regions are defective in building a functional kinetochore and do not properly recruit spindle assembly checkpoint proteins [Citation41]. Also, re-integration of damaged chromatin from micronuclei into the main nucleus, which occurs with almost 40% of the micronuclei, triggers replication problems, genomic instability and extensive genomic rearrangements involving chromotripsis [Citation37,Citation42,Citation43]. Finally, the surrounding membrane of micronuclei is more fragile when compared to the membrane surrounding the main nucleus. As a consequence, the nuclear membrane of micronuclei is prone to rupture, which results in the release of chromatin into the cytosol [Citation44].
Inactivation of multiple DNA repair factors has been shown to result in formation of micronuclei [Citation45]. For instance, inactivation of the HR factors BRCA1, BRCA2 or Rad51 leads to chromosome segregation failure with a range of consequences, including micronucleus formation [Citation46–Citation48]. These effects are exaggerated when HR-deficient cells are treated with genotoxic agents, including PARP inhibitors [Citation34]. Similarly, defects in removal of ribonucleotides from DNA leads to mitotic failures. During normal DNA replication, ribonucleotides may be incorporated into DNA, making DNA more susceptible to mutagenesis and strand breaks [Citation49]. Ribonucleotide excision repair (RER) functions to remove aberrantly incorporated ribonucleotides and thereby maintains genome stability. Conversely, inactivation of the RER nuclease RNaseH2, which also functions in removing RNA:DNA hybrids (R-loops) that arise during transcription, interferes with maintenance of genome stability [Citation50,Citation51]. Importantly, inactivating mutations in RNASEH2A lead to cytoplasmic DNA, as a result of micronuclei formation [Citation50].
Of note, because the presence of micronuclei reflects the accumulation of persistent DNA lesions or chromosome mis-segregation, micronucleus formation is established as a reliable method for toxicological assessment of the clastogenic or aneugenic effects of compounds [Citation52].In line with DNA repair defects leading to micronuclei that are prone to rupture, increased amounts of cytoplasmic DNA have been observed in various contexts of DNA repair deficiency, including ATM, ERCC1 and BRCA1 deficiency [Citation53,Citation54].
Genomic instability can also lead to the release of DNA into the cytoplasm through mitosis-independent mechanisms (, left panel). At stalled replication forks, the presence of ssDNA activates the checkpoint kinase ATR to prevent entry into mitosis with under-replicated regions [Citation55]. Subsequent restart of stalled replication forks depends on degradation of nascent DNA by MRE11 [Citation56]. However, unsuccessful restoration of replication forks leads to release of ssDNA parts into the cytosol, a process that is stimulated by the BLM helicase and EXO1 exonuclease [Citation57] and can be prevented by binding of RPA and Rad51 to stretches of ssDNA [Citation58]. Recently, the dNTPase SAMHD1 was shown to promote DNA resection capacity, and in conjunction with MRE11 prevents the release of ssDNA into the cytosol [Citation59–Citation61]. In line with these findings, mutations in SAMHD1 increase the release of DNA into the cytoplasm that occur during replicating errors [Citation59].
Response to cytoplasmic DNA: cgas/sting signaling
As soon as double-stranded DNA (dsDNA) or ssDNA enters the cytosol, it is recognized by pattern recognition receptors, including the DNA sensing molecule cyclic GMP-AMP synthase (cGAS). This response is part of the innate immune response, the first-line defense against a range of pathogens, including viruses and bacteria. The basis of this innate response is that no free DNA should be present in the cytoplasm (, right panel).
cGAS can bind various DNA substrates but has the highest affinity for dsDNA, of which the length strongly influences the potential to activate cGAS [Citation62,Citation63]. Once cGAS is in complex with DNA, it is able to catalyze the synthesis of cyclic GMP-AMP (cGAMP), which in turn binds the ER-membrane adaptor protein stimulator of interferon genes (STING) [Citation64,Citation65]. Activated STING subsequently recruits and activates the TBK1 kinase, which phosphorylates the transcription factor IRF3. STING also leads to activation of both canonical and non-canonical signaling of the NF-κB transcription factor by indirect degradation of its inhibitor IkB [Citation66,Citation67]. Activation of both IRF3 and NF-κB transcription results in the expression of type-I interferon (IFN) genes and pro-inflammatory cytokines, which instigates a cell-intrinsic innate immune response [Citation68,Citation69]. Importantly, positive feedback regulation leads to type-I IFN-induced cGAS expression due to the presence of IFN response elements in the cGAS promoter [Citation70]. This feedback loop is further regulated by cleavage of cGAS and IRF3 by the apoptotic caspase-3 [Citation71].
The recognition of cytosolic DNA does not only occur through cGAS. Various other DNA sensors were identified to be present in the cytoplasm; however, their ability to activate STING-dependent IFN responses appears to be limited. Besides cGAS, the most prominent DNA sensors that are able to induce IFN signaling in response to cytoplasmic DNA appear to be “AIM2-like receptors” (ALRs), including IFI16 and AIM2 [Citation72]. In conjunction with ATM and PARP-1, IFI16 forms a complex with STING upon nuclear DNA damage and triggers NF-κB signaling, independently of cGAS [Citation73,Citation74]. AIM2 forms an ‘inflammasome; in response to cytoplasmic DNA, and thereby promotes secretion of pro-inflammatory cytokines via caspase-1 [Citation75–Citation78]. Although multiple DNA sensors seem to possess DNA-binding capacities, the downstream activation of STING seems to be crucial to ultimately initiate innate immune responses [Citation79].
In addition to cytoplasmic DNA, also RNA has been demonstrated to enter the cytoplasm. Cytoplasmic RNA is predominantly recognized by the RNA sensors Retinoic acid-inducible gene-I protein (RIG-I) and melanoma differentiation-associated protein-5 (MDA5) [Citation80]. Detection of RNA species in the cytoplasm also triggers the production of inflammatory cytokines, including type-I IFN. However, this process depends on mitochondrial antiviral-signaling protein (MAVS) and is independent of cGAS [Citation81]. Although STING was proposed to function in the cellular response to cytoplasmic RNA, this role is not entirely clear [Citation79]. Also, IFN signaling in response to sensing of cytoplasmic RNA appears more relevant for anti-viral responses against RNA virus infections rather than cancer-associated genomic instability [Citation82].
cGAS/STING activation in situations of genomic instability
Sensing of cytoplasmic DNA as a mechanism to respond to pathogens is based on the premise that the “own” DNA of the cell is retained within the nucleus. Clearly, in situations where cytoplasmic DNA arises due to genomic instability or genotoxic treatment, cGAS/STING signaling will be activated by “self” DNA and leads to a sterile inflammatory response.
Indeed, various conditions in which persistent DNA damage is induced have been linked to inflammatory signaling, although the underlying mechanisms initially remained elusive. Irradiation, for instance, was shown to induce pro-inflammatory cytokines secretion [Citation83,Citation84]. Only recently, the induction of cytosolic DNA after irradiation was shown to trigger cGAS/STING signaling, which was shown to be responsible for the observed inflammatory response [Citation85,Citation86]. Similarly, DNA damage repair defects, as for instance induced by loss of BRCA1, BRCA2 or ATM lead to micronuclei formation and cGAS/STING-dependent IFN signaling [Citation46,Citation47,Citation53,Citation87]. Likewise, DNA lesions as a result of telomere erosion [Citation88,Citation89] or oncogenic stress were shown to activate cGAS/STING signaling [Citation86]. Additionally, aberrant RNA:DNA hybrids were reported to trigger cGAS/STING signaling [Citation90]. Specifically, mutations in genes encoding RNase H2 subunits lead to the autoimmune disorder Aicardi-Goutières syndrome (AGS), which is characterized by increased production of type-I IFN [Citation91,Citation92]. Of note, the observed inflammatory response in AGS was recently demonstrated to depend on cGAS/STING signaling, which in part may be instigated by micronuclei formation [Citation50,Citation93]. Defective processing of stalled replication forks can also lead to cGAS/STING-dependent inflammatory signaling. Under physiological conditions, the resection capacity of SAMHD1 prevents the release of ssDNA from stalled replication forks into the cytosol. Conversely, SAMHD1 deficiency leads to accumulation of cytoplasmic ssDNA and thereby triggers a cGAS/STING-induced cytokine response [Citation59]. Besides resection capacity, SAMHD1 also prevents the induction of a cGAS/STING-induced IFN response upon viral infection, and limits anti-viral T cell responses in vivo [Citation94].
Cells are able to degrade DNA, which has aberrantly reached the cytoplasm. TREX1, a cytoplasmic exonuclease – originally described as DNAse-III – can degrade ssDNA in the cytoplasm [Citation95,Citation96]. As a consequence, TREX1 deficiency, analogous to RNase H2 or SAMHD1 inactivation, triggers a cell-intrinsic inflammatory response, which requires cGAS [Citation97]. In line with this notion, the cGAS-dependent IFN response triggered by cytoplasmic HIV-1 derived ssDNA is suppressed by TREX1 [Citation98,Citation99]. Clearly, cells with defects in the function of cytoplasmic nucleases fail to clear cytoplasmic DNA, which will result in similar cell-intrinsic inflammatory responses [Citation100].
Taken together, genomic DNA can trigger pro-inflammatory responses when genome maintenance is defective, while various nucleases, both in the nucleus (e.g. RNase H2 and SAMHD1) and cytoplasm (e.g. TREX1), can prevent accumulation of cytoplasmic DNA and therefore dampen innate inflammatory responses.
Consequences of inflammatory signaling induced by genomic instability
Early on, the secretion of pro-inflammatory cytokines was recognized as an important feature of senescence, a state of permanent growth arrest. Senescence can be triggered by multiple cues including telomere erosion, in which critical shortening of telomeres instigate DNA damage signaling.
The array of cytokines that is secreted by senescent cells – known as the senescence-associated secretory phenotype (SASP) – has been described as a consequence of DNA damage and NF-κB signaling [Citation83,Citation101]. The secretion of SASP cytokines facilitates immune cell recruitment, as part of an attempt to eliminate possibly pre-malignant cells, thereby providing a cell-intrinsic surveillance mechanism with tumor-suppressive capacity [Citation102,Citation103].
Recently, it was found that the cGAS/STING pathway promotes SASP and regulates senescence both in vitro and in vivo [Citation86,Citation89,Citation104]. In good agreement with this notion, different treatments that induce senescence, including irradiation, CDK4/6 inhibition or oncogene expression, were able to engage the cGAS/STING pathway [Citation86]. Specifically, due to the presence of chromatin fragments in the cytoplasm of senescent cells, activation of cGAS/STING – and thus SASP – maintains paracrine senescence [Citation86]. Indeed, also telomere damage that occurs during replicative crisis was shown to result in cytosolic DNA fragments, which trigger cGAS/STING-dependent autophagy [Citation105,Citation106]. The observations that senescence was STING- and cGAS-dependent, suggest that cGAS/STING signaling plays an important role in regulating SASP and maintenance of a senescence state [Citation86,Citation104]. Indeed, cells lacking cGAS or STING were able to escape replicative crisis and continue proliferation, underscoring the notion that the inability to initiate cell-intrinsic inflammatory signaling may allow oncogenic transformation of genomically instable cells [Citation106].
Instead of apoptotic cell death, cells that undergo replicative crisis show characteristics of autophagy, including vacuolization and lysosomal protein expression [Citation106]. Gui et al. recently showed that cGAMP triggers STING translocation to the endoplasmatic reticulum and Golgi, where it supports the formation of autophagosomes. Through this mechanisms, cytosolic DNA is targeted for destruction, independently of the canonical cGAS/STING effector TBK1 and inflammatory cytokine release [Citation107]. Similarly, cytosolic DNA originating from micronuclei in RNase H2 mutant cells is targeted by autophagy. Inhibition of autophagy, as a consequence, aggravated the IFN response [Citation50]. These findings illustrate that autophagy plays a role in limiting the amounts of cytoplasmic DNA and through this mechanism determines cell fate in situations of genomic instability.
cGAS/STING signaling in the tumor microenvironment
The secretion of cytokines upon cGAS/STING signaling serves many paracrine functions (). Type-I IFN plays an important role in shaping the innate immune response towards tumor cells. The impact of IFN in this context is illustrated by the finding that mice in which dendritic cells cannot respond to type-I IFN due to lack of the IFNAR receptor or its downstream signaling molecule STAT1, are unable to clear tumor cells and show defects in antigen cross-presentation towards CD8+ T cells [Citation108,Citation109]. Furthermore, IFN signaling in antigen-presenting cells (APCs) is essential for the accumulation of CD8+ dendritic cells in the tumor and for tumor cell recognition [Citation109]. Also, expression of cytokines that are secreted upon STING activation, including CCL5 and CXCL10, has been shown to correlate with high tumor infiltration of CD8+ T cells [Citation110]. Conversely, CD8+ T cell priming is severely impaired in STING- or IRF3-deficient mice and results in the failure to reject immunogenic tumors [Citation111]. Likewise, STING-induced IFN secretion in prostate cancer cells due to loss of the MUS81 endonuclease triggers macrophage-dependent phagocytosis of tumor cells [Citation112]. STING activation in tumor cells enhances the expression of several proteins, such as Suppressor of Cytokine Signaling-1 (SOCS1) in Epstein-Barr virus-associated carcinoma cells and myeloid cells. As a result, production of GM-CSF and IL-6 is inhibited, leading to a decrease in activation of myeloid-derived suppressor cells (MDSCs) and thereby lowering its immunosuppressive functions [Citation113]. Also, STING activation in tumor cells as a result of DNA damage and ensuing cytoplasmic DNA triggers the expression of NKG2D receptor ligands, which promotes NK cell-dependent tumor cell killing [Citation114,Citation115]. Finally, Type I IFNs and STAT1 activation have been described to induce polarization of M1 macrophages [Citation116,Citation117], a specific macrophage subtype that is known for its anti-tumor responses [Citation118]. These findings support an important role of inflammatory signaling and secreted cytokines upon STING activation in tumor cells on infiltration and activation of surrounding immune cells to trigger anti-tumor responses.
Figure 2. cGAS/STING signaling serves multiple paracrine functions in the tumor microenvironment.
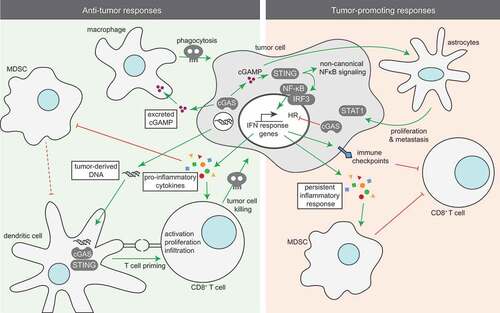
cGAS/STING signaling not only originates tumor cell-intrinsically. STING signaling can also be initiated in the tumor microenvironment. Specifically, tumor cell-derived DNA can be taken up by antigen-presenting cells (APCs) in which it triggers STING signaling. Indeed, in vitro and in vivo data showed that when tumor-derived DNA is taken up by APCs, it enters the cytosol and triggers cGAS, leading to phosphorylation of TBK1, IRF3 and STING-induced IFNβ production [Citation111]. Indeed, release of tumor-derived DNA triggered by irradiation led to uptake of tumor DNA by dendritic cells and resulted in a cGAS/STING type-I IFN response and induction of an adaptive anti-tumor response [Citation119].
Based on other studies, cGAMP was shown to exert its function in a paracrine fashion. cGAMP is able to migrate through gap junctions to activate STING in neighboring cells and thereby provides a soluble “warning signal” [Citation120]. In a more recent study, NK cells from STING-deficient mice failed to generate effective anti-tumor responses, in contrast to NK cells from cGAS-deficient mice [Citation121]. Specifically, in cGAS-deficient mice, injection of cGAS-proficient tumor cells that were able to produce cGAMP led to rejection of tumor cells via STING activation in NK cells [Citation121]. These findings support the importance of STING activation in response to paracrine cGAMP to trigger anti-tumor responses in the tumor microenvironment (, left panel) [Citation122]. In line with these observations, the paracrine actions of cGAMP are being explored as a target for possible treatment strategies.
Tumor-promoting features of cGAS/STING signaling
In contrast to the observed STING-induced anti-tumor responses, cGAS/STING signaling also has tumor-promoting features (, right panel). For instance, cGAMP produced by cancer cells in the brain and transferred to astrocytes via gap-junctions was shown to promote cancer growth [Citation123]. Specifically, in response to cGAMP, astrocytes activated STING-signaling and produced cytokines, including IFN and TNF, which in turn activated STAT1 and NF-κB signaling in brain cancer cells to induce growth, chemoresistance and eventually promoted metastasis [Citation123].
As described above, cGAS/STING signaling elicits secretion of pro-inflammatory cytokines, which facilitate the recruitment of immune cells as part of an innate immune response. However, contradicting observations have been done in this context. Whereas STING signaling was demonstrated to inhibit activation of MDSCs to promote anti-tumor immune activation [Citation113], another study reported that STING signaling in response to irradiation promotes tumor infiltration of myeloid-derived suppressor cells, leading to resistance of cancer cells towards irradiation [Citation124]. Also, STING activation in tumors characterized by low antigenicity, promoted tumor growth via indoleamine-2,3-dioxygenase (IDO) activation [Citation125].
Important to realize in this context is that acute and chronic IFN responses lead to differential downstream effects. Whereas early type-I IFN responses promote elimination of tumor cells [Citation108], persistent inflammation, which is also accompanied by production of pro-inflammatory cytokines, promotes tumor growth and metastatic properties in established tumors [Citation126]. In good agreement with these findings, chronic STAT-1-mediated IFN responses trigger immune checkpoint activation and resistance towards anti-PD1, anti-PD-L1 or anti-CTLA4-targeted immune checkpoint blockade due to increased expression of T cell inhibitory receptors and exhausted T cells [Citation127]. Furthermore, genetic or pharmacological interference with tumor-induced IFN signaling through JAK inactivation improved responses of immune checkpoint therapy-resistant tumors [Citation127]. Of note, two CRISPR/Cas9-based genetic screens identified IFN-gamma signaling as a key requirement for successful T cell-based immunotherapies [Citation128,Citation129]. Based on these latter studies, one would argue against using inhibitors of interferon signaling in combination with immune checkpoint inhibitors.
In line with the observed tumor-promoting effects of a chronic IFN response, chromosomally instable tumor cells were shown to continuously trigger STING signaling due to their micronuclei, which promoted metastatic capacity [Citation130]. Surprisingly, in these tumor cells, cGAS/STING activation did not result in canonical downstream events, including TBK1/IRF3 phosphorylation, canonical NF-κB activation and type-I IFN secretion. Rather, chronic cGAS/STING activation was found to install non-canonical NF-κB activation, which was independent of TBK1 [Citation130]. In line with these findings, analysis of TCGA samples revealed a correlation between high chromosomal instability and expression of non-canonical NF-κB target genes in breast cancer [Citation130].
The observation that the downstream consequences of cGAS/STING are not generic and can be skewed towards non-canonical pro-tumorigenic effects resembles findings in senescent cells. Whereas cGAS/STING activation in senescent cells leads to secretion of pro-inflammatory cytokines, p38-MAPK signaling can prevent excretion of IFN, altering the SASP [Citation89]. In line with these findings, senescence has been demonstrated to exert pro-tumorigenic effects, including metalloproteinase-mediated remodeling of the extracellular matrix, which facilitates migration of tumor cells [Citation131,Citation132]. Also, SASP components, especially CXCL12, have been attributed to attract and promote the survival of cancer-associated fibroblasts (CAFs) [Citation133]. CXCL12, which is also excreted by CAFs, stimulates proliferation of tumor cells and promotes angiogenesis [Citation133,Citation134]. Combined, besides leading to permanent cell cycle arrest of damaged cells, the inflamed state of senescent cells promotes aggressive tumor behavior and is associated with poor prognosis [Citation135,Citation136].
In summary, cGAS/STING activation can lead to differential downstream effects in tumor cells (). In general, induction of an IFN response triggers the immune system to clear tumor cells. In contrast, non-canonical NF-κB activation triggered by chronic IFN responses preferentially leads to tumor growth and metastasis. These dual effects, including tumor-promoting features, might explain why cGAS and/or STING are hardly ever lost or mutated in cancer. Yet, it remains unclear how tumor cells deal with the tumor-eradicating effects of STING signaling. Further complicating these observations, cGAS itself was recently also described to have non-canonical functions in DNA repair, where it inhibits HR and may promote genomic instability and tumor progression [Citation137,Citation138].
How do genomic instable tumors escape cGAS/STING dependent immune clearance
cGAS/STING signaling clearly plays an important role in anti-tumor immune responses and promotes immune clearance of tumor cells. Yet, genomic instability is a common feature of cancer and a continuous source of cytoplasmic DNA, either through the formation of micronuclei or leakage of DNA fragments from aberrantly processed stalled replication forks [Citation139,Citation140]. As a consequence, tumor cells continuously produce intrinsic cues that activate cGAS/STING activation and subsequent inflammatory signaling. Indeed, it has been shown that high STING expression correlates with higher expression of pro-inflammatory genes in both cancer cell lines and multiple human cancers from database analyses [Citation89].
The notion that tumor cells frequently display cGAS/STING activation implies that during transformation of normal cells into malignant cells, cells evolve mechanisms to suppress the tumor cell-clearing effects of STING signaling to allow tumor formation (). How tumor cells achieve this, remains unclear.
Figure 3. Mechanisms by which tumor cells can escape anti-tumor effects of cGAS/STING signaling.
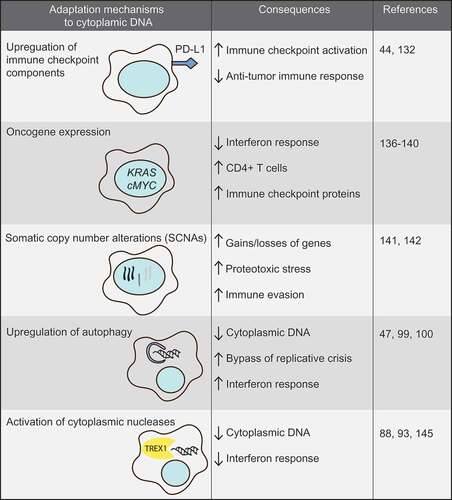
Suppression of STING signaling in tumor cells has been demonstrated, for instance in colorectal cancer cell lines and melanoma cells [Citation141,Citation142]. The level of STING suppression appeared functional, since it altered the cellular responses to virus-mediated therapies [Citation141,Citation142]. Furthermore, database analyses showed that STING signaling may be suppressed in tumors due to loss-of-function mutations in TMEM173, the gene encoding STING, or epigenetic silencing of CGAS/TMEM173, although the frequencies of these events were low [Citation142,Citation143].
In line with cGAS/STING signaling remaining intact in cancer cells, breast cancers with DNA repair defects showed cytoplasmic DNA, constitutive activation of cGAS/STING signaling and increased T cell infiltration, but did not trigger effective anti-tumor immune responses [Citation47]. The lack of an anti-tumor T cell response in these tumor cells could be explained by DNA damage-induced STING activation and subsequent upregulation of the immune checkpoint component PD-L1 [Citation47,Citation144]. Thus, although cGAS/STING signaling in tumor cells is activated, the consequent anti-tumor immune response can be counterbalanced, for instance, through increased expression of immune-checkpoint proteins.
Suppression of the anti-tumor cGAS/STING signaling cascade might also be achieved by oncogene overexpression. MYC, encoding the transcription factor c-MYC, is frequently found amplified in multiple cancer types and is an established oncogene [Citation145]. In tumors that are characterized by high genomic instability, e.g. high-grade serous ovarian cancers and triple negative breast cancers, more than half show amplification of MYC [Citation146,Citation147]. C-MYC overexpression is not only a critical oncogenic driver of tumor growth but also has inflammation modulatory effects. In a KRAS-driven tumor model, c-MYC expression was shown to contribute to both immunosuppressive and inflammatory phenotypes in the tumor microenvironment, with the CCL9 and IL-23-mediated tumor-promoting effects [Citation148]. Conversely, c-MYC inactivation in models of lymphoma and B cell leukemia lead to alterations in cytokine release and increased numbers of CD4+ T cells within the tumor microenvironment, which mediated tumor regression [Citation149]. Furthermore, c-MYC inactivation lead to down-regulation of the PD-L1 immune checkpoint protein on tumor cells, further underscoring a role for c-MYC in shaping immune responses in the tumor microenvironment [Citation150]. Similarly, also the KRAS oncogene was recently shown to modulate inflammatory responses. Specifically, KRAS inhibits IRF2 and thereby down-regulates IFN responses, resulting in increased resistance towards immune checkpoint inhibition [Citation151]. Likewise, expression of the viral HPV oncogenes E1A and E7 in cervical cancer were described to interact with STING to inhibit DNA sensing and prevent activation of the cGAS/STING pathway [Citation152]. These combined data support a model in which oncogene activation not only drives proliferation but simultaneously alters the expression of immune checkpoints on tumor cells and the subsequent presence and activation of immune cells to ultimately escape anti-tumor immunity.
Alternatively, tumors with high levels of genomic instability may evolve karyotypes that go along with immune evasion. Specifically, tumors with high levels of aneuploidy showed a reduction in cytotoxic infiltrating immune cells and conversely, an increased expression of cell proliferation markers [Citation153]. Although it remains elusive how aneuploidy results in immune evasion mechanistically, high levels of somatic copy number alterations (SCNAs) were predictive for poor response to CTLA-4-mediated immunotherapy and could serve as a biomarker in this context [Citation153,Citation154].
Another mechanism by which tumor cells can adapt to deal with inflammatory signaling that is triggered by cytoplasmic DNA, is autophagy upregulation. Autophagy is a catabolic process that involves self-digestion of organelles and has been shown to affect multiple aspects of tumor cell biology, including tumor suppression [Citation155,Citation156]. However, elevated levels of autophagy were recently shown to allow bypass of replicative crisis and enhanced survival of genomically instable cells [Citation106]. Of note, DNA in the cytoplasm can trigger autophagy to mediate clearance of cytoplasmic DNA in a manner that depends on STING but is independent of IFN secretion [Citation107]. In line with these findings, inhibiting autophagy aggravated the IFN response, whereas induction of autophagy leads to bypass of replicative crisis to continue proliferation [Citation50,Citation106].
Finally, multiple nucleases, including TREX1, are able to clear cytoplasmic DNA and thereby prevent cell-intrinsic immunity [Citation95,Citation100]. Tumor cells utilize this mechanism to dampen the cellular response to cytoplasmic DNA. For instance, TREX1 is induced in tumor cells upon irradiation to degrade irradiation-induced cytoplasmic DNA [Citation157]. This response prevents activation of cGAS/STING-induced IFN secretion and subsequent activation of surrounding immune cells. Possibly, tumor cells with high expression levels of such nucleases may be less susceptible to therapies that induce DNA damage and cGAS/STING activation.
Targeting the inflammatory signaling in genomically instable cancers
cGAS/STING signaling as a determinant of anti-cancer therapy response
Similar to other features of cancer cells, the presence of cytoplasmic DNA in tumor cells appears to be a determinant of tumor behavior and treatment outcome and might turn out to be an actionable vulnerability of tumor cells.
The induction of micronuclei has for long been recognized as a consequent of radiotherapy as well as genotoxic chemotherapeutics [Citation158–Citation160]. Treatment-induced micronuclei formation has been linked to adaptation to a G2/M cell cycle arrest. Similarly, treatment with genotoxic chemotherapeutics or radiotherapy was shown to increase IFN signaling [Citation161,Citation162]. Increasingly, we realize that the treatment-induced interferon response that goes along with micronuclei formation is not merely a bystander effect, but also a determinant of treatment outcome. For instance, irradiation-induced secretion of Type-I IFN triggers both innate and adaptive immune mechanisms that target tumor cells [Citation163]. In line with these findings, intra-tumoral administration of type-I IFN could mimic the effects of irradiation on tumor regression [Citation163]. Furthermore, the STING-dependent inflammatory response in tumor cells is linked to the abscopal effects on distinct lesions and sensitivity to anti-CTLA4 treatment [Citation85]. Similarly, inhibition of colony-stimulating factor-1 receptor (CSF-1R) resulted in enhanced IFN signaling in breast cancer and led to an increased sensitivity to chemotherapy [Citation164].
Also, the anti-neoplastic effects of the anti-mitotic drug paclitaxel have been related to inflammatory micronucleus formation [Citation165]. For long, the cytotoxic effects of the microtubule drug paclitaxel were related to its ability to arrest cells in mitosis. However, paclitaxel treatment was also shown to induce aberrant mitotic exit and extensive micronucleation [Citation166,Citation167]. Importantly, the paclitaxel-induced micronucleus formation went along with DNA damage induction, but not apoptosis induction per se. Conversely, the ability of cancer cells to induce IFN signaling in response to DNA damage was shown to confer treatment resistance. Specifically, in a TREX1 deficient background, breast cancer cells became resistant to radiotherapy [Citation57]. This was attributed to the role of TREX1 in clearance of irradiation-induced cytoplasmic DNA, which is in part caused by the formation of ssDNA fragments [Citation57]. In line with this notion, irradiation was shown to be more effective in repeated low-doses compared to high dose to prevent induction of TREX1 and to effectively trigger IFN production [Citation157]. The expression of certain nucleases in tumor cells might therefore serve as a marker to guide irradiation dose and fractioning.
PARP inhibitors have been shown to effectively target tumors with BRCA1/2 defects and are described to target HR-defective tumors based on synthetic lethality [Citation168]. Currently, several PARP inhibitors are approved for treatment of BRCA1/2-mutant high-grade serous ovarian cancer, breast cancer, and pancreatic cancer. Recently, effective killing of HR-deficient tumor cells upon PARP inhibitor treatment was shown to involve defects in mitosis, leading to micronucleation and mitotic catastrophe [Citation34]. In line with these observations, PARP inhibitor treatment was shown to trigger an anti-tumor immune response via tumor-derived cGAMP which activated STING signaling in immune cells in a BRCA1-deficient tumor model [Citation169]. Furthermore, treatment with PARP inhibitors upregulated PD-L1 expression on tumor cells and a combination with anti-PD-1 enhanced the survival of BRCA1-tumor bearing mice [Citation169,Citation170]. Importantly, treatment with PARP inhibitor also triggered the accumulation of cytoplasmic DNA and thus cGAS/STING activation independent of BRCA1/2 mutation status [Citation171]. Finally, the effectiveness of PARP inhibitor treatment, especially in HR-deficient tumors, seemed to be dependent on tumor infiltration of CD8+ T cells [Citation172]. These data further support the rational of combining PARP inhibitors with immune checkpoint therapies.
In good agreement with inflammatory signaling being a determinant of therapy response, expression of a set of IFN-induced genes in cancer cell lines was shown to correlate with chemotherapy or radiotherapy resistance and could be used to separate high-from low-risk patients [Citation173]. Specifically, a panel of seven of these IFN-induced genes could identify resistance to chemo- and radiotherapy in breast cancer patients. Silencing of these IFN-induced genes could subsequently reverse the resistance of triple negative breast cancer cells to chemo- and radiotherapy, again underscoring that IFN is not a bystander effect but is causally involved in treatment outcome [Citation174]. Similarly, activation of IFN/STAT1 signaling was shown to predict chemotherapy response in ER-negative breast cancer [Citation175]. These studies indicate further that IFN signaling plays an important role in therapy sensitivity, immune cell activity and underscores the potential value to target this response in tumor cells.
Therapeutic activation of STING signaling
The importance of STING-induced IFN signaling in tumor responses to genotoxic agents, might be of use to therapeutically activate STING intra-tumorally and thereby enhancing innate immune responses. The flavonoid DMXAA was shown to function as a mouse-specific STING ligand and has anti-tumor effects in solid tumors [Citation176,Citation177]. Intra-tumoral injection of DMXAA or human STING-specific cyclic dinucleotide derivates induced regression of established tumors as well as metastatic lesions [Citation178]. Specifically, intra-tumoral injection of STING agonists in multiple cancer mouse models improved anti-tumor CD8+ T cell responses, which were further enhanced by immune checkpoint inhibition [Citation179,Citation180]. Surprisingly, type I IFN production in one of these studies was shown to come from tumor-associated endothelial cells rather than tumor cells or dendritic cells [Citation179]. In this context, administration of liposomal nanoparticle-delivered cGAMP was shown to be more effective than soluble cGAMP, circumventing the need for intra-tumoral injections [Citation181]. Nanoparticle delivery of cGAMP was effective in different tumor models resistant to PD-L1 checkpoint blockade, whereas the observed tumor regression was lost in STING- or IFNAR-deficient mice [Citation181]. In good agreement with the described roles of irradiation on STING-induced IFN responses, cGAMP treatment in combination with irradiation further increased anti-tumor CD8+ T cell responses, in a STING-dependent fashion [Citation119].
Targeting innate immune checkpoints
The described effects of cGAS/STING pathway activation on innate immunity suggest a prominent role for immune checkpoint inhibition in genomically instable tumors. cGAS and STING protein levels were shown to correlate with PD-L1 levels in ovarian cancer cell lines and PD-L1 levels were further enhanced by cGAMP treatment [Citation182]. Furthermore, combined treatment of cGAMP with anti-PD-L1 increased the anti-tumor effects of in vivo injected melanoma cell lines, which was attributed to enhanced STING-dependent tumor antigen cross-presentation in dendritic cells [Citation183]. PD-L1 expression was also increased upon induction of DNA DSBs, through activation of the ATM, ATR and Chk1 kinases, and was further increased upon loss of DNA repair proteins, including BRCA2 or Ku70/80 [Citation144]. Thus, combination treatment of agents that induce DSBs while inhibiting cell cycle checkpoint inhibitors (e.g. ATM, ATR or Chk1), might therefore prevent increase of PD-L1 expression and thus decrease response to immune checkpoint inhibitors [Citation184].
Conclusions and outlook
Vertebrates have evolved an elegant system by which detection of foreign DNA in the cytosol triggers an innate immune response. This same mechanism is also triggered by cytoplasmic self DNA, a frequently occurring feature of tumor cells due to their genomic instability or induced by genotoxic treatments. The response to cytoplasmic DNA in tumor cells has gained enormous attention over the past few years, because cGAS/STING signaling was shown to be activated upon cytoplasmic DNA, which established a direct link between genomic instability and inflammatory signaling. The subsequent type-I IFN response plays important roles in tumor growth, immune evasion and determines treatment outcome.
The increasing knowledge on the impact of cGAS/STING signaling on anti-tumor immunity has led to increasing endeavors to target this pathway therapeutically. STING agonists have been developed, including synthetic cGAMP, and are used to boost infiltration and activation of immune cells into the tumor microenvironment. However, cGAMP administration alone might not be sufficient, as STING activation by cGAMP on its own resembles immune cells with low cross-priming activity [Citation119]. Combining cGAMP treatment with genotoxic therapies, such as irradiation, could enhance these responses through recruitment of multiple immune cells and engagement of several DNA damage response pathways.
However, caution should be taken regarding cGAMP treatment in tumors which are not chromosomal instable, as has been shown that cGAMP increases invasion and migration of cells with low chromosomal instability, probably due to the tumor-promoting effects of non-canonical NF-κB activation [Citation130,Citation185]. Also, treatment schedule and dosing may be of impact on the effectiveness of cGAMP treatment. Repeated treatments and high dosages were found to be unfavorable for long-term tumor-specific T cell responses [Citation180]. Important in this context is the notion that the induction of STING-mediated inflammatory signaling has both pro-tumorigenic and anti-tumorigenic effects. Currently, it is unclear how tumor cells have adapted to dealt with STING activation and shape the downstream effects into effects that promote growth and evasion of immune clearance. Multiple non-exclusive mechanisms may be responsible, including increased autophagy and non-canonical effects of oncogene activation.
Acknowledgments
F.T. and M.A.T.M.v.V. are supported by the Dutch Cancer Society/Alpe D’huzes (Grant EMCR2014-7048). M.A.T.M.v.V. is supported by the European Research Council (ERC CoS Grant 682421).
Disclosure statement
M.A.T.M.v.V. has acted on the scientific advisory board of Repare Therapeutics.
Additional information
Funding
Related Research Data
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117–1130.
- Li G-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98.
- Hampel H, Frankel WL, Martin E, et al. Screening for the lynch syndrome (Hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005;352:1851–1860.
- Koopman M, Kortman GAM, Mekenkamp L, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer. 2009;100:266–273.
- Latham A, Srinivasan P, Kemel Y, et al. Microsatellite instability is associated with the presence of lynch syndrome pan-cancer. J Clin Oncol. 2019;37:286–295.
- Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12:801–817.
- Maciejowski J, de Lange T. Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol. 2017;18:175–186.
- von Morgen P, Maciejowski J. The ins and outs of telomere crisis in cancer. Genome Med. 2018;10:89.
- Maciejowski J, Li Y, Bosco N, et al. Chromothripsis and kataegis induced by telomere crisis. Cell. 2015;163:1641–1654.
- Kotsantis P, Petermann E, Boulton SJ. Mechanisms of oncogene-induced replication stress: jigsaw falling into place. Cancer Discov. 2018;8:537–555.
- Schoonen PM, Guerrero Llobet S, van Vugt MATM. Replication stress: driver and therapeutic target in genomically instable cancers. Adv Protein Chem Struct Biol. 2019;115:157–201.
- Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009;220:292–296.
- Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–465.
- Macheret M, Halazonetis TD. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature. 2018;555:112–116.
- Poli J, Tsaponina O, Crabbé L, et al. dNTP pools determine fork progression and origin usage under replication stress. Embo J. 2012;31:883–894.
- Bester AC, Roniger M, Oren YS, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446.
- Neelsen KJ, Zanini IMY, Herrador R, et al. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol. 2013;200:699–708.
- Jones, RM, Mortusewicz O, Afzal I, et al. Increased replication initiation and conflicts with transcription underlie cyclin E-induced replication stress. Oncogene. 2013;32:3744–3753.
- Hirao A, Kong, YY, Matsuoka S, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–1827.
- Kastan MB, Onyekwere O, Sidransky D, et al. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311.
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability — an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–228.
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319(80):1352–1355.
- Gorgoulis VG, Vassiliou LVF, Karakaidos P et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913.
- Kandoth C, McLellan, MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339.
- Synnott NC, Murray A, McGowan PM, et al. Mutant p53: a novel target for the treatment of patients with triple-negative breast cancer? Int J Cancer. 2017;140:234–246.
- Cole A J, Dwight T, Gill AJ, et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci Rep. 2016;6:26191.
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
- De Witt Hamer PC, Mir SE, Noske D, et al. WEE1 kinase targeting combined with DNA-damaging cancer therapy catalyzes mitotic catastrophe. Clin Cancer Res. 2011;17:4200–4207.
- Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429.
- Naim V, Wilhelm T, Debatisse M, et al. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol. 2013;15:1008–1015.
- Minocherhomji S, Ying S, Bjerregaard VA, et al. Replication stress activates DNA repair synthesis in mitosis. Nature. 2015;528:286–290.
- Ying S, Minocherhomji S, Chan KL, et al. MUS81 promotes common fragile site expression. Nat Cell Biol. 2013;15:1001–1007.
- Schoonen PM, Talens F, Stok C, et al. Progression through mitosis promotes PARP inhibitor-induced cytotoxicity in homologous recombination-deficient cancer cells. Nat Commun. 2017;8:15981.
- Gisselsson D, Pettersson L, Höglund M, et al. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci. 2000;97:5357–5362.
- Janssen A, van der Burg M, Szuhai K, et al. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–1898.
- Crasta K, Ganem NJ, Dagher R, et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–58.
- Sheltzer JM, Blank HM, Pfau SJ, et al. Aneuploidy drives genomic instability in yeast. Science. 2011;333:1026–1030.
- Liu S, Kwon M, Mannino M, et al. Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature. 2018;561:551–555.
- Terradas M, Martín M, Genescà A. Impaired nuclear functions in micronuclei results in genome instability and chromothripsis. Arch Toxicol. 2016;90:2657–2667.
- Soto M, García-Santisteban I, Krenning L, et al. Chromosomes trapped in micronuclei are liable to segregation errors. J Cell Sci. 2018;131:jcs214742.
- Bakhoum SF, Cantley LC. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell. 2018;174:1347–1360.
- Zhang C-Z, Spektor, A, Cornils H, et al. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522:179–184.
- Hatch EM, Fischer AH, Deerinck TJ, et al. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell. 2013;154:47–60.
- Kalsbeek D, Golsteyn RM. G2/M-Phase Checkpoint Adaptation and Micronuclei Formation as Mechanisms That Contribute to Genomic Instability in Human Cells. Int J Mol Sci. 2017;18:2344.
- Heijink AM, Talens F, Jae LT, et al. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat Commun. 2019;10:100.
- Parkes E E, Walker SM, Taggart LE, et al. Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. J Natl Cancer Inst. 2017;109:djw199.
- Laulier C, Cheng A, Stark JM. The relative efficiency of homology-directed repair has distinct effects on proper anaphase chromosome separation. Nucleic Acids Res. 2011;39:5935–5944.
- Williams JS, Kunkel TA. Ribonucleotides in DNA: origins, repair and consequences. DNA Repair (Amst). 2014;19:27–37.
- Bartsch K, Knittler K, Borowski C, et al. Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum Mol Genet. 2017;26:3960–3972.
- Cornelio DA, Sedam HNC, Ferrarezi JA, et al. Both R-loop removal and ribonucleotide excision repair activities of RNase H2 contribute substantially to chromosome stability. DNA Repair (Amst). 2017;52:110–114.
- Fenech M. The cytokinesis-block micronucleus technique: a detailed description of the method and its application to genotoxicity studies in human populations. Mutat Res. 1993;285:35–44.
- Härtlova A, Erttmann SF, Raffi FA, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. 2015;42:332–343.
- Chabanon RM, Soria J-C, Lord CJ, et al. Beyond DNA repair: the novel immunological potential of PARP inhibitors. Mol Cell Oncol. 2019;6:1585170.
- Flynn RL, Zou L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci. 2011;36:133–140.
- Hashimoto Y, Ray Chaudhuri A, Lopes M, et al. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol. 2010;17:1305–1311.
- Erdal E, Haider S, Rehwinkel J, et al. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by trex1. Genes Dev. 2017;31:353–369.
- Wolf C, Rapp A, Berndt N, et al. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat Commun. 2016;7:11752.
- Coquel F, Silva MJ, Técher H, et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature. 2018;557:57–61.
- Beloglazova N, Flick R, Tchigvintsev A, et al. Nuclease activity of the human SAMHD1 protein implicated in the aicardi-goutieres syndrome and HIV-1 restriction. J Biol Chem. 2013;288:8101–8110.
- Daddacha W, Koyen AE, Bastien AJ, et al. SAMHD1 promotes DNA end resection to facilitate DNA repair by homologous recombination. Cell Rep. 2017;20:1921–1935.
- Luecke S, Holleufer, A, Christensen MH, et al. cGAS is activated by DNA in a length‐dependent manner. EMBO Rep. 2017;18:1707–1715.
- Kranzusch PJ, Lee AS-Y, Berger JM, et al. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 2013;3:1362–1368.
- Wu J, Sun L, Chen X, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339(80):826–830.
- Sun L, Wu J, Du F, et al. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science. 2013;339(80):786–791.
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678.
- Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol. 2014;88:5328–5341.
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792.
- Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103.
- Ma F, Li B, Liu SY, et al. Positive feedback regulation of type I IFN production by the IFN-inducible DNA sensor cGAS. J Immunol. 2015;194:1545–1554.
- Ning X, Wang Y, Jing M, et al. Apoptotic caspases suppress type I interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol Cell. 2019;74:19–31.e7.
- Unterholzner L, Keating SE, Baran M, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997–1004.
- Dunphy G, Flannery SM, Almine, JF, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol Cell. 2018;71:745–760.e5.
- Veeranki S, Choubey D. Interferon-inducible p200-family protein IFI16, an innate immune sensor for cytosolic and nuclear double-stranded DNA: regulation of subcellular localization. Mol Immunol. 2012;49:567–571.
- Ponomareva L, Liu H, Duan X, et al. AIM2, an IFN-inducible cytosolic DNA sensor, in the development of benign prostate hyperplasia and prostate cancer. Mol Cancer Res. 2013;11:1193–1202.
- Schroder K, Muruve DA, Tschopp J. Innate immunity: cytoplasmic DNA sensing by the AIM2 inflammasome. Curr Biol. 2009;19:R262–R265.
- Fernandes-Alnemri T, Yu J-W, Datta P, et al. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513.
- Hornung V, Ablasser A, Charrel-Dennis M, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518.
- Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. 2013;14:19–26.
- Zhao Y, Ye X, Dunker W, et al. RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat Commun. 2018;9:4841.
- Chan YK, Gack MU. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol. 2016;14:360–373.
- Franz KM, Neidermyer WJ, Tan Y-J, et al. STING-dependent translation inhibition restricts RNA virus replication. Proc Natl Acad Sci U S A. 2018;115:E2058–E2067.
- Rodier F, Coppé JP, Patil CK, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979.
- Di Maggio F, Minafra L, Forte GI, et al. Portrait of inflammatory response to ionizing radiation treatment. J Inflamm. 2015;12:14.
- Harding S M, Benci JL, Irianto J, et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466–470.
- Glück S, Guey B, Gulen MF, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19:1061–1070.
- Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215:1287–1299.
- Chen Y-A, Shen, YL, Hsia HY, et al. Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway. Nat Struct Mol Biol. 2017;24:1124–1131.
- Dou Z., Ghosh K, Vizioli MG, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–406.
- Mankan AK, Schmidt T, Chauhan D, et al. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. Embo J. 2014;33:2937–2946.
- Crow YJ, Manel N. Aicardi–goutières syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15:429–440.
- Crow YJ, Leitch A, Hayward BE, et al. Mutations in genes encoding ribonuclease H2 subunits cause aicardi-goutières syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910–916.
- Mackenzie KJ, Carroll P, Lettice L, et al. Ribonuclease H2 mutations induce a cGAS/STING‐dependent innate immune response. Embo J. 2016;35:831–844.
- Maelfait J, Bridgeman A, Benlahrech A, et al. Restriction by SAMHD1 limits cGAS/STING-dependent innate and adaptive immune responses to HIV-1. Cell Rep. 2016;16:1492–1501.
- Yang Y-G, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131:873–886.
- Rice GI, Rodero MP, Crow YJ. Human disease phenotypes associated with mutations in TREX1. J Clin Immunol. 2015;35:235–243.
- Ablasser A, Hemmerling I, Schmid-Burgk JL, et al. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol. 2014;192:5993–5997.
- Yan N, Regalado-Magdos AD, Stiggelbout B, et al. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol. 2010;11:1005–1013.
- Gao D, Wu J, Wu YT, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906.
- Stetson DB, Ko JS, Heidmann T, et al. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598.
- Chien Y, Scuoppo C, Wang X, et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–2136.
- Kang T-W, Yevsa T, Woller N, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551.
- Sagiv A, Krizhanovsky V. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology. 2013;14:617–628.
- Yang H, Wang H, Ren J, et al. J. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A. 2017;114:E4612–E4620.
- Comb WC, Cogswell P, Sitcheran R, et al. IKK-dependent, NF-κB-independent control of autophagic gene expression. Oncogene. 2011;30:1727–1732.
- Nassour J, Radford R, Correia A, et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature. 2019;565:659–663.
- Gui X, Yang H, Li T, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;1. DOI:10.1038/s41586-019-1006-9
- Diamond MS, Kinder M, Matsushita H, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003.
- Fuertes MB, Kacha AK, Kline J, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–2016.
- Muthuswamy R, Berk E, Junecko BF, et al. NF-κB hyperactivation in tumor tissues allows tumor-selective reprogramming of the chemokine microenvironment to enhance the recruitment of cytolytic T effector cells. Cancer Res. 2012;72:3735–3743.
- Woo S-R., Fuertes MB, Corrales L, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842.
- Ho SSW, Zhang WY, Tan NYJ, et al. The DNA structure-specific endonuclease MUS81 mediates DNA sensor STING-dependent host rejection of prostate cancer cells. Immunity. 2016;44:1177–1189.
- Zhang C, Ye SB, Ni JJ, et al. STING signaling remodels the tumor microenvironment by antagonizing myeloid-derived suppressor cell expansion. Cell Death Differ. 2019. DOI:10.1038/s41418-019-0302-0
- Lam AR, Le Bert N, Ho SS, et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res. 2014;74:2193–2203.
- Gasser S, Orsulic S, Brown EJ, et al. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–1190.
- Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761.
- Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. 2014;5:614.
- Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer. 2017;117:1583–1591.
- Deng L, Liang H, Xu M, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843–852.
- Ablasser A, Schmid-Burgk, JL Hemmerling I, et al. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. 2013;503:530–534.
- Marcus A, Mao AJ, Lensink-Vasan M, et al. Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity. 2018;49:754–763.e4.
- Sundararaman SK, Barbie DA. Tumor cGAMP Awakens the Natural Killers. Immunity. 2018;49:585–587.
- Chen Q, Boire A, Jin X, et al. Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493–498.
- Liang H, Deng L, Hou Y, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun. 2017;8:1736.
- Lemos H, Mohamed E, Huang L, et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 2016;76:2076–2081.
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867.
- Benci JL, Xu B, Qiu Y, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016;167:1540–1554.e12.
- Manguso RT, Pope H.W., Zimmer, M.D., et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413–418.
- Patel SJ, Sanjana NE, Kishton RJ, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548:537–542.
- Bakhoum SF, Ngo B, Laughney AM, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553:467–472.
- Krtolica A, Parrinello S, Lockett S, et al. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98:12072–12077.
- Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67:3117–3126.
- Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348.
- Begley L, Monteleon C, Shah RB, et al. CXCL12 overexpression and secretion by aging fibroblasts enhance human prostate epithelial proliferation in vitro. Aging Cell. 2005;4:291–298.
- Milanovic M, Fan DN, Belenki D, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018;553:96–100.
- Demaria M, O’Leary MN, Chang J, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7:165–176.
- Liu H, Zhang H, Wu X, et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature. 2018.
- Jiang H, Panda S, Xue X, et al. The innate immune DNA sensor cGAS is a negative regulator of DNA repair hence promotes genome instability and cell death. bioRxiv. 2018;465401. DOI:10.1101/465401
- Gisselsson D, Björk J, Höglund M, et al. Abnormal nuclear shape in solid tumors reflects mitotic instability. Am J Pathol. 2001;158:199–206.
- Ng KW, Marshall EA, Bell JC, et al. L. cGAS–STING and cancer: dichotomous roles in tumor immunity and development. Trends Immunol. 2018;39:44–54.
- Xia T, Konno H, Ahn J, et al. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016;14:282–297.
- Xia T, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 2016;76:6747–6759.
- Konno H, Yamauchi S, Berglund A, et al. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene. 2018;37:2037–2051.
- Sato H, Niimi A, Yasuhara T, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. 2017;8:1751.
- Jain M, Arvanitis C, Chu K, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297:102–104.
- Zeng M, Kwiatkowski NP, Zhang T, et al. Targeting MYC dependency in ovarian cancer through inhibition of CDK7 and CDK12/13. Elife. 2018;7:1–20.
- Horiuchi D, Kusdra L, Huskey NE, et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med. 2012;209:679–696.
- Kortlever RM, Sodir NM, Wilson CH, et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell. 2017;171:1301–1315.e14.
- Rakhra K, Bachireddy P, Zabuawala T, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell. 2010;18:485–498.
- Casey SC, Tong L, Li Y, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;9935(80):1–9.
- Hänggi K, Ruffell B. Oncogenic KRAS Drives Immune Suppression in Colorectal Cancer. Cancer Cell. 2019;35:535–537.
- Lau A, Gray EE, Brunette RL, et al. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science. 2015;350(80):568–571.
- Davoli T, Uno H, Wooten EC, et al. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. 2017;355(6322):eaaf8399.
- Budczies J, Seidel A, Christopoulos P, et al. Integrated analysis of the immunological and genetic status in and across cancer types: impact of mutational signatures beyond tumor mutational burden. Oncoimmunology. 2018;7:e1526613.
- Karantza-Wadsworth V, Patel S, Kravchuk O, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–1635.
- Mathew R, Kongara S, Beaudoin B, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381.
- Vanpouille-Box C, Alard A, Aryankalayil MJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618.
- Lewis CW, Golsteyn RM. Cancer cells that survive checkpoint adaptation contain micronuclei that harbor damaged DNA. Cell Cycle. 2016;15:3131–3145.
- Hermine T, Jones NJ, Parry JM. Comparative induction of micronuclei in repair-deficient and -proficient Chinese hamster cell lines following clastogen or aneugen exposures. Mutat Res. 1997;392:151–163.
- Kubara PM, Kernéis-Golsteyn, S., Studény, A., et al. Human cells enter mitosis with damaged DNA after treatment with pharmacological concentrations of genotoxic agents. Biochem J. 2012;446:373–381.
- Khodarev NN, Beckett M, Labay E, et al. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A. 2004;101:1714–1719.
- Sistigu A, Yamazaki T, Vacchelli E, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20:1301–1309.
- Burnette BC, Liang H, Lee Y, et al. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011;71:2488–2496.
- Salvagno C, Ciampricotti M, Tuit S, et al. Therapeutic targeting of macrophages enhances chemotherapy efficacy by unleashing type I interferon response. Nat Cell Biol. 2019;21:511–521.
- Mitchison TJ, Pineda J, Shi J, et al. Is inflammatory micronucleation the key to a successful anti-mitotic cancer drug? Open Biol. 2017;7:170182.
- Jordan MA, Wendell K, Gardiner S, et al. Mitotic block induced in heLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 1996;56:816–825.
- Shi J, Orth JD, Mitchison T. Cell Type Variation in Responses to Antimitotic Drugs that Target Microtubules and Kinesin-5. Cancer Res. 2008;68:3269–3276.
- Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355(80):1152–1158.
- Ding L, Kim HJ, Wang Q, et al. PARP inhibition elicits STING-dependent antitumor immunity in brca1-deficient ovarian cancer. Cell Rep. 2018;25:2972–2980.e5.
- Jiao S, Xia W, Yamaguchi H, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23:3711–3720.
- Shen J, Zhao W, Ju Z, et al. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019;79:311–319.
- Pantelidou C, Sonzogni O, Taveira MDO, et al. PARP inhibitor efficacy depends on CD8+ T cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. 2019;9(6):722–737.
- Weichselbaum RR, Ishwaran H, Yoon T, et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci. 2008;105:18490–18495.
- Boelens MC, Wu TJ, Nabet BY, et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. 2014;159:499–513.
- Legrier M-E., Bièche I, Gaston J, et al. Activation of IFN/STAT1 signalling predicts response to chemotherapy in oestrogen receptor-negative breast cancer. Br J Cancer. 2016;114:177–187.
- Conlon J, Burdette DL, Sharma S, et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol. 2013;190:5216–5225.
- Kim S, Li L, Maliga Z, et al. Anticancer flavonoids are mouse-selective STING agonists. ACS Chem Biol. 2013;8:1396–1401.
- Corrales L, Glickman LH, McWhirter SM, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11:1018–1030.
- Demaria O, De Gassart A, Coso S, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci. 2015;112:15408–15413.
- Sivick KE, Desbien AL, Glickman LH, et al. Magnitude of therapeutic STING activation determines CD8+ T cell-mediated anti-tumor immunity. Cell Rep. 2018;25:3074–3085.e5.
- Cheng N, Watkins-Schulz R., Junkins RD, et al. A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1–insensitive models of triple-negative breast cancer. JCI Insight. 2018;3(22):e120638.
- Grabosch S, Bulatovic M, Zeng F, et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene. 2018;38(13):2380–2393.
- Wang H, Hu S, Chen X, et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A. 2017;114:1637–1642.
- Mouw KW, Konstantinopoulos PA. From checkpoint to checkpoint: DNA damage ATR/Chk1 checkpoint signalling elicits PD-L1 immune checkpoint activation. Br J Cancer. 2018;118:933–935.
- Wang J, Yi S, Zhou J, et al. The NF-κB subunit RelB regulates the migration and invasion abilities and the radio-sensitivity of prostate cancer cells. Int J Oncol. 2016;49:381–392.