
ABSTRACT
Aurora-A is a serine/threonine kinase, which is overexpressed in multiple human cancers and plays a key role in tumorigenesis and tumor development. In this study, we found that the receptor of activated C-kinase1 (RACK1), an important regulator of biological functions, interacted with Aurora-A and co-localized with Aurora-A at centrosomes. Moreover, RACK1 induces the auto-phosphorylation of Aurora-A in vitro and in vivo. Depletion of RACK1 impaired the activation of Aurora-A in late G2 phase, then inhibited the mitotic entry and leaded to multi-polarity, severe chromosome alignment defects, or centrosome amplification. Taken together, these results suggest that RACK1 is a new partner of Aurora-A and play a critical role in the regulation of the Aurora-A activity during mitosis, which may provide a basis for future anticancer studies targeting Aurora-A.
KEYWORDS:
Introduction
Aurora-A is a serine/threonine kinase in the Aurora kinase family [Citation1,Citation2]. The three members of this kinase family identified to date are referred to as Aurora-A, -B, and – C and are known to be involved in the regulation of centrosome function, bipolar spindle assembly, and chromosome segregation processes [Citation1,Citation2]. Aurora-A localizes to centrosomes and spindle microtubules proximal to centrosomes during mitosis [Citation2]. The expression and the activity of Aurora-A kinase are regulated in a cell cycle-dependent manner; levels are low in G1/S, upregulated during G2/M, and rapidly reduced after mitosis [Citation3–Citation5]. It is well known that activation of Aurora-A in the late G2 phase of the cell cycle is essential for recruitment of the cyclin B1-Cdk1 complex to centrosomes, which is required for mitotic entry [Citation6,Citation7]. The ectopic overexpression of Aurora-A induces oncogenic transformation [Citation2,Citation7], and inhibition of Aurora kinase activity in mammalian cells leads to abnormal cell growth and polyploidy centrosome dysfunction [Citation8]. Therefore, recent research has focused on developing drugs that inhibit aberrant activation of Aurora-A kinase, and several selective inhibitors of Aurora-A, including MLN 8237 and MLN 8054, have been developed and tested in clinical trials [Citation9,Citation10].
Aurora-A kinase has a C-terminal catalytic domain (Cd) that is highly conserved within the Aurora kinase family and is regulated by phosphorylation in a cell cycle-dependent manner. This phosphorylation, which is required for kinase activity, occurs on a conserved residue, threonine 288, within the activation loop (aa 274–299) of the kinase’s catalytic domain and significantly increases enzymatic activity [Citation11,Citation12]. Upon activation, Aurora-A phosphorylates many substrates, such as Cdc25B, histone H3, Lats2, to promote progression through the stages of mitosis [Citation13,Citation14]. Failure of Aurora-A activation results in G2 arrest or a defective entry into mitosis, marked by failure of centrosomes to separate and associated monopolar mitotic spindles, and consequent defects in chromosome alignment [Citation15,Citation16]. Aurora-A interacts with a number of proteins that are required for commitment to mitosis and cell-cycle checkpoints including TPX2, TACC, FBXL7, FoxO1 CBF-B, and PLK1 [Citation4,Citation17,Citation18].
Receptor of activated protein kinase C (RACK1, GNB2L1) is a 36-kDa cytosolic protein with a propeller-like structure of 7 WD40 repeats. Because of its association with a large number of signaling proteins such as protein kinase C (PKC), Src, FAK and PDE4D5 [Citation19]. RACK1 has been identified as an adaptor protein in multiple intracellular signal transduction pathways. As to the role of RACK1 in cell cycle, it was involved in the coordination of cell growth and movement by inhibiting Src activity at G1 and mitotic cell-cycle checkpoints and cell survival pathways [Citation20,Citation21]. It is showed that the RACK1 is required for cytokinesis, germline membrane organization, and the recruitment of RAB-11–labeled recycling endosomes to the peri-centrosomal region and spindle [Citation22]. Given the important roles of RACK1 in the mitotic process, we examined whether Aurora-A is functionally engaged with RACK1 in mitotic progression.
We now provide molecular insight into the regulation of mitotic entry in human cells. We identified RACK1 as a new partner that binds to Aurora-A, and this interaction was shown to result in the auto-phosphorylation and consequent activation of Aurora-A. The auto-phosphorylation of Aurora-A facilitated by interaction with RACK1 is required for the commitment of cells to mitosis. Collectively, we firstly found that Rack1 is a new partner of Aurora-A and is required in the mitosis by regulation of Aurora-A.
Results
RACK1 interacts with Aurora-A
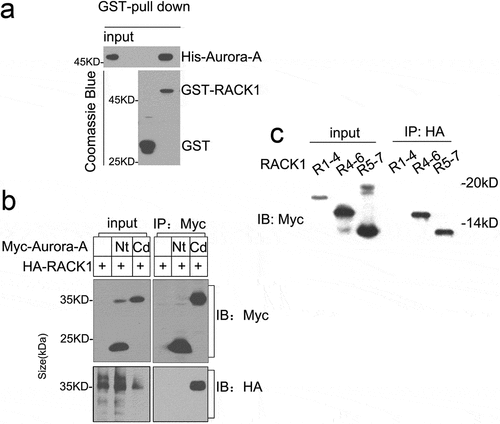
To explore the functional partner of Aurora-A, yeast two-hybrid assay was performed using Aurora-A as bait. Besides Ajuba, a previously reported Aurora-A association protein [Citation6], RACK1 was also found to interact with Aurora-A (data not shown). To validate the interaction between RACK1 and Aurora-A in vitro, His-tag Aurora-A and GST-tag RACK1 were expressed in E. coli and purified as described in methods, respectively. Then, the purified His-Aurora-A was incubated with GST-RACK1 or GST and pulled down by glutathione-Sepharose beads. As shown in , His-Aurora-A was precipitated by GST-RACK1, not GST only, suggesting that Aurora-A interacts with RACK1 directly in vitro. We constructed Myc-tagged Aurora-A’s N-terminal regulatory domain (aa 1–128, Nt) and the C-terminal catalytic domain (aa 129–403, Cd). Myc-Nt or Myc-Cd was co-transfected 293T cells with HA-tag RACK1, respectively. The lysates were subjected to immunoprecipitation with antibodies to Myc. As shown in , HA-RACK1 was detected in immunoprecipitates of Myc-Cd. RACK1 has seven WD repeats, to delineate the region of RACK1 responsible for its association with Aurora-A, we generated pCMV-Myc 1-4WD (aa 1–178), 4-6WD (aa 125–260), and 5–7WD (aa 181–318) and co-transfected 293T cells with HA-tag Aurora-A, respectively. Lysates of cells were then subjected to immunoprecipitation with antibodies to HA. Immunoblot analysis of the resulting precipitates revealed the presence of Myc RACK1 4-6WD and 5-7 WD in the precipitates of Aurora-A-HA [Citation23]. Taken together, these results indicate that RACK1 is a new partner of Aurora-A and the region of Aurora-A responsible for its association with RACK1 was C terminal region. We also demonstrated that both RACK1 4–7 WDs mediate the interaction with the Aurora-A.
Figure 1. RACK1 forms a complex with Aurora-A. (a) RACK1 associates with Aurora A directly in vitro. His-Aurora A was incubated with GST or GST-RACK1 for 2 h, then added glutathione-Sepharose beads and incubated for additional 1 h at 4°C. The beads were spinned down and washed with PBS for 3 times, and the bound proteins were analyzed by immunoblot. (b) RACK1 interacts with Aurora-A-Cd in vivo. 293T cells were transfected with Myc-Nt plus HA-RACK1 or Myc-Cd plus HA-RACK1 and cell lysates were analyzed by immunoprecipitation (IP) and western blotting with anti-Myc or HA antibodies, respectively. Whole-cell lysates were blotted and shown as the input. (c) RACK1 4–7 WDs interact with Aurora-A in vivo. 293T cells were transfected with Aurora-HA plus Myc-tagged RACK1 (1–4) WD, RACK1 (4–6)WD, RACK1(5–7)WD, respectively. The cell lysates were analyzed by IP against HA antibody and western blotting with anti-Myc.
RACK1 co-localized with Aurora-A at centrosome
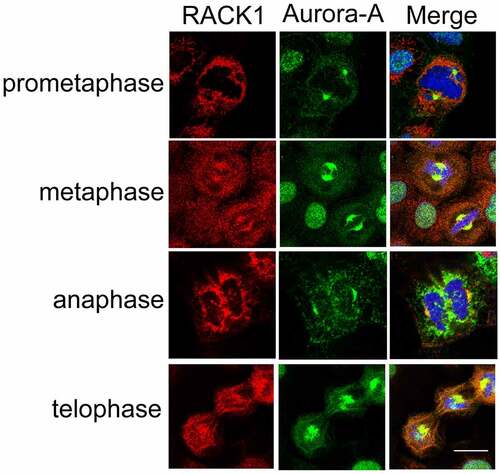
RACK1 interacts with Aurora-A, which localizes mainly to the centrosomes [Citation5], raising the possibility that RACK1 is co-localized with Aurora-A at the centrosomes during the cell cycle. To address the hypothesis, the subcellular localization of RACK1 and Aurora-A were examined in HeLa cells by immunofluorescence using anti-RACK1 and anti-Aurora-A antibodies, respectively. As shown in , RACK1 was co-localized with Aurora-A in the duplicated centrosomes as the cells progressed through mitosis from prophase to cytokinesis. Interestingly, there is also evidence showing that RACK1 localizes to the centrosomes during the cell cycle in C. elegans embryo [Citation24]. This finding suggests that RACK1 co-localized with Aurora-A on the centrosome during G2-M phase and may play an important role in cell-cycle regulation.
Figure 2. RACK1 co-localized with Aurora-A at centrosome. The subcellular localization of RACK1 and Aurora-A were examined by using anti- Aurora-A polyclonal antibody and anti- RACK1 monoclonal antibody in mitotic HeLa cells, respectively. Scale bar: 10 µm.
RACK1 regulates the kinase activity of Aurora-A
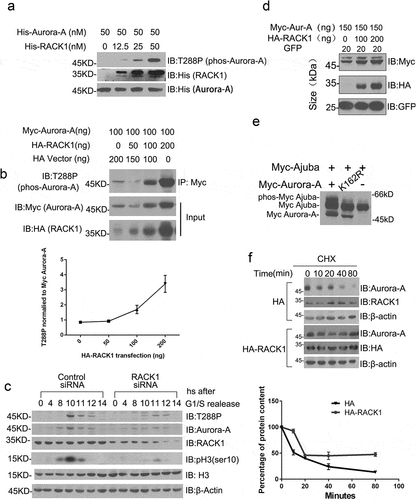
RACK1 has been reported to associate with several kinases and regulate their kinases activation [Citation25–Citation27]. Based on our co-immunoprecipitation and co-localization data, we speculated that RACK1 may also regulate the kinase activity of Aurora-A. We examined this possibility first in a cell-free system. Purified His-tag Aurora-A was incubated with various concentrations of His-tag RACK1 in a kinase reaction buffer. The auto-phosphorylation of Aurora-A on Thr288, as revealed by western blot using phos-T288 antibody, increased in a manner dependent on the amount of His-RACK1 (). Then, Myc-Aurora-A was co-transfected with various concentrations of HA-RACK1 into HeLa cells, and the auto-phosphorylation statues of Aurora-A was examined by western bolt using phos-T288 antibody. Consistent with the in vitro kinase assay, the expression of RACK1 increased the auto-phosphorylation of Aurora-A in cells (). Given that Histone H3 is a major substrate of Aurora-A and can be phosphorylated on Ser10 by Aurora-A during mitosis. We also detected the Aurora-A activity by detecting with antibodies specific for histone H3 phosphorylated on Ser10 at the indicated time after double thymidine block using HeLa cells treated with control siRNA or RACK1 siRNA. As shown in , depletion of RACK1 greatly inhibited the phosphorylation of its well-established downstream effectors, histone H3. Furthermore, we examined whether RACK1 can be phosphorylated by Aurora-A. 293T cells were transfected with expression vectors for both HA-RACK1 and either Myc-tagged wild-type Aurora-A or kinase-inactive mutants thereof (K162R). We did not notice the lower-mobility form of RACK1 as Ajuba appeared in cells co-expressing wide-type Aurora-A (), indicating that RACK1 is not phosphorylated by Aurora-A. These results indicated that RACK1 facilitates the auto-phosphorylation of Aurora-A in vitro and in vivo.
Figure 3. RACK1 regulates the kinase activity and protein stability of Aurora-A. (a) RACK1 induces the auto-phosphorylation of Aurora-A in vitro. Purified His-tag Aurora A was incubated with various concentrations of His-tag RACK1 for 30 min at 30°C. After the reaction, equal volume of 2× SDS sampling buffer was added and was boiled for 10 min. These samples were subjected to SDS-PAGE and immunoblotted by using indicated antibodies. (b) RACK1 induces the auto-phosphorylation of Aurora-A in vivo. Myc-Aurora A were co-transfected with HA vector or HA-RACK1 into HeLa cells. 48 hours later, the cells were harvested and the cell lysates were immunoprecipitated by using anti-Myc antibody, then analyzed by western blot using anti-phos-Aurora-A antibody. Whole-cell lysates were blotted using anti-HA and anti-Myc antibodies. (c) RACK1 depletion impaired the stability and the activation of Aurora A. HeLa cells treated with control siRNA or RACK1 siRNA were synchronized by using double thymidine block, and then harvested at various time points, and analyzed by western blot using indicated antibodies. (d) Myc-Aurora-A was co-transfected with gradient levels of HA-RACK1 into HeLa cells. eGFP-N1 was co-transfected as a transfection efficiency control. 48 hours later, the cells were harvested and the cell lysates were immunoblotted with anti-Myc and anti-HA antibodies. (e) Myc-Ajuba was co-transfected with wide-type Aurora-A or the kinase-inactive mutant thereof (K162R) into HeLa cells. 48 h later, the cells were harvested and the cell lysates were immunoblotted with anti-Myc antibody. (f) Lower degradation of endogenous Aurora-A with overexpression of RACK1. HeLa cells transfected with HA empty vector of HA-RACK1 were treated 20 ug/ml cycloheximide for 10, 20, 40, and 80 min. The levels of Aurora-A, endogenous RACK1, HA-RACK1 were analyzed using western blotting. Actin levels are shown as loading controls.
RACK1 regulates the stability of Aurora-A
We also noticed that the protein level of Myc-Aurora-A was also increased when co-transfected with HA-RACK1 (), suggesting that RACK1 may also regulate the stability of Aurora-A. To further confirm that, we examined the protein level of Aurora-A in RACK1-depletion cells. HeLa cells treated with control siRNA or RACK1 siRNA were synchronized by using double thymidine block, then harvested at various time points, and analyzed by western blot using indicated antibodies. Consistent with previous reports, the expression of Aurora-A and its auto-phosphorylation was increased and peaked at 10 h after release from thymidine block in control cells. However, the expression of Aurora-A and its auto-phosphorylation was dramatically suppressed in RACK1 depletion cells ().
To determine whether the increase of Aurora-A protein level by RACK1 is involved in the ubiquitin-proteasome pathway, de novo protein synthesis was blocked with the protein synthesis inhibitor CHX under normal culture conditions. The stability of short-lived Aurora-A with overexpression of HA-RACK1 or HA only was monitored for comparison. Treatment of 293T cells with 20 μg/ml CHX resulted in a time-dependent reduction of the Aurora-A levels, and only 20% of its initial level remained after exposure to CHX for 40 min. By contrast, the level of Aurora-A remained constant initially with overexpression of RACK1 and was then reduced by ~40% after treatment with CHX for 40 min (). These results indicated that RACK1 also regulate the stability of Aurora-A, although the underlying mechanism needs to be further investigated.
RACK1 is essential for mitotic entry and bipolar spindle assembly
RACK1 regulates the stability and kinase activity of Aurora-A, which is essential for mitotic entry, suggesting that RACK1 may also regulate the mitotic entry. To further examine the involvement of RACK1 in mitotic entry and progression, HeLa cells treated with control siRNA or RACK1 siRNA were synchronized by using double thymidine block, then harvested at various time points, and cell-cycle progression was monitored by flow cytometry using propidium iodide staining. As shown in , RACK1 siRNA treated cells progressed the G2/M phase much more slowly than control siRNA treated cell. These results indicate that RACK1 plays an important role in a mitotic entry like Aurora-A. To verify that G2/M arrest induced by RACK1 siRNA was due to RACK1 knock-down, a pcDNA3.1 tagged siRNA-resistant, RACK1 replacement vector (termed as RACK1-MUT) was generated by mutating nucleotide base pairs to alternative nucleotides within the RACK1 siRNA2 target region without altering the amino acid sequences, which would be expected to rescue the knockdown phenotype if RACK1 was responsible. HeLa cells were then stably integrated with pcDNA3.1 empty vector, RACK1-WT or RACK1-MUT (). The HeLa cells stably expressing pcDNA3.1, RACK1-WT or RACK1-MUT were synchronized and then transfected with either control siRNA or RACK1 siRNA2 in the interval between the two thymidine blocks. 13hr after the release, half of the cells were analyzed by western blot to detect endogenous RACK1 and exogenous RACK1 proteins expressed from siRNA-resistant cDNA constructs or wide-type RACK1 using the RACK1 antibody. Both endogenous RACK1 and wild-type Myc-RACK1 were knocked down to below 10% using actin as a control. In contrast, expression of Myc-RACK1 produced by the RACK-MUT vector containing four silent mutations was resistant to RACK1 siRNA 2 as expected. Another half of the cells were analyzed by flow cytometry. As expected, the introduction of RACK1 siRNA2 caused G2/M arrest in RACK1-WT clone, and reintroduction of siRNA-resistant cDNA RACK1-MUT rescued the defect ().
Figure 4. RACK1 is essential for mitotic entry and bipolar spindle assembly. (a) RACK1 is essential for mitotic entry. HeLa cells treated with control siRNA, RACK1 siRNA or Aurora-A siRNA were synchronized by using double thymidine block, and then harvested at various time points, and cell-cycle progression was monitored by flow cytometry using propidium iodide staining. (b) Exemplar western blot of RACK1 in the HeLa cells stably expressing pcDNA3.1, RACK-WT or RACK-MUT. Exemplar western blot of endogenous RACK1 and Myc-RACK1 in the stable lines mentioned above with treatment of either control siRNA or RACK1 siRNA. (c) The HeLa cells stably expressing pcDNA3.1, RACK-WT or RACK-MUT were synchronized and then transfected with either control siRNA or RACK1 siRNA in the interval between the two thymidine blocks.13hr after the release, half of the cells were analyzed by western blot using the RACK1 antibody. Another half of the cells were analyzed by flow cytometry. (d) RACK1 is essential for bipolar spindle assembly. Hela cells treated with control siRNA or RACK1 siRNA were synchronized by using double thymidine block, and then analyzed by Immunofluorescence staining using indicated antibodies. The percentages of cells with abnormal mitotic spindles were quantified. Scale bar: 10 µm. (e) The overexpression of RACK1 can rescue the defect of abnormal spindle assembly. The HeLa cells stably expressing pcDNA3.1, RACK-WT or RACK-MUT were synchronized and then transfected with either control siRNA or RACK1 siRNA in the interval between the two thymidine blocks. The percentages of cells with abnormal mitotic spindles were quantified after immunofluorescence staining.
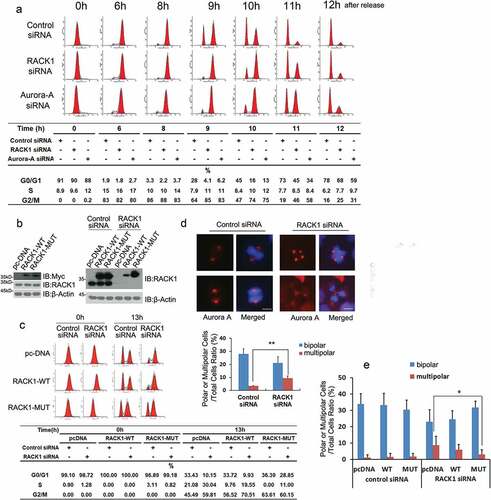
As Aurora-A is involved in the regulation of centrosome function, bipolar spindle assembly, and chromosome segregation processes [Citation28], the effects of RACK1 depletion on spindle organization and chromosome alignment were also examined by immunofluorescence staining using anti-Aurora-A antibody. The control siRNA transfected cells displayed normal bipolar mitotic spindles. However, cells that transfected with RACK1 siRNA showed a variety of spindle organization defects, including multi-polarity, and centrosome amplification ( and Figure S3). The percentage of cells with abnormal mitotic spindles in RACK1 siRNA transfection is increased by around 10%. As expected, the introduction of WT RACK1 and siRNA-resistant cDNA RACK1-MUT rescued the defect (). These results suggested that RACK1 play an important role in the regulation of spindle organization.
In conclusion, we found that RACK1 interacted with Aurora-A and co-localized with Aurora-A at centrosomes, then regulates both the kinase activity and stability of Aurora-A. Like Aurora-A, RACK1 is essential for mitotic entry and bipolar spindle assembly of cells. These findings may provide a basis for understanding the regulation of Aurora-A and future anticancer studies targeting Aurora-A.
Discussion
Aurora-A plays an important role in the regulation of spindle formation and mitosis. In this context, we have identified RACK1, the receptor for activated C-kinase1, as a novel partner and activator of Aurora-A kinase. Overexpression of RACK1 activated Aurora-A and knockdown of RACK1 in HeLa cells delayed the entry of most cells into the mitotic phase presumably due to its role in the activation of Aurora-A.
In addition to RACK1, there are many other proteins lacking enzymatic activity, such as cofactors Tpx2 [Citation29], HEF1 [Citation30], Ajuba [Citation6,Citation8], FAK [Citation31,Citation32], and Bora [Citation33] or even protein phosphatases, may also regulate the initial activation of Aurora-A. Our study adds RACK1 to the list of physiological upstream activators of Aurora-A. Interestingly, among them; Ajuba, HEF1, and FAK are all focal adhesion proteins. For example, Ajuba is involved in cell adhesion and cell–cell contacts [Citation34] and we also found that Ajuba was among the RACK1 and Aurora-A precipitates (Figure S1). How do these proteins transfer from the membrane to centrosome and interact with Auroroa-A? It is reported that RACK1 is involved in regulating trafficking of many proteins. RACK1 is a PK1 shuttling protein, whose intracellular localization changes in response of cell signal. (Dorit Ron et al., J. Biol. Chem. 1999). RACK1 regulates the plasma membrane localization of some proteins [Citation35,Citation36]. RACK1 may play an essential role not only in activating Aurora-A for mitotic commitment, but also recruitment of the activators to centrosome. We also observed that RACK1 can interact with Ajuba, an essential Aurora-A activator (data not shown). Therefore, the simplest interpretation of these data would be that recruitment of RACK1 to centrosome is the major regulatory step for the activation of Aurora-A. Ajuba, HEF1, and FAK1 may come in contact in the cytoplasm and then, the complex is recruited to the centrosome. However, this hypothesis needs more evidence. The discovered interaction between oncogene Aurora-A and RACK1 as well as the functional importance of their interaction for activation of Aurora-A and mitosis progress should help understand their respective roles in physiology and therapeutic potentials in many pathophysiological settings.
Conclusion
We presented a novel activator of Aurora-A, and the activation of Aurora-A by RACK1 is essential for the mitotic entry. Data from this study allow a better understanding of regulation of Aurora-A and offer insights into the molecular mechanism involved in regulating the catalytic activity of Aurora-A.
Materials and methods
Plasmids, antibodies, and other materials
Full-length Aurora-A (NCBI Accession No: NP_940838.1) and RACK1 (NCBI Accession No: NC_000005.10) were cloned into indicated vectors to generate constructs encoding Myc-tagged Aurora-A, HA-tagged RACK1, His-tagged Aurora-A, and GST-tagged RACK1, respectively.
The siRNA-resistant RACK–MUT construct was generated by mutating four nucleotide base pairs to alternative nucleotides within the siRNA target region without altering the amino acid sequences. The sequences of all constructs were confirmed by DNA sequencing. Cycloheximide (CHX) was purchased from Millipore (Billerica, MA, USA) and dissolved in ethanol at 100 mg/ml.
Cell culture and treatment
HeLa cells were obtained from Shanghai cell bank of Chinese academy of sciences (Shanghai, China). Cells were cultured in DMEM medium with 10% fetal bovine serum at 37°C in a humidified incubator with 5% CO2. For synchronization, HeLa cells were blocked for 16 h with thymidine (2mM), washed three times with PBS (phosphate-buffered saline) and then incubated with fresh medium. After 8 h, thymidine was added again, and the cells were incubated for an additional 16 h. Plates were then washed, and fresh medium was added. The extent of synchronization was controlled by flow cytometric analysis of DNA. For transfection, 293T or HeLa cells at 80% confluence were transfected with plasmids using Lipofectamine (Invitrogen) in serum free medium according to manufacturer’s instructions. After 5 h of incubation, the medium was replaced with fresh complete medium. At 48-h posttransfection, cells were subject to immunoblot or immunofluorescence microscopy.
Immunofluorescence microscopy
Cells grown on coverslips were fixed in 4% formaldehyde in PBS for 10 min and resolved by 0.2% Triton X-100 for 15 min at room temperature. The permeabilized cells were blocked with 10% normal horse serum plus 1% bovine serum albumin (BSA) for 1 h. A drop of the diluted monoclonal antibody against RACK1 (1:500 Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and polyclonal antibody against Aurora-A (1:1000 Cell Signaling Technology, Inc., Beverly, MA, USA) was placed on the coverslips overnight and were then incubated with Alexa 546 (red fluorescence)-conjugated goat anti-mouse and Alexa 488 (green fluorescence)-conjugated goat anti-rabbit secondary antibodies (Life Technologies) for 1 h. The nuclei were stained with 0.1 mg/ml 4ʹ,6-diamidino-2-phenylindole (DAPI) for 10 min at room temperature in the dark. The coverslips were then mounted onto glass slides using Aqua Polymount (Polysciences, Warrington, PA, USA). Fluorescence images were captured at room temperature using an LSM 710 confocal microscope equipped with an Axiovert 100 M microscope, a 63x oil-immersion lens (NA 1.4) with Carl Zeiss 4.2 software (all from Carl Zeiss, Inc., Thornwood, NY, USA). Confocal images of each antigen and of DAPI staining were combined using ImageJ (National Institutes of Health) software without any other manipulations.
RNAi
The siRNA sequences (Genepharma, Shanghai, China) targeting RACK1 were as follows: RACK1 siRNA #1: 5ʹ-CCAUCAAGCUAUGGAAUACTT-3ʹ (sense) and 5ʹ- GUAUUCCAUAGCUUGAUGGTT-3ʹ (antisense); RACK1 siRNA #2: 5ʹ- CUGACCAGGGAUGAGACCTT-3ʹ (sense) and 5ʹ- GGUCUCAUCCCUGGUCAGTT-3ʹ (antisense); The siRNA sequences targeting Aurora-A were as follows: Aurora-A siRNA #1: 5ʹ-AUUCUUCCCAGCGCGUUCCTT-3ʹ (sense) and 5ʹ-GGAACGCGCUGGGAAGAAUTT-3ʹ (antisense); Aurora-A siRNA #2: 5ʹ-AUGCCCUGUCUUACUGUCA-3ʹ (sense) and 5ʹ-UGACAGUAAGACAGGGCAU-3ʹ (antisense). The siRNA-resistant wild-type RACK1 was generated by replacing ATTA to GCTC in RACK1 siRNA #1 targeting region. RACK1 siRNA #1 could not down-regulate R1 mutant of RACK1 since R1 mutant does not contain the RACK1 siRNA #1 targeting region.
Western blotting
Protein extracts were separated by 8–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. After blocking, the membranes were incubated with primary antibodies against different proteins at 4°C overnight, followed by incubation with horseradish peroxidase-conjugated secondary antibody. Immunoreactivity was visualized by enhanced chemiluminescence (Santa Cruz Biotechnology, Inc.).
Anti-HA (ab18181) and Anti-GAPDH (ab8245) antibodies were purchased from Abcame. Anti-Myc (sc-20,179) and anti-RACK1 (sc-17,754) were purchased from Santa Cruz Biotechnology, Inc. Anti-Phospho-Aurora-A (Thr288) (2914) and anti- Aurora-A (3092) were purchased from Cell Signaling Technology, Inc.
GST pull-down assay
His-tag Aurora-A and GST-tag RACK1 were expressed in E. coli and purified according to the instruction. 100 ng His- Aurora-A was incubated with 100 ng GST or GST-RACK1 for 2 h, then added glutathione-Sepharose beads and incubated for additional 1 h at 4°C. The beads were spinned down and washed with PBS for 3 times, and then bound proteins were analyzed by immunoblot.
In vitro kinase assay
For the kinase assay, purified His-Aurora-A or His-Aurora-A-Cd and indicated concentrations of His RACK1 were incubated in 30 μl kinase buffer containing 10 mM ATP for 30 min at 30°C. For His-Aurora-A and His-Aurora-A-Cd is highly phosphorylated in bacteria, the protein should be dephosphorylated with Protein Phosphatase 1 (New England Biolabs) before the kinase assay. After the reaction, an equal volume of 2× SDS sampling buffer was added, and then the mixture was boiled for 10 min. This sample was subjected to 12% SDS-PAGE and was immunoblotted with anti-Phospho-Aurora-A (Thr288) antibody.
Flow cytometry
The synchronized cells were washed in PBS (pH 7.0) and fixed in cold 70% ethanol overnight at 4°C. After being washed again with PBS, cells were incubated with 50 mg/ml propidium iodide (Sigma) and 100 mg/ml RNase A (Sigma) for 30 min at room temperature. Fluorescence from the propidium iodide–DNA complex was measured using a flow cytometer, and the percentage of cells in each cell-cycle phase was quantified using the Modifit program.
Immunoprecipitation
Mitotic cells were collected 9 h after the release from double thymidine block. For immunoprecipitation, cells were lysed with cell lysis buffer (20 mM Tris–HCl, pH 7.5,150 mM NaCl, 1% Triton, 1 mM EGTA, 1 mM Na2EDTA, 2.5 mM sodium pyrophosphate, 1 mM b-glycerophosphate, 1 mM Na3VO4and 1 µg/ml leupeptin) and the lysate was centrifuged (12,000g for 20 min). The supernatant was precleared with protein A/G beads (Sigma, USA), followed by incubation with 2 µl antibody for 2 h and thereafter with protein A/G beads for 2 h, all at 4°C. Pellets were washed five times with lysis buffer and resuspended in sample buffer and analyzed by SDS-PAGE.
Author’s contributions
M.B., S. S., H. F., and Y. L. performed most experiments. Y.Z. and W.Y. provided technical support for various assays. M.B., L.Y.. and J.W. designed the experiments and wrote the manuscript. All the authors read and approved the final manuscript.
Supplemental Material
Download MS Word (305.8 KB)Acknowledgments
We thank Yongjun Dang at School of Basic Medical Science, Fudan University for fruitful scientific discussions and sharing some reagents.
Disclosure statement
No potential conflict of interest was reported by the authors.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
Related Research Data
References
- Marumoto T, Zhang D, Saya H. Aurora-A - A guardian of poles. Nat Rev Cancer. 2005;5:42–50.
- Yan M, Wang C, He B, et al. Aurora-A kinase: a potent oncogene and target for cancer therapy. Med Res Rev. 2016;36:1036–1079.
- Barros TP, Kinoshita K, Hyman AA, et al. Aurora A activates D-TACC-Msps complexes exclusively at centrosomes to stabilize centrosomal microtubules. J Cell Biol. 2005;170:1039–1046.
- Kinoshita K, Noetzel TL, Pelletier L, et al. Aurora A phosphorylation of TACC3/maskin is required for centrosome-dependent microtubule assembly in mitosis. J Cell Biol. 2005;170:1047–1055.
- Liu Q, Ruderman JV. Aurora A, mitotic entry, and spindle bipolarity. Proc Natl Acad Sci U S A. 2006;103:5811–5816.
- Hirota T, Kunitoku N, Sasayama T, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114:585–598.
- Bruinsma W, Aprelia M, Garciá-Santisteban I, et al. Inhibition of Polo-like kinase 1 during the DNA damage response is mediated through loss of Aurora A recruitment by Bora. Oncogene. 2017;36:1840–1848.
- Bai M, Ni J, Wu J, et al. A novel mechanism for activation of Aurora-A kinase by Ajuba. Gene. 2014;543:133–139.
- Nair JS, Schwartz GK. MLN-8237; A dual inhibitor of aurora A and B in soft tissue sarcomas. Oncotarget. 2016;7:12893–12903.
- Kobayashi A, Hashizume C, Dowaki T, et al. Therapeutic potential of mitotic interaction between the nucleoporin Tpr and aurora kinase A. Cell Cycle. 2015;14:1447–1458.
- Zhang Y, Ni J, Huang Q, et al. Identification of the auto-inhibitory domains of Aurora-A kinase. Biochem Biophys Res Commun. 2007;357:347–352.
- Afonso O, Figueiredo AC, Maiato H. Late mitotic functions of Aurora kinases. Chromosoma. 2017;126:93–103.
- Wike CL, Graves HK, Hawkins R, et al. Aurora-A mediated histone H3 phosphorylation of threonine 118 controls condensin I and cohesin occupancy in mitosis. Elife. 2016;5:e11402.
- Yabuta N, Mukai S, Okada N, et al. The tumor suppressor Lats2 is pivotal in Aurora A and Aurora B signaling during mitosis. Cell Cycle. 2011;10:2724–2736.
- Bai M, Ni J, Shen S, et al. Aurora-A kinase-inactive mutants disrupt the interaction with Ajuba and cause defects in mitotic spindle formation and G2/M phase arrest in HeLa cells. BMB Rep. 2014;47:631–636.
- Gallini S, Carminati M, De Mattia F, et al. NuMA phosphorylation by Aurora-A orchestrates spindle orientation. Curr Biol. 2016;26:458–469.
- Zorba A, Buosi V, Kutter S, et al. Molecular mechanism of Aurora A kinase autophosphorylation and its allosteric activation by TPX2. Elife. 2014;3:e02667.
- Kamran M, Long ZJ, Xu D, et al. Aurora kinase A regulates Survivin stability through targeting FBXL7 in gastric cancer drug resistance and prognosis. Oncogenesis. 2017;6:e298.
- Serrels B, Sandilands E, Serrels A, et al. A complex between FAK, RACK1, and PDE4D5 controls spreading initiation and cancer cell polarity. Curr Biol. 2010;20:1086–1092.
- Mamidipudi V, Dhillon NK, Parman T, et al. RACK1 inhibits colonic cell growth by regulating Src activity at cell cycle checkpoints. Oncogene. 2007;26:2914–2924.
- Mamidipudi V, Zhang J, Lee KC, et al. RACK1 Regulates G1/S Progression by Suppressing Src Kinase Activity. Mol Cell Biol [Internet]. 2004;24:6788–6798. doi:10.1128/MCB.24.15.6788-6798.2004.
- Ai E, Skop AR. Endosomal recycling regulation during cytokinesis. Commun Integr Biol. 2009;2:444–447.
- Shu F. Control of the cell cycle and its relation with aurora kinases (d). Shanghai: Fudan University; 2002 (Ph.D. thesis, in Chinese). Available from: http://cdmd.cnki.com.cn/Article/CDMD-10246-2003125296.htm
- Adams DR, Ron D, Kiely PA. RACK1, A multifaceted scaffolding protein: structure and function. Cell Commun Signal. 2011;9:22.
- López-Bergami P, Habelhah H, Bhoumik A, et al. RACK1 mediates activation of JNK by protein kinase C [corrected]. Mol Cell [Internet]. 2005;19;309–320. Available from: http://www.sciencedirect.com/science/article/pii/S1097276505014280
- Li JJ, Xie D. RACK1, a versatile hub in cancer. Oncogene. 2014;34:1890–1898.
- Sato T, Takahashi H, Hatakeyama S, et al. The TRIM-FLMN protein TRIM45 directly interacts with RACK1 and negatively regulates PKC-mediated signaling pathway. Oncogene. 2015;34:1280–1291.
- Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003;3:51–62.
- Bayliss R, Sardon T, Vernos I, et al. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol Cell. 2003;12:851–862.
- Hamze-Komaiha O, Sarr S, Arlot-Bonnemains Y, et al. SHIP2 regulates lumen generation, cell division, and ciliogenesis through the control of basolateral to apical lumen localization of Aurora A and HEF 1. Cell Rep. 2016;17:2738–2752.
- Mahankali M, Henkels KM, Speranza F, et al. A non-mitotic role for aurora kinase A as a direct activator of cell migration upon interaction with PLD, FAK and Src. J Cell Sci [Internet]. 2014;128;516–526. Available from: http://jcs.biologists.org/content/128/3/516.full
- Peng H, Gong P-G, Li J-B, et al. The important role of the receptor for activated C kinase 1 (RACK1) in nasopharyngeal carcinoma progression. J Transl Med [Internet]. 2016;14:131. doi:10.1186/s12967-016-0885-x.
- Seki A, Coppinger JA, Jang CY, et al. Bora and the kinase Aurora A cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008;320:1655–1658.
- Marie H, Pratt SJ, Betson M, et al. The LIM protein ajuba is recruited to cadherin-dependent cell junctions through an association with α-catenin. J Biol Chem. 2003;278:1220–1228.
- Ikebuchi Y, Ito K, Takada T, et al. Receptor for activated C-kinase 1 regulates the cell surface expression and function of ATP binding cassette G2. Drug Metab Dispos [Internet]. 2010;38;2320–2328. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20858845
- Ikebuchi Y, Takada T, Ito K, et al. Receptor for activated C-kinase 1 regulates the cellular localization and function of ABCB4. Hepatol Res [Internet]. 2009;39;1091–1107. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19674157