
ABSTRACT
Gallbladder cancer (GBC) is the common malignancy of the bile tract system with extremely poor clinical outcomes, owing to its metastatic property and intrinsic resistance to the first-line drugs. Although it is well-established that cholesterol abnormity contributes to gallstone formation, a leading risk factor for GBC, the link of cholesterol homeostasis with GBC has not been investigated. The present study systematically examined the genes implicated in cholesterol homeostasis, and revealed altered gene expressions of de novo cholesterol biosynthesis and sterol sulfonation (SULT2B1), reduced bile acid synthesis (CYP7B1 and CYP39A1) and impaired sterol efflux (ABCA1, ABCG5, LCAT, and CETP) in GBC tissues. Suppression of cholesterol biosynthesis by lovastatin inhibited GBC cell proliferation possibly through attenuating the DNA repair process. Further investigation revealed lovastatin sensitized GBC cells to cisplatin-induced apoptosis and suppressed the activation of CHK1, CHK2, and H2AX during DNA damage response. By using chemically distinct statins, HMGCR depletion or supplementing mevalonate, the product of HMGCR, we showed the inhibitory effects on DNA repair process of lovastatin were due to the blockage of the mevalonate pathway. Subcutaneous xenograft mice model suggested lovastatin promoted the therapeutic efficacy of cisplatin, and significantly prolonged the survival times of tumor-bearing mice. Moreover, HMGCR ablation repressed tumor growth in vivo, which can be rescued partially by restored expression of HMGCR, suggesting the on-target effects of lovastatin. Therefore, our study provides the clinical relevance of cholesterol homeostasis with GBC progression, and highlights a novel intervention of combined use of lovastatin and cisplatin for GBC.
Introduction
Cholesterol is an essential component of cell membranes for maintaining cellular homeostasis, and a precursor for steroid hormones and bile acids synthesis [Citation1]. It is also enriched in lipid rafts and is implicated in many intracellular signaling pathways [Citation2,Citation3]. Epidemiologic findings suggest a positive link between elevated cholesterol level and a higher risk for certain cancer types [Citation4–Citation6]. Cholesterol homeostasis is tightly controlled by a series of physiological processes, which involves its import, synthesis, export, metabolism, and esterification [Citation7]. 3-hydroxy-3-methyl-glutaryl coenzyme A (HMGCR) is the first committed rate-limiting enzyme of the mevalonate pathway for de novo cholesterol biosynthesis. Statins, the HMGCR inhibitor, is commonly used to reduce serum cholesterol and the incidence of cardiovascular events by preventing the formation of mevalonate from HMG-CoA and thereby inhibiting cholesterol synthesis as well as the downstream isoprenoids [Citation8]. Functionality of the mevalonate pathway is thought to have critical cellular functions such as membrane integrity, cell signaling, cell cycle progression and immune response [Citation9–Citation11].
As part of a growing effort to repurpose clinically approved drugs to treat cancer, statins have also been used to prevent cancer and facilitate therapeutic efficacy. Accumulating studies among large scale of cancer patients have revealed statins use have shown to reduce mortality in multiple cancer types [Citation12], suggesting targeting cholesterol metabolism is a promising intervention for cancer treatment.
Gallbladder cancer (GBC) is the sixth leading malignancy of the digestive tract with extremely poor prognosis [Citation13]. The 5-year survival rate is only 5% to 10%, owing to its metastatic property and intrinsic resistance to the first-line drugs [Citation14,Citation15]. Gallstone was considered to be the leading high risk factor for GBC, as more than 90% of patients with gallbladder cancer were found concomitantly with gallstone, which result from abnormalities in cholesterol metabolism [Citation16,Citation17]. However, the clinical relevance of cholesterol homeostasis with GBC has not yet determined. It has also not investigated whether targeting cholesterol metabolism by statins would provide therapeutic significance for GBC. More importantly, the biological characteristics of innate chemoresistance of GBC compromise the therapeutic options and efficacy. Overcoming chemoresistance by use of established drugs is a promising strategy to improve the outcome of anticancer therapy in the short term. Therefore, it is not clear whether statins would facilitate the chemotherapeutic efficacy.
The chemotherapeutic responsiveness is influenced by numerous factors including DNA damage response (DDR) [Citation18]. DNA damage response (DDR) engages two distinct kinase signaling cascades, the ATM-Chk2 and ATR-Chk1 pathways that coordinated cellular mechanisms for regulating the recognition of DNA damage, the activation of DNA repair factors, the initiation and coordination of DNA repair pathways, transit through the cell cycle and apoptosis [Citation19]. Phosphorylation of H2A.X (γ-H2AX) by ATM or ATR plays a critical role in the activation of repair or damage-signaling factors to the sites of DNA damage [Citation20]. Disruption of DNA damage and replication checkpoint responses was demonstrated to enhance tumor cell killing by diverse genotoxic agents [Citation21,Citation22].
In the present study, we provide systematic examination of cholesterol homeostasis, and revealed that elevated de novo cholesterol biosynthesis, decreased bile acid synthesis and sterol efflux processes were found in GBC tissues as compared to the adjacent tissues, whereas cholesterol importing in GBC was not altered, suggesting the aberrant de novo cholesterol biosynthesis is the major cause for cholesterol homeostasis. Depletion of the mevalonate pathway by lovastatin (LVST) significantly promotes GBC cell apoptosis. Furthermore, we found combined stimulation of GBC with LVST and cisplatin (DDP) markedly improved chemotherapeutic efficacy both in vitro and in vivo. RNA-sequencing of GBC cells with statins stimulation revealed compromised DNA repair and G2M checkpoint signaling. Statins use indeed attenuated the DDR by suppressing CHK1 and CHK1 signaling, and inhibited the activation of γ-H2AX to the sites of DNA break. Restoration of the mevalonate pathway by the addition of mevalonic acid rescued the DDR and conferred the resistance to cisplatin. Thus, our study for the first time provides the clinical relevance of cholesterol homeostasis with GBC, and lay basis for the use of statins and cisplatin for GBC treatment.
Materials and methods
Samples and patients
The GBC tumor tissues and adjacent tissues were collected between 2005 and 2014 from the department of Biliary-Pancreatic Surgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University. GBC patients were provided with written informed consent. All manipulations were performed under the approval of the Research Ethics Committee of Renji Hospital, School of Medicine, Shanghai Jiao Tong University.
In vivo xenograft model
For in vivo evaluation of drug efficacy, a total of 2 × 106 NOZ cells in 100μL saline were injected subcutaneously in nude mice. When the tumor volume reached approximately 200 mm3, tumor-bearing mice were randomly divided into indicated groups. Mice were then given indicated drug through intraperitoneal injection (cisplatin, 5mg/kg weekly) or dietary addition of lovastatin (50 mg/kg/day) for 3 weeks. Mice receiving saline and fed with regular chow were used as control. Tumor size was examined weekly. After 4 weeks, the mice were scarified. The tumors were either harvested for tumor weight determination or embedded into paraffin blocks for histological analysis. The study was approved by the Research Ethics Committee of Renji Hospital, School of Medicine, Shanghai Jiao Tong University.
Cell culture and lentiviral preparation
Human gallbladder cancer cell NOZ, GBC-SD, and SGC-996 were obtained from the Cell Bank of Shanghai Institutes of Biological Sciences, Chinese Academy of Sciences. ZCF-1701 was isolated from primary tumor of a GBC patient. GBC cells were maintained in DMEM high glucose medium in 5% CO2 at 37°C. Oligonucleotides encoding hairpin precursors of HMGCR (5’- GGTTCTAAAGGACTAACATAA-3’) were used for generating short interference RNA (siRNA) constructs into lentiviral vector pGIPZ. HMGCR cDNA was cloned into pLX304-Blast-V5. Lentiviral production was prepared as previously described [Citation23]. Briefly, 293FT cells with approximately 30% confluence were co-transfected with 10 µg target plasmid, 7.5 µg psPAX2 packaging plasmid, and 2.5 µg pMD2.G envelope plasmid. For 6–12 h, replace with 15 ml fresh medium and incubate cells for 48 h. Medium containing virus particles was collected by spinning at 4000 g for 5 min. For stable cell line selection, cells were infected with lentivirus for 24 h and further selected with puromycin (5µg/mL) for 48 h.
Chemosensitivity, clonogenic formation, and apoptosis assays
For drug sensitivity, 8 × 103 cells were plated into 96-well plates in 100 μL suspension medium overnight, and were treated with indicated concentrations of statin or cisplatin for 48 h. 10 μL CCK8 solution in 100 µL medium was added into each well and incubated for 1–2 h. Absorbance at 450 m was detected by Microplate reader (Epoch, BioTek). For colony formation, 5 × 102 cells were plated into 6-well or 12-well plates overnight and treated with indicated drugs for 24 h. Plates were replaced with fresh medium and were incubated for additional 2 weeks. Plates were then washed gently twice with PBS, fixed with 4% methanol for 30 min and stained with crystal violet staining solution. The number of colonies were counted and statistically analyzed. For cell apoptosis assays, cells were treated with indicated dose of statin or cisplatin for 24 h, collected for FITC Annexin V and PI staining. Flow cytometry was carried out on a FACS caliber flow cytometer (BD Biosciences, San Jose, CA, USA). Data acquisition and analysis were carried out using Cell Quest (BD Biosciences).
Immunofluorescence and TUNEL assay
GBC cells were seeded on slides overnight and treated with 5μM lovastatin, 0.5μg/mL cisplatin or DMSO for 12 h. Slides were then washed in PBS and fixed in 4% paraformaldehyde for 4 h. Slides were blocked in PBS containing 0.1% Triton X-100 and 5% normal donkey serum at room temperature for 1 h. After phosphor-H2A.X(1:500) antibody incubation at 4°C overnight, signals were developed with Alexa Fluor secondary antibodies at room temperature for 30 min. Before mounting, cells were counterstained with DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride). Slides were examined by fluorescence microscopy (Olympus DP72) or laser confocal microscopy (Olympus FV-1200), as indicated. For TUNEL assay, cells were treated as described above, slides were then stained with TUNEL Apoptosis Assay Kit according to the protocol provided by the manufacturer.
RNA isolation and quantitative real-time PCR
Fresh GBC tumor tissues and adjacent tissues were grinded and homogenized in Trizol (Invitrogen) for total RNA extraction. Extracted RNA was reversed transcribed with the first-strand cDNA synthesis kit. qRT-PCR was performed using SYBR® Premix Ex Taq™ according to the manual protocol provided by TaKaRa. The primers for each gene are listed in of Supplementary Material and Method.
Table 1. Primers used for real-time PCR following reverse transcription
Reagents and antibodies
Lovastatin (HY-N0504), Simvastatin (HY-17502), Atorvastatin (HY-B0589) were purchased from MedchemExpress (Shanghai, China). Mevalonic acid lithium salt (50838) was obtained from Sigma-Aldrich. Antibodies against phosphor-CHK1 (133D3), phosphor-CHK2 (C13C1), and phosphor-CDC2 (10A11) were purchased from Cell Signaling Technology. CDC25A (55031-1-AP) and CDC25C (16485-1-AP) antibodies were obtained from Proteintech (Wuhan, China). Phosphor-H2A.X (SAB4504428) antibody was purchased from Sigma-Aldrich. β-actin (60008-1-Ig) was obtained from Proteintech (Wuhan, China).
Statistical analysis
Statistical analysis was performed with SPSS 20 for Windows (SPSS Inc). The Pearson χ2 test or Fisher’s exact test, the Student t test and Bonferroni’s post hoc test were used for comparing the differences between the variables. Kaplan-Meier analysis was used to determine survival. Log-rank test was used to compare survival between groups. All statistical tests were two-sided, and p value ≤ 0.05 was considered statistically significant.
Result
Cholesterol homeostasis is aberrant in GBC
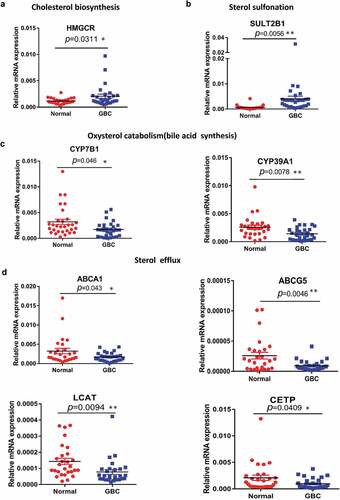
Cholesterol homeostasis involves its import, synthesis, export, metabolism, and esterification. To figure out the clinical relevance of cholesterol homeostasis with GBC, we performed qPCR analysis to examine the mRNA levels of key genes in cholesterol homeostasis. Increased mRNA expression of HMGCR in de novo cholesterol biosynthesis and sterol sulfonation (SULT2B1) was found (), whereas cholesterol catabolism for bile acid synthesis through the mevalonate pathway was significantly repressed as key genes like CYB7B1 and CYB39A1 were downregulated in GBC tissues ()). The genes involved in sterol efflux (ABCA1, ABCG5, LCAT, and CETP) were remarkably decreased in GBC tissues compared with the adjacent tissues ()). Other processes including HLD-sterol influx (SRB1), sterol esterification (ACAT1, ACAT2) was not significantly affected (Supplemental Figure 1(a–e)). These observations suggested that elevated de novo cholesterol biosynthesis, impaired sterol catabolism (bile acid synthesis) and sterol efflux pathways might result in sterols accumulation in GBC tissues.
Figure 1. Aberrant expression of genes implicated in cholesterol biosynthesis homeostasis. (a–d) Quantitative RT-PCR analysis of the mRNA levels for genes involved in cholesterol homeostasis of 30 paired samples of human GBC tissues and adjacent normal tissues. Statistical analysis was performed with a two-tailed ratio t test; Each dot represents the mean of relative mRNA levels. * p < 0.05, ** p < 0.01, *** p < 0.001
Blockage of cholesterol biosynthesis suppresses GBC cell growth and induces apoptosis
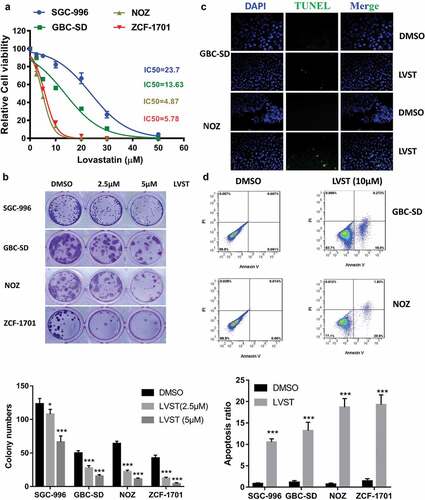
Given the increased cholesterol accumulation in GBC tissues, we reasoned that blocking cholesterol biosynthesis might suppress GBC progression. Inhibition of cholesterol biosynthesis by lovastatin attenuates cell growth in several GBC cell lines including SCG-996, GBC-SD, NOZ and ZCF-1701 ()). The half maximal inhibitory concentration (IC50) ranges from 4 μM to 25 μM, respectively, suggesting GBC cells with higher aggressiveness appear to be sensitive to lovastatin. Evaluation of cell viability by colony formation assay indicates lovastatin represses GBC cell survival in a dose-dependent manner ()). Furthermore, lovastatin induces cell apoptosis and DNA fragmentation as revealed Fluorescence-Activated Cell Sorting (FACS) using Annexin V-FITC and TUNEL assay (). These results here demonstrated that by blocking cholesterol biosynthesis using lovastatin, GBC cell growth is suppressed and cell apoptosis is promoted.
Figure 2. Suppression of cholesterol synthesis significantly facilitate GBC cell apoptosis. (a) Lovastatin inhibits GBC cell growth. Indicated cell lines were treated with indicated dose of lovastatin for 48 h. Cell viability was evaluated by CCK8 assays. (b) Lovastatin suppresses colony formation ability. 5 × 102 cells were plated into 6-well or 12-well plates overnight and treated with lovastatin (LVST, 10μM) or DMSO for 24 h. Plates were replaced with fresh medium and were incubated for an additional 2 weeks. Plates were then washed gently twice with PBS, fixed with 4% methanol for 30 min and stained with crystal violet staining solution. The number of colonies were counted and statistically analyzed. (c) Lovastatin induces DNA fragmentation as revealed by TUNEL. GBC cells were seeded on slides overnight and were treated with 5μM lovastatin for 12 h. Slides were then stained with TUNEL Apoptosis Assay Kit according to the protocol provided by the manufacturer. Before mounting, cells were counterstained with DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride). Slides were examined by fluorescence microscopy (Olympus DP72) or laser confocal microscopy (Olympus FV-1200), as indicated. Representative images are showed. TUNEL, Terminal deoxynucleotidyl transferase dUTP nick end labeling. (d) Lovastatin suppresses colony formation ability. 5 × 102 cells were plated into 6-well or 12-well plates overnight and treated with lovastatin (LVST, 10μM) or DMSO for 24 h. Plates were replaced with fresh medium and were incubated for an additional 2 weeks. Plates were then washed gently twice with PBS, fixed with 4% methanol for 30 min and stained with crystal violet staining solution. The number of colonies were counted and statistically analyzed. (e) Lovastatin causes significant cell apoptosis. Indicated cells were treated with indicated dose of statin for 24 h or DMSO, collected for FITC Annexin V and PI staining and analyzed by Flow cytometry
Suppression of cholesterol biosynthesis impairs DNA damage response
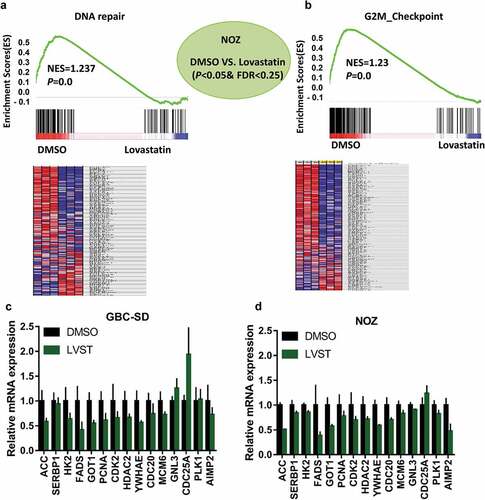
To understand the molecular basis through which cholesterol insufficiency leading to GBC growth inhibition, RNA-sequencing analysis was used to identify the potential signaling cascades in the NOZ cells following lovastatin treatment. Using GSEA, pathways including DNA repair, G2M checkpoint, MYC target, E2F target, PI3K_AKT/mTOR signaling, and apoptosis were found significantly enriched in GBC cells with lovastatin stimulation () and supplemental Figure 2A-2E). We next selected several genes differentially expressed genes related to PI3K_AKT/mTOR signaling and DNA repair pathways. qPCR analysis showed genes including ACC, SERBP1, HK2, PCNA, PLK1, and CDC25A were indeed regulated in GBC-SD and NOZ cells with lovastatin treatment (). Given that the relevance of cholesterol with DNA repair or G2M checkpoint has not been investigated, we next ask whether suppression of cholesterol biosynthesis would facilitate cell apoptosis and repress DNA repair upon cisplatin (DDP) treatment.
Figure 3. RNA-sequencing analysis identified cholesterol depletion modulates DNA repair process. GSEA and heat map analysis revealed DNA repair (a) and G2M checkpoint (b) pathways were remarkably enriched in GBC cells with lovastatin treatment. NOZ cells were treated with DMSO or 10μM lovastatin (LVST) for 24 h. The cells were then harvested and subjected to RNA-sequencing analysis. Gene set enrichment analysis (GSEA) was performed on the Broad Institute Platform and statistical significance (false discovery rate, FDR) was set at 0.25. Characteristic gene sets from DMSO and LVST stimulation were analyzed according to the genes presenting the strongest enrichment scores for each gene set. Lovastatin stimulation modulates PI3K/mTOR and DNA repair-related genes in GBC cells (c) and NOZ cells (d). GBC-SD and NOZ cells were treated with DMSO or lovastatin (10μM) for 24 h and subjected to qPCR analyses. Candidate genes for PI3K/mTOR (ACC, SERBP1, HK2, FADS, and GOT1) and G2M checkpoint (PCNA, CDK2, HDAC2, YWHAE, CDC20, MCM, GNL3, CDC25A, PLK1 and AIMP2) were selected to verify the results of RNA-sequencing. * p≤ 0.05, ** p≤ 0.01, *** p≤ 0.001
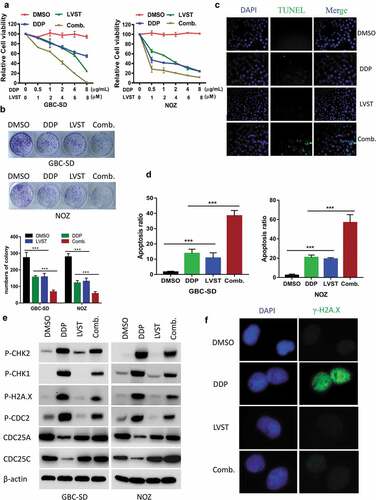
It was demonstrated that GBC-SD as well as NOZ with both lovastatin and cisplatin led to increased cell death compared with lovastatin or cisplatin alone ()). Similar effects were observed by evaluating the colony formation ability and DNA fragmentation and cell apoptosis (–)). In lines with this observation, depletion of cholesterol by lovastatin dramatically reduced the activation of CHK1, CHK2, and γ-H2AX induced by cisplatin stimulation, suggesting impaired DNA damage response. Moreover, tyrosine-15 phosphorylation of CDC2, CDC25A, and CDC25C, the downstream targets of CHK1 and CHK2 were also downregulated ()). Immunofluorescence (IF) also indicated that lovastatin treatment attenuated the activation of γ-H2AX ()). Taken together, these data here suggested cholesterol biosynthesis is engaged in DNA repair to suppress cell apoptosis.
Figure 4. Cholesterol depletion synergistically sensitized GBC cells to cisplatin through impaired DNA damage. (a) Lovastatin promotes cisplatin sensitivity of GBC cells. 8 × 103 GBC cells or NOZ cells were plated into 96-well plates and treated with indicated concentrations of statin or cisplatin (DDP) for 48 h, analyzed by CCK8 assay. (b) Lovastatin elicits impaired the clonogenic viability of GBC cells induced by cisplatin (DDP). 5 × 102 GBC cells or NOZ cells were plated into 12-well plates overnight and treated with 10μM lovastatin (LVST), DDP (1μg/mL) or DMSO for 24 h. Plates were replaced with fresh medium and were incubated for an additional 2 weeks. Plates were then washed gently twice with PBS, fixed with 4% methanol for 30 min and stained with crystal violet staining solution. The number of colonies were counted and statistically analyzed. (c) Lovastatin facilitates cisplatin-induced DNA fragmentation of GBC cells. GBC cells were seeded on slides overnight and were treated with 5μM lovastatin for 12 h. Slides were then stained with TUNEL Apoptosis Assay Kit according to the protocol provided by the manufacturer. Before mounting, cells were counterstained with DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride). Slides were examined by fluorescence microscopy (Olympus DP72) or laser confocal microscopy (Olympus FV-1200), as indicated. Representative images are showed. TUNEL, Terminal deoxynucleotidyl transferase dUTP nick end labeling. (d) Lovastatin enhances cisplatin-induced cell apoptosis. Lovastatin GBC-SD and NOZ cells were treated with 10μM lovastatin, 1μg/mL cisplatin or in combination for 12 h. Cells were then prepared and stained with FITC Annexin V and PI staining solutions and were determined by Fluorescence-activated cell sorting (FACS). (e) Lovastatin-enhanced chemosensitivity was associated with compromised DNA Repair. GBC cells were incubated with lovastatin (10μM), cisplatin (1μg/mL) or a combination of lovastatin and cisplatin for 24 h. Cell lysates were prepared and subjected to western blot analysis using indicated antibodies. (f) Lovastatin attenuates H2A.X activation during DNA damage. Cells were treated with indicated drugs and were immunostained with phospho-H2AX (Green) and DAPI (blue)
The DDR suppressive effect of lovastatin is due to on-target inhibition of the mevalonate pathway
To address the effect of lovastatin in DDR inhibition, other chemically distinct statins used to verify this observation similarly enhanced the efficacy of cisplatin ()). Furthermore, ablation of HMGCR by shRNA lenti-viral infection significantly suppresses GBC cell growth ()). HMGCR silencing markedly promotes cisplatin-induced cell death ()). On the other hand, supplementing GBC-SD or NOZ cells with mevalonic acid, the product of HMGCR activity, almost abolished the enhanced effect by lovastatin ()). Evaluation of DDR pathways suggested mevalonate acid supplementation in GBC cells restored the activation of CHK1, CHK1, and their downstream targets, suggesting rescued DNA repairs and cell cycle arrest ()). IF analysis of the H2A.X shows a similar result ()). These findings indicated that the inhibitory effects on DNA repair processes of lovastatin are dependent on the blockage of the mevalonate pathway.
Figure 5. Lovastatin-enhanced chemosensitivity is due to on-target inhibition of the mevalonate pathway. (a) Chemical distinct statin simvastatin and atorvastatin promotes therapeutic efficacy of cisplatin. 5 × 102 GBC cells or NOZ cells were plated into 12-well plates overnight and treated with indicated doses of statins or DMSO for 24 h. Plates were replaced with fresh medium and were incubated for an additional 2 weeks. Plates were then washed gently twice with PBS, fixed with 4% methanol for 30 min and stained with crystal violet staining solution. The number of colonies were counted and statistically analyzed. Representative images are showed. (b) HMGCR knockdown impairs GBC cell proliferation. 2 × 103 NOZ or GBC-SD cells were seeded in 96-well plates overnight. The proliferative activity was determined using Cell Counting Kit 8 for 5 days. (c) HMGCR ablation sensitizes GBC cells to cisplatin treatment. Indicated cell lines were treated with DMSO, DDP (1μg/mL) alone or combined administration. Representative images are showed. (d) Incubation of GBC cells with mevalonic acid abolishes the chemosensitivity upon cisplatin and lovastatin cotreatment. 5 × 102 GBC cells or NOZ cells were plated into 12-well plates, and were treated with (10μM) statin or cisplatin (1μg/mL) for 24 h. After 2 weeks’ incubation, the colonies were fixed and stained with 4% formaldehyde in PBS containing 0.05% crystal violet. The number of colonies were counted and statistically analyzed. Representative images are showed. (e) Supplementing of mevalonic acid reverses lovastatin induced DNA repair inhibition. GBC cells were incubated with lovastatin (10μM), cisplatin (1μg/mL), mevalonic acid (1mM) or indicated combination for 24 h. Cell lysates were prepared and subjected to western blot analysis using indicated antibodies. (f) Mevalonic acid restores H2A.X activation in the presence of lovastatin during DNA damage. GBC cells were seeded on slides overnight and were treated with lovastatin (10μM), cisplatin (1μg/mL), mevalonic acid (1mM) or indicated combination for 24 h. Slides were then phospho-H2AX (Green). Before mounting, cells were counterstained with DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride). Slides were examined by fluorescence microscopy (Olympus DP72) or laser confocal microscopy (Olympus FV-1200), as indicated. Representative images are showed
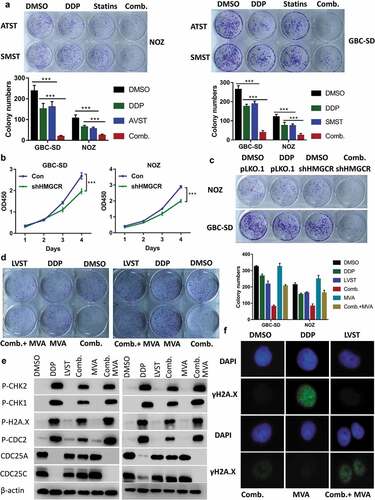
Cholesterol biosynthesis repression promotes the therapeutic efficacy of cisplatin
To provide in vivo evidence supporting the lovastatin effects, we established subcutaneous xenograft node mice using NOZ cell line, followed by cisplatin and lovastatin treatment alone or in combination. Mice receiving either cisplatin or lovastatin therapy showed significantly reduced tumor growth both in tumor size and weight compared with saline control group, suggesting cholesterol ablation indeed elicits antitumor activity against GBC cells ()). Co-treatment by cisplatin and lovastatin in mice resulted in notably enhanced antitumor efficacy in contrast to either cisplatin or lovastatin therapy alone ()). More importantly, mice given combined treatment dramatically prolonged the survival times compared with cisplatin or lovastatin administration alone ()). Histological analysis revealed apparently more necrosis, reduced Ki-67 reactivity and increased apoptosis in tumors with combined cisplatin and lovastatin in relation to control group, cisplatin or lovastatin treatment. Consistently, evaluation of γ-H2AX, a sensor of DNA damage response also supported compromised DNA damage response in combined group relative to cisplatin therapy ()).
Figure 6. Inhibition of the cholesterol biosynthesis pathway promotes the therapeutic efficacy of cisplatin in nude mice. (a) Lovastatin synergistically suppresses the tumor growth in vivo. Left, the tumor growth curve of indicated group. Right, demonstration of tumor xenograft. Lower, illustration of tumor size in indicated group. (b) Lovastatin enhances tumor necrosis, cell apoptosis and attenuates H2A.X activation in the xenograft of indicated group. Ki-67, proliferation marker protein Ki-67. TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling. (c) A combined therapy of lovastatin and cisplatin remarkably improves the survival times of mice. *, **, *** means p < 0.05, p < 0.01, and p < 0.001. (d) HMGCR silencing inhibits tumor proliferation, which can be partially rescued by restored expression of HMGCR. Res, rescued expression of HMGCR
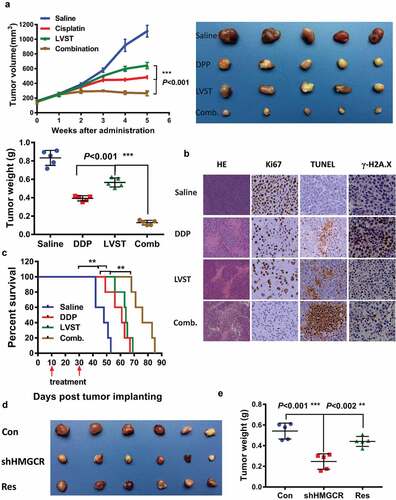
To verify that this effect is not due to off-target inhibition by lovastatin, we also examined the tumor growth in GBC cells with HMGCR depletion. Expectedly, HMGCR knockdown led to decreased tumor growth, which could be rescued by restored expression of HMGCR, at least partially, suggesting the functionality of cholesterol biosynthesis is required for GBC cell proliferation (). Collectively, these observations demonstrated that suppression of the mevalonate pathway facilitates the therapeutic efficacy of cisplatin, and prevents DNA damage response.
Discussion
Accumulating evidence has suggested cholesterol metabolism is widely involved tumor initiation and progression [Citation24–Citation27]. Suppression of cholesterol homeostasis, commonly by statin use, revealed a link between statin use and reduced cancer-related mortality in multiple cancer types [Citation12]. It has been well-established that the gallstone, which ranks the top high-risk factor for GBC, is correlated with abnormal cholesterol homeostasis [Citation28]. More than 90% of patients with GBC were diagnosed with gallstone, suggesting possible link between cholesterol homeostasis and GBC development [Citation16,Citation29,Citation30]. However, how cholesterol homeostasis affects GBC progression has not yet determined. In the present study, we performed systematic evaluation of genes implicated in cholesterol homeostasis pathways. HMGCR and SULT2B1, which are involved in de novo cholesterol biosynthesis and sterol sulfonation, were upregulated in GBC tissues. In contrast, genes encoding CYP7B1 and CYP39A1 for cholesterol catabolism (bile acid synthesis) were downregulated. The process of sterol efflux including genes like ABCA1, ABCG5, LCAT, and CETP significantly decreased in GBC tissues compared with the adjacent counterparts. No abnormity of processes such as sterol influx and sterol esterification were found, which was different from the observation in liver cancer showing the sterol catabolism and sterol efflux pathways are impaired [Citation31]. This discrepancy suggests gallbladder cancer addicts to cholesterol through de novo biosynthesis, opening avenue for statins implication for GBC. However, whether gene expression alterations in cholesterol homeostasis indeed lead to cholesterol accumulation in GBC tissues warrants further examination.
As widely prescribed in other cancer types [Citation32–Citation35], statins treatment indeed remarkably inhibits GBC cell growth and induce cell death. Particularly, this observation was supported by a recent retrospective study demonstrating a link between statin use and reduced risk of biliary tract cancers [Citation36]. Nevertheless, we observed a very different IC50 values among GBC cell lines, suggesting differential response to statin treatment. However, it is unknown which are determinants for statin sensitivity. p53 deficiency was recently defined as a critical activator of the mevalonate pathway and determines the sensitivity of statin in liver cancer [Citation37], which may explain the cell type-specific diverse effects of statin. To provide mechanical insight, we performed RNA-sequencing and GSEA analysis to identify potential signaling pathways. DNA repair, G2M checkpoint, MYC target, E2F target, and PI3K_AKT/mTOR signaling were enriched in GBC cells with lovastatin treatment. MYC target, E2F target, and PI3K_AKT/mTOR pathways were crucial for tumor cell proliferation [Citation38–Citation42]. DNA repair and G2M checkpoint signaling cascades are implicated in genomic instability, protecting cell survival from DNA damage. Given that the mevalonate pathway produces many metabolites that are indispensable for many biological processes [Citation1], perturbation of the mevalonate pathway by the usage of statins has given rise to unexpected cholesterol-independent pleiotropic effects. Inhibition of the mevalonate pathway was found to overcome acquired anti-HER2 treatment resistance in breast cancer and was associated with EGFR-TKIs (tyrosine kinase inhibitors) efficacy through multiple mechanisms [Citation43,Citation44], reinforcing the notion that cancer cells addict to the mevalonate pathway for survival and progression [Citation9–Citation11]. In this study, we uncovered that the mevalonate pathway crosstalks with the DNA damage repair signaling, an event closely correlated with chemotherapy. We further verified suppression of the mevalonate pathway indeed facilitates the therapeutic efficacy of cisplatin, a first-line drug for GBC treatment. Mechanistically, it was demonstrated that lovastatin abrogated activation of CHK1, CHK2 and H2A.X, critical mediators for G2M checkpoint and DNA repair process, and impaired cell cycle arrest for proper DNA damage response. It is currently unclear how lovastatin treatment connects the mevalonate pathway to the suppression of CHK1, CHK2, and H2A.X. We noted that mTOR signaling, which links growth signaling to DNA damage response, was compromised upon lovastatin treatment. It seems likely that lovastatin treatment integrates mTOR pathway to impair DNA damage response, leading to reduced tumor growth. We also provide solid data suggesting the tumor-suppressive function of lovastatin was due to on-target inhibition of HMGCR and the mevalonate pathway in vitro and in vivo, further indicating the mevalonate pathway is an effective therapeutic target, of particular in terms of a combined treatment. However, these evidences are collected in the preclinical model. The future work should evaluate the therapeutic efficacy of statin combined with chemotherapy in a randomized controlled trial. Taken together, our study uncovered the aberrant genes expression profile in cholesterol homeostasis, and demonstrated that targeting cholesterol biosynthesis improves the therapeutic efficacy of cisplatin both in vitro and in vivo through compromising DNA repair process.
Supplemental Material
Download Zip (4.2 MB)Acknowledgments
This work was supported by the National Natural Science Foundation of China (81702864, 81773184, and 81472240) and Shanghai Sailing Program for Young Talents (17YF1411200). This work was also supported by the Medical-Engineering Joint Fund of Shanghai Jiaotong University (YG2016QN51), Shanghai Outstanding Academic Leaders Plan, Innovative Research Team of High-level Local Universities in Shanghai and the Research Fund of Shanghai Tongren Hospital, Shanghai Jiaotong University School of Medicine.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Spann NJ, Glass CK. Sterols and oxysterols in immune cell function. Nat Immunol. 2013;14:893–900.
- Castellano BM, Thelen AM, Moldavski O, et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science. 2017;355:1306–1311.
- Gong X, Qian H, Zhou X, et al. Structural insights into the Niemann-Pick C1 (NPC1)-mediated cholesterol transfer and Ebola infection. Cell. 2016;165:1467–1478.
- Pelton K, Freeman MR, Solomon KR. Cholesterol and prostate cancer. Curr Opin Pharmacol. 2012;12:751–759.
- Jacobs EJ, Stevens VL, Newton CC, et al. Plasma total, LDL, and HDL cholesterol and risk of aggressive prostate cancer in the cancer prevention Study II nutrition cohort. Cancer Causes Control. 2012;23:1289–1296.
- Shafique K, McLoone P, Qureshi K, et al. Cholesterol and the risk of grade-specific prostate cancer incidence: evidence from two large prospective cohort studies with up to 37 years’ follow up. BMC Cancer. 2012;12:25.
- Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol. 2008;9:125–138.
- Otto CM. Statins for primary prevention of cardiovascular disease. BMJ. 2016;355:i6334.
- Baek AE, Yu YA, He S, et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun. 2017;8:864.
- Perucha E, Melchiotti R, Bibby JA, et al. The cholesterol biosynthesis pathway regulates IL-10 expression in human Th1 cells. Nat Commun. 2019;10:498.
- Wang Z, Wu Y, Wang H, et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc Natl Acad Sci U S A. 2014;111:E89–98.
- Nielsen SF, Nordestgaard BG, Bojesen SE. Statin use and reduced cancer-related mortality. N Engl J Med. 2012;367:1792–1802.
- Agopian VG, Hiatt JR. The timing of reoperation for incidental gallbladder cancer: sooner or later? JAMA Surg. 2017;152:149.
- Kasumova GG, Tabatabaie O, Najarian RM, et al. Surgical management of gallbladder cancer: simple versus extended cholecystectomy and the role of adjuvant therapy. Ann Surg. 2017;266:625–631.
- Aleksandrova K, Boeing H, Nothlings U, et al. Inflammatory and metabolic biomarkers and risk of liver and biliary tract cancer. Hepatology. 2014;60:858–871.
- Muszynska C, Lundgren L, Lindell G, et al. Predictors of incidental gallbladder cancer in patients undergoing cholecystectomy for benign gallbladder disease: results from a population-based gallstone surgery registry. Surgery. 2017;162:256–263.
- Baez S, Tsuchiya Y, Calvo A, et al. Genetic variants involved in gallstone formation and capsaicin metabolism, and the risk of gallbladder cancer in Chilean women. World J Gastroenterol. 2010;16:372–378.
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745.
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078.
- Kinner A, Wu W, Staudt C, et al. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694.
- Dent P, Tang Y, Yacoub A, et al. CHK1 inhibitors in combination chemotherapy: thinking beyond the cell cycle. Mol Interv. 2011;11:133–140.
- Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429.
- Zhang Y, Liu Y, Duan J, et al. Hippocalcin-like 1 suppresses hepatocellular carcinoma progression by promoting p21(Waf/Cip1) stabilization by activating the ERK1/2-MAPK pathway. Hepatology. 2016;63:880–897.
- Kory N, Grond S, Kamat SS, et al. Mice lacking lipid droplet-associated hydrolase, a gene linked to human prostate cancer, have normal cholesterol ester metabolism. J Lipid Res. 2017;58:226–235.
- Stopsack KH, Gerke TA, Sinnott JA, et al. Cholesterol metabolism and prostate cancer lethality. Cancer Res. 2016;76:4785–4790.
- McDonnell DP, Park S, Goulet MT, et al. Obesity, cholesterol metabolism, and breast cancer pathogenesis. Cancer Res. 2014;74:4976–4982.
- Warner M, Gustafsson JA. On estrogen, cholesterol metabolism, and breast cancer. N Engl J Med. 2014;370:572–573.
- Di Ciaula A, Wang DQ, Garruti G, et al. Therapeutic reflections in cholesterol homeostasis and gallstone disease: a review. Curr Med Chem. 2014;21:1435–1447.
- Rai R, Tewari M, Kumar M, et al. Expression profile of cholecystokinin type-A receptor in gallbladder cancer and gallstone disease. Hepatobiliary Pancreat Dis Int. 2011;10:408–414.
- Trevino F, Carter O. Gallstone size and the risk of gallbladder cancer. JAMA. 1984;251:3080–3081.
- Lu M, Hu XH, Li Q, et al. A specific cholesterol metabolic pathway is established in a subset of HCCs for tumor growth. J Mol Cell Biol. 2013;5:404–415.
- Baandrup L, Dehlendorff C, Friis S, et al. Statin use and risk for ovarian cancer: a Danish nationwide case-control study. Br J Cancer. 2015;112:157–161.
- Bockorny B, Dasanu CA. HMG-CoA reductase inhibitors as adjuvant treatment for hematologic malignancies: what is the current evidence? Ann Hematol. 2015;94:1–12.
- Mayor S. Statin use is associated with lower risk of liver cancer, study shows. BMJ. 2015;350:h1034.
- Wu BU, Chang J, Jeon CY, et al. Impact of statin use on survival in patients undergoing resection for early-stage pancreatic cancer. Am J Gastroenterol. 2015;110:1233–1239.
- Liu ZW, Alsaggaf R, McGlynn KA, et al. Statin use and reduced risk of biliary tract cancers in the UK clinical practice research datalink. Gut. 2019;68:1458–1464.
- Moon SH, Huang CH, Houlihan SL, et al. p53 represses the mevalonate pathway to mediate tumor suppression. Cell. 2019;176:564–580 e19.
- Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35.
- Hsieh AL, Walton ZE, Altman BJ, et al. MYC and metabolism on the path to cancer. Semin Cell Dev Biol. 2015;43:11–21.
- Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703.
- Guerrero-Zotano A, Mayer IA, Arteaga CL. PI3K/AKT/mTOR: role in breast cancer progression, drug resistance, and treatment. Cancer Metastasis Rev. 2016;35:515–524.
- Karki R, Man SM, Malireddi RK, et al. NLRC3 is an inhibitory sensor of PI3K-mTOR pathways in cancer. Nature. 2016.
- Ali A, Levantini E, Fhu CW, et al. CAV1-GLUT3 signaling is important for cellular energy and can be targeted by Atorvastatin in non-small cell lung cancer. Theranostics. 2019;9:6157–6174.
- Sethunath V, Hu H, De Angelis C, et al. Targeting the mevalonate pathway to overcome acquired anti-HER2 treatment resistance in breast cancer. Mol Cancer Res. 2019.