
ABSTRACT
Autophagy is cellular recycling process that plays a complex role in cancer. Pre-clinical studies indicating a pro-tumorigenic role of autophagy have led to the launch of dozens of clinical trials combining autophagy inhibition with other standard of care therapies in different tumor types. A recent publication utilized a novel, acute, CRISPR/Cas9 assay to identify cancer cell lines that are exquisitely sensitive to loss of core autophagy genes within the first 7 days. However, weeks later, rare populations of originally autophagy dependent cells were found that could circumvent autophagy inhibition. Analysis of these rare clones revealed that in the process of circumventing loss of autophagy, the cells upregulated NRF2 signaling to maintain protein homeostasis and consequently become more sensitive to proteasome inhibition as well as knock down of NRF2. This review highlights recent publications regarding the role of autophagy in cancer and potential mechanisms cancer cells may be able to commandeer to circumvent autophagy inhibition. We hope to make significant clinical advances by understanding if and when cancer cells will become resistant to autophagy inhibition, and pre-clinical studies may be able to provide insight into the best combinatorial therapies to prevent tumor relapse while on autophagy inhibitors.
Macroautophagy
Autophagy is a cellular recycling process utilized by most cells in the body to maintain homeostasis by removing damaged or excess proteins and organelles [Citation1]. Macroautophagy is a multi-step process that involves over 20 core autophagy proteins and many additional regulatory proteins, most of which were first identified in yeast, but are conserved through mammals [Citation2]. A double membrane phagophore structure is initiated by the ULK complex which includes the protein kinases ULK1 and ULK2, ATG13, ATG101, and RB1CC1 (otherwise known as FIP200). This complex is regulated by nutrient sensing pathways including mTOR complexes. When nutrients are plentiful, mTORC1/2 phosphorylate and deactivate the ULK kinases [Citation3]. However, when nutrients are depleted mTOR can no longer phosphorylate the complex, allowing activation of ULK1/2 and subsequent phosphorylation of ATG13 and FIP200. These events are necessary to initiate phagophore nucleation which is followed by activation of the Beclin complex. The BH3 domain-containing protein, Beclin1 (BECN1), interacts with phosphatidylinositol 3-kinase (VPS34) [Citation4]. Additional core autophagy proteins in this complex include UVRAG, Bif-1, AMBRA1 and ATG14 [Citation5]. Moreover, other BCL2 family proteins also interact with Beclin1: some activate the complex while others inhibit downstream function [Citation6]. Once initiated the phagophore elongates to engulf cytoplasmic material and membrane elongation and closure is dependent on two ubiquitin-like conjugation systems. First, ATG5 is conjugated to ATG12 via the E1-like enzyme, ATG7, and the E2-like enzyme, ATG10. Subsequently, ATG7 interacts with another E2-like enzyme, ATG3, and together with the ATG5-12 conjugate, in a complex with ATG16L1, acting as an E3-like enzyme, these complexes facilitate the conjugation of phosphatidylethanolamine (PE) to the GABARAP/LC3 family of proteins [Citation1]. Most notable of this family is LC3B which is first cleaved by the cysteine protease family, ATG4A-D, prior to conjugation with PE [Citation7]. The final LC3B-PE (also known as LC3-II) conjugate is incorporated into the forming autophagosome membrane and thus frequently used as a marker of autophagosomes. LC3-II is critical for efficient autophagosome formation and closure, however, in its absence autophagosomes can still be formed but at a significantly decreased rate [Citation8,Citation9]. LC3 also plays a role in interacting with key autophagy cargo receptors that facilitate delivery of material to autophagosomes and many proteins that are selectively targeted to the autophagosome for degradation contain LC3-interacting domains or LIRs.
After the double membrane autophagosome containing cargo is formed, SNARE proteins like STX17 and SNAP29, mediate fusion with lysosomes. The fused organelle is termed an autophagolysosome, where an acidic pH allows cathepsins and proteases to degrade the autophagosome contents and generate amino acids and other building blocks that are recycled back into cellular metabolism and protein translation. Importantly, LC3-II is also degraded in the autophagolysosomes because it is incorporated into the phagophore membrane [Citation1].
In addition to macro-autophagy, other forms of autophagy have been characterized that facilitate direct delivery of proteins and other cellular material to the lysosome. For example, chaperone-mediated autophagy utilizes LAMP2A and Hsc70 to target proteins containing the conserved motif, KFERQ, directly to the lysosome [Citation10]. Micro-autophagy also involves direct invagination of the lysosome [Citation11]. While autophagy can be an unselective bulk degradation process, there are numerous forms of selective autophagy and intricate signaling pathways can induce the degradation of specific organelles. The most well characterized include selective degradation of mitochondria, ribosomes, bacteria, ferritin, endoplasmic reticulum, and peroxisomes: A.K.A mitophagy, ribophagy, xenophagy, ferritinophagy, ERphagy, and pexophagy [Citation12].
Autophagy and cancer
Autophagy plays a complex and context specific role in cancer[Citation13]. In normal cells, autophagy promotes cellular homeostasis by maintaining nutrient availability, DNA stability and decreasing chronic tissue damage and reactive oxygen species (ROS) [Citation14–Citation17]. Aberrations of autophagy in normal cells has been shown to increase tumorigenesis. Beclin1 in particular, and its interacting proteins, have been identified as haplo-insufficient tumor suppressors [Citation18–Citation22]. Loss of Beclin1 is associated with increased tumor incidence in mouse models and patients. However, in humans, BECN1 is adjacent to the potent tumor suppressor, BRCA1, and it has also been reported that BECN1 loss on its own does not cause tumorigenesis without the loss of BRCA1 in combination [Citation23]. Additionally, Beclin1 has several other autophagy-independent functions that could contribute to tumor suppression [Citation24]. However, despite such complexities, it is widely viewed that autophagy can reduce tumor initiation. For example, studies in pancreatic ductal adenocarcinoma (PDAC) and lung cancer models have shown that genetic deletion of the conjugation machinery, specifically ATG5 or ATG7, causes an increase in pre-neoplastic lesions [Citation25–Citation28].
In established tumors, autophagy promotes tumor growth and in the same studies that showed loss of ATG5 or ATG7 caused an increase in pre-neoplastic lesions, long term analysis showed these lesions were far less likely to develop into invasive carcinomas [Citation25–Citation28]. Actively proliferating tumor cells must survive in harsh environments that are often nutrient poor and hypoxic, and cancer cells are often dependent on autophagy to support nutrient availability. Many studies in cultured cells and a variety of mouse models have shown that deletion or depletion of core autophagy genes reduces tumor cell viability and increases life span. These effects have been attributed to tumor cell autonomous mechanisms that include blocked tumor growth and increased tumor cell death mediated by an accumulation of dysfunctional mitochondria, reduced fatty acid oxidation, reduced glycolytic capacity, elevated DNA damage, and impaired tumor cell metabolism [Citation25,Citation29,Citation30,Citation31,Citation32,Citation33,Citation34,Citation35].
Recently, non-tumor cell-autonomous mechanisms have emerged that also contribute to the tumor promotional roles of autophagy. Inhibition of autophagy in the tumor cells can promote an immunogenic response and cytokine mediated attraction of natural killer (NK) cells into the tumor microenvironment (TME), ultimately reducing tumor growth [Citation36,Citation37]. Additionally, autophagy in the immune cells themselves is important and inhibition of autophagy in CD8+ T-cells or tumor associated macrophages can promote tumor rejection in mice [Citation38,Citation39]. LC3-associated phagocytosis (LAP) utilizes some of the autophagy machinery to facilitate the conjugation of LC3 to phagosomal membranes. Inhibition of LAP, but not canonical autophagy, in tumor associated macrophages (TAMs) causes an M1 (anti-tumor) phenotype that induces a STING-dependent type-1 interferon and anti-tumor T-cell response [Citation40]. Inhibition of autophagy in other supporting cells within the TME has also affected tumor growth independent of the immune system. For example, inhibition of autophagy in pancreatic stellate cells causes a decrease in secreted alanine which is critical for pancreatic ductal adenocarcinoma (PDAC) cell growth [Citation41]. Even distant organs can effect non-cell autonomous autophagy mediated tumor growth, and loss of autophagy in the liver causes a decrease in circulating arginine and dramatically effects tumor growth [Citation42].
A variety of pre-clinical studies have also shown that the addition of autophagy inhibition to standard of care chemotherapy regiments increases tumor cell killing [Citation43,Citation13]. Together these pre-clinical studies have led to the launch of over 60 clinical trials utilizing autophagy inhibitors in patients [Citation44]. While there are a variety of pharmacological agents in development that target the autophagy pathway at different stages and work in a variety of mechanisms; the only autophagy-inhibitors that are currently approved for use in patients are the lysomotropic agents, chloroquine (CQ) and hydroxychloroquine (HCQ) [Citation43]. These lysosome inhibitors were originally approved to treat malaria and rheumatoid arthritis in patients and have been repurposed to target autophagy for either single agent use or in combination with other chemotherapy regiments or targeted therapies. Such systemic drugs will in theory inhibit autophagy in all cells and thus could affect both tumor cell-autonomous and non–autonomous functions of autophagy. Indeed, in the first wave of clinical trials with CQ/HCQ in combination with other therapies some studies have showed promising results. Importantly, target doses could be reached with minimal toxicity [Citation44]. While these early phase trials were not designed to test efficacy, some anti-tumor effects were also observed. In melanoma, 41% of patients treated with CQ and the alkylating agent, temozolomide (TZD) showed a partial response or stable disease. In another study treating patients with brain metastasis derived from a variety of solid tumors, 84% of patients treated with CQ and radiation reported a one-year survival rate compared to 55% of patients treated with radiation alone [Citation45,Citation46]. A study in glioblastoma patients reported a median survival of 33 months in patients treated with CQ and TZD, compared to control patients who had a median survival of 11 months [Citation47]. While these studies have provided hints of clinical success, when taking all the studies together, autophagy inhibition still has a long way to go before it could be considered a beneficial therapeutic. This is likely due to a variety of factors including no identified biomarkers that have been demonstrated to identify the patients most likely to benefit from treatment, a need for more specific and potent autophagy inhibitors, as well as a lack in understanding of potential mechanisms of resistance.
Identifying autophagy dependent cancer cells
One critical hurtle to overcome as autophagy inhibition moves forward in the clinic is the identification of autophagy dependent cancers or subtypes. In vitro and pre-clinical animal studies have shown that cancer cells vary in their dependence upon autophagy so that some cancer cell lines are acutely sensitive to genetic inhibition of autophagy but others show very little change in viability in the absence of autophagy. Tumors with RAS-RAF-MEK-ERK pathway activation are often, but not always, hyper-sensitive to either genetic or pharmacological autophagy inhibition. For example, pancreatic ductal carcinomas (PDAC), where RAS activation is common, show a significant decrease in tumor cell viability after autophagy inhibition [Citation26,Citation48,Citation31]. Constitutive BRAF(V600E) mutations have also been associated with tumor cell dependence on autophagy and pediatric brain cancers that harbor the mutation are more sensitive to autophagy inhibition than tumor cells with wild type BRAF [Citation49,Citation50]. TP53 has been linked to autophagy dependence and co-deletion of p53 in a humanized PDAC mouse model with KRAS activation abolished the anti-tumor effects of genetic autophagy inhibition [Citation28]. However, subsequent studies revealed these effects may be isolated to germ line deletion of TP53 because somatic loss of heterozygosity of TP53, which is the more common form of p53 deletion in human tumors, did not have the same effect [Citation26]. A direct relationship between RAS mutation and autophagy dependence has yet to be found, and in a panel of lung cancer cell lines, RAS mutation did not correlate with CQ sensitivity [Citation51]. Moreover, tetracycline-induced HRASG12V increased autophagy but not sensitivity to CQ, in fact the opposite effect was seen, where the mutant cells were somewhat protected from CQ induced cell death [Citation51].
Cancer cells without RAS pathway mutations have also been identified as extremely autophagy dependent in vitro [Citation52,Citation53]. A study conducted in a panel of breast cancer cells subject to shRNAs that target ATG5, ATG7, and BECN1 found that some lines were exquisitely sensitive to KD of core autophagy genes. Triple negative breast cancer cell lines in particular, including the MDA-MB-231, HCC-1937, BT549, and MDA-MB-468 cells, showed a decrease in cell viability and an increase in cell death after autophagy inhibition. These results were attributed to elevated autophagy-mediated STAT3 signaling in triple negative lines [Citation52].
Despite this work and many other in vitro studies showing that some, but not all, cancer cells have decreased viability after genetic autophagy depletion, a recent, highly-cited study called these results into question. Specifically, an shRNA screen performed in multiple cancer cell lines showed no dropout of shRNAs targeting 3 autophagy proteins and complete inactivation of the autophagy regulator ATG7 using CRISPR/Cas9 had no effect on tumor cell growth and survival [Citation54]. This study raised a serious contradiction for the field and led to suggestions that the autophagy pathway is not a suitable therapeutic target in cancer. A possible explanation for such discrepancies is that tumor cells may undergo selection to avoid the effects of inactivation of an otherwise essential gene. To test this hypothesis, we recently developed a more robust and quantitative assay to test if autophagy is more essential in some cancer cells than others [Citation53].
Barcoded shRNA and CRISPR screens using genome wide or other large scale libraries have been used to identify essential genes but are susceptible to caveats such as a lack of validated reagents, i.e. gRNAs and/or shRNAs that have not been experimentally tested to ensure that they inactivate their targets [Citation55,Citation56,Citation57]. Samples from such screens are usually harvested at a single timepoint giving a snapshot picture of the over/under represented barcodes and thus cannot account for the possibility that “essentiality” may be more dynamic when a process is studied for which compensatory mechanisms could exist. To address these concerns and identify the essentiality of autophagy genes in cancer cells, we developed a quantitative, live-cell gene essentiality assay using CRISPR/Cas9 to monitor cell viability within hours of editing and with no selection [Citation53]. Specifically, in-vitro transcribed guide RNAs that target a gene of interest are co-transfected into double labeled mCherry+/GFP+ (mCh+/GFP+) cells where loss of GFP is an accurate surrogate for loss of the other co-targeted gene of interest. Live-cell imaging is then used to monitor viability of the edited GPP- population within hours after editing. Extensive proof of principle studies showed efficient loss of GFP in greater than 90% of the cells and co-deletion of GFP and a gene of interest. Importantly, the assay accurately identified known essential genes; i.e. genes required for critical processes like DNA replication, mRNA transcription, and protein translation.
The majority of studies in the autophagy field, whether in vitro or in vivo, usually only test 1–3 genes in a given experiment. However, many studies have shown that the core autophagy machinery can be used in other cellular processes making it difficult to rule out autophagy-independent functions [Citation58]. We therefore tested the essentiality of 12 core ATGs, in an arrayed manner, that are needed for different steps in the autophagy process. We included genes important for phagophore initiation and nucleation like ULK2, ATG13, AMBRA, and RB1CC1 (FIP200), BECN1, and VPS34 as well as genes important for LC3 conjugation which mediates phagophore elongation and closure including ATG3, ATG5, ATG7, and ATG12. Finally, we included genes that affect the later steps of the process like autophagosome fusion with lysosomes and lysosomal function like STX17 and LAMP2, respectively. We tested the essentiality of all 12 genes in a panel of 8 cancer cell lines across different tumor types and mutational backgrounds [Citation53].
To quantify the essentiality of each gene and compare across cell lines, we generated a CRISPR growth score (CGS). Within every experiment guide RNAs were included that target established essential genes, such as POLR2A, PCNA, or eEF2 and established non-essential genes like PTEN or FOXO3a. The area under the curve was calculated for the GFP- population after co-transfection with guide RNAs targeting GFP and each gene of interest including the control essential and non-essential genes. The CGS was calculated and normalized such that the area under the curve for the essential gene and non-essential genes ran in tandem for each experiment were 0 and 1, respectively. The CGS for the 12 autophagy genes were normalized accordingly in each experiment and assigned scores ranging between 0 and 1. Occasionally some ATGs would generate a CGS less than 0 indicating that gene was more essential than the control essential gene as measured in the assay [Citation53].
A critical advantage to this assay is the ability to begin monitoring the effect of KO on viability immediately after transfection with live cell imaging every 1–10 hours for 7 days. Importantly, there is no antibiotic selection, so any loss in viability is presumably from the targeted guide RNAs transfected. This is in contrast to traditional pooled CRISPR or shRNA screens where there is usually a 48 hour antibiotic selection that kills uninfected cells followed by several days before cell harvest and analysis of shRNA or gRNA abundance changes. This could potentially be enough time for cells to adapt to loss of a particular gene allowing for false negative hits. This is especially concerning in cancer cells with a high propensity for adaption. It is also important to note that for each of the 12 autophagy genes tested as well as the 5 control genes, five guide RNAs were invitro transcribed and then tested invitro for their ability to cut their target DNA sequences. Two guides for each gene that could efficiently cut in the invitro assay were chosen and transfected in with 2 guides that target GFP. The capability to confirm every guide RNA used in the assay also provides an extra level of rigor for these experiments.
With this assay, we identified 4 out of 8 cell lines where 10 or more of the 12 autophagy genes tested scored a CGS <0.5, and we classified these lines as autophagy dependent [Citation53]. This includes the breast cancer cell lines, BT549 and MDA-MB-468, the lung cancer line, H292, and the pediatric brain cancer line MA-794. We also identified two cell lines whose viability was generally unaffected when autophagy genes were knocked out, and at least 6 of the 12 autophagy genes scored a CGS > 0.5. These cell lines were classified as autophagy independent and included the HCT116 colon cancer cell line and the MCF7 breast cancer line. There were also cell lines with an intermediate phenotype, defined by 3–5 genes scoring a CGS > 0.5, and included the HT1080 and SJSA sarcoma cell lines.
There was a large variation in the CRISPR growth scores across the cell lines, for example, in the autophagy independent MCF7 cells, some of the conjugation machinery genes received CG scores of ~0.8 while the highly autophagy dependent lines like the BT549, MDA-MB-468, and MA-794 lines received scores below zero for the same genes – indicating the conjugation machinery scored as more essential than DNA transcription or protein translation in these cells. Moreover, within a cell line we identified outlier genes with a CGS out of the range of the other genes in the pathway. For example, in the autophagy independent HCT116 cells the majority of the autophagy genes scored above 0.5; however, ULK2 received a constantly low CGS of less than 0.2 [Citation53]. These data suggest that ULK2 may have autophagy independent functions in this cell line. Consistent with this hypothesis, previous studies have identified autophagy independent functions for ULK2. In neurons ULK1/2 interact with SEC16A to mediate ER-to-Golgi trafficking, and this interaction does not require other core autophagy proteins, like ATG13 [Citation59]. The ULK kinases have also been implicated in other vesicular trafficking events as well as stress granule disassembly [Citation60,Citation61]. These results emphasize the need to manipulate multiple autophagy genes and highlight the capabilities of our assay to identify autophagy independent functions.
Importantly, our studies confirmed previously published data on some cell lines. For example, with shRNA studies Maycotte et al showed that the MCF7 cells were more resistant to loss of core ATGs than the MDA-MB-468 and BT549 cells [Citation52]. Our study also included additional cell lines that have not previously been tested for autophagy dependence, and we identified a new highly autophagy dependent cell line, the lung cancer H292 line. Interestingly, neither mutations in the RAS-RAF-MEK-ERK pathway nor TP53 mutations associated with autophagy dependence in our studies. The autophagy independent HCT116 and MCF7 cells have WT P53, however the highly autophagy dependent H292 and MA-794 lines as well as the intermediate SJSA line also have WT p53. Similarly, HCT116 cells harbor an activating RAS mutation, yet the MCF7 cells have no RAS-RAF-MEK-ERK aberrations and the MA-794 cells also have a BRAF(V600E) mutation. While 8 cell lines is a relatively small sample size, within our cohort, no mutational patterns were observed that could explain autophagy dependence.
Since out of the 8 cell lines we tested, our CRISPR/Cas9 assay coupled with live cell imaging identified 4 cancer cell lines that are dependent on autophagy for survival within the first 7 days of editing, these results conflict with the idea that no cancer cells are dependent on autophagy for survival [Citation54]. Our study does however further support the numerous in vitro and in vivo studies showing decreased cancer cell viability after autophagy inhibition in specific contexts [Citation25,Citation52,Citation26,Citation29,Citation31,Citation32, Citation33,Citation62,Citation50,Citation34,Citation27,Citation28,Citation63].
Cancer cells can circumvent autophagy inhibition
We next asked if any of the autophagy-dependent cells could adapt and eventually grow, despite showing a severe decrease in viability in the first seven days after editing. To do this, we continued to maintain the autophagy dependent BT549 and H292 lines treated with ATG7 targeted guide RNAs [Citation53]. After a few weeks of continued nutrient replenishment, the cells began to grow enough to clone out the GFP- population. Surprisingly, in both the BT549 and H292 lines, we identified rare clones with complete loss of ATG7 and a corresponding loss of LC3 conjugation and autophagic flux. These ATG7−/- clones, derived from the originally autophagy dependent cells, maintained growth rates equal to WT cells, even under autophagy inducing conditions including nutrient starvation, hypoxia, and forced oxidative respiration with galactose. These data suggest the ATG7−/- clones circumvented loss of LC3 conjugation and acquired new mechanisms to replace the essential functions of autophagy. As expected, the ATG7−/- cells were also resistant to pharmacological autophagy inhibition with CQ, both in vitro and in xenograft studies in nude mice. We again performed the CRISPR assay targeting all 12 core autophagy genes and found the ATG7−/- clones were not only resistant to guide RNAs targeting ATG7 but also significantly more resistant to guide RNAs targeting most of the other autophagy genes as well. This further supports the conclusion that the KO clones acquired a mechanism to circumvent the entire autophagy pathway ().
Figure 1. A flow chart describing how rare, autophagy-dependent cancer cells that circumvent autophagy inhibition have acquired new targetable susceptibilities.
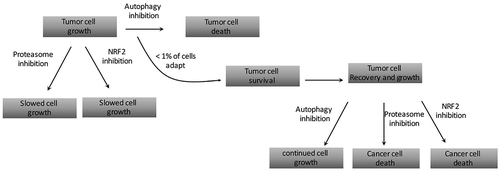
These results suggest that a patient’s tumor that is initially sensitive to autophagy inhibition may eventually become resistant while still on therapy. To date there have not been any studies to directly test this possibility; however, preliminary results from the first wave of clinical trials with CQ/HCQ have shown hints of acquired resistance. In a Phase I trial in dogs treated with HCQ and doxorubicin, two canine patients showed a complete initial response but were subsequently removed from the study due to progressive disease [Citation64]. In another phase I study, a patient with brain metastasis who initially responded to CQ and temozolomide eventually presented with persistent growth after 4 months on therapy [Citation46].
Further analysis of the ATG7−/- clones in our studies also revealed the encouraging finding that adaptation to autophagy inhibition also leads to increased susceptibilities to other targeted agents. Specifically, we showed the ATG7−/- cells had defective proteasomes, and consequently were more sensitive to pharmacological proteasome inhibition with bortezomib (). Previous studies have linked the ubiquitin proteasome system (UPS) and autophagy showing that autophagy can act as compensatory degradation system when proteasome mediated degradation is impaired [Citation65, Citation66]. Moreover, previous studies have shown that the addition of autophagy inhibition can increase cancer cell killing with bortezomib [Citation67–Citation71]. These studies concluded that autophagy was a compensatory mechanism for impaired UPS functioning. However, our study was the first to show that when autophagy is inhibited, and damaged proteasomes therefore cannot be degraded via autophagy [Citation72,Citation73], cancer cells are more sensitive to proteasome inhibition.
Our results combined with previous studies suggest that a combination strategy with CQ and bortezomib may be a more viable therapeutic strategy than either single agent alone. In 2014, Dr. Amaravadi’s group conducted a clinical trial in refractory multiple myeloma patients utilizing this combination [Citation74]. They showed the combination therapy was feasible and well tolerated, with no adverse effects due to dose-limiting toxicity. However, the patient responses were minimal at best, with 3 out of the 22 patients showing a very good partial response and greater than 50% of the patients had either stable disease or a minor response. The study was designed to test the hypothesis that autophagy compensates for impaired UPS and therefore 64% of the patients enrolled in the study had received bortezomib as a prior therapy, of which 44% of those patients were refractory to the single agent. Interestingly, the results did not support the original hypothesis, and the patients that were refractory to prior treatment with bortezomib were still largely unresponsive to the combination. This suggest that if a tumor is already resistant to proteasome inhibition adding in autophagy inhibition won’t help. Our recent study suggests a slightly different strategy. We hypothesize that it would be better to start in patients with autophagy dependent tumors that are also sensitive to proteasome inhibition. In those tumors, we would predict that combination treatment with proteasome inhibition and autophagy inhibition would create a situation whereby the tumor cannot adapt to avoid one of the interventions without becoming more sensitive to the other. While it is too early to consider such studies in humans (because we have no good way to identify autophagy-dependent tumors at this time), pre-clinical studies to test such ideas are feasible.
Autophagy and NRF2
The transcription factor nuclear factor erythroid 2-related factor 2 (NFE2L2), commonly known as NRF2, is a master regulator of cellular homeostasis and many different cytoprotective pathways. Most notably, NRF2 regulates redox homeostasis and directly regulates γ-glutamyl cysteine ligase – the rate limiting enzyme in glutathione (GSH) biosynthesis, as well as other enzymes that maintain GSH in its’ reduced state. Critical NADPH generating enzymes, including malic enzyme 1 (ME1), isocitrate dehydrogenase 1 (IDH1), glucose-6-phosphate dehydrogenase (G6PD), and 6-phosphogluconate dehydrogenase (PGD), are all transcriptionally regulated by NRF2 [Citation75,Citation76]. Accordingly, NRF2 activity inversely correlates with the intracellular level of reactive oxygen species (ROS) [Citation77]. Tissues in NRF2 knock out mice have increased ROS production compared to WT mice [Citation78]. It can also affect mitochondrial respiration and loss of NRF2 causes a decrease in mitochondrial membrane potential. NRF2 can also affect protein homeostasis and most of the proteasomal subunits are direct transcriptional targets [Citation79–Citation81].
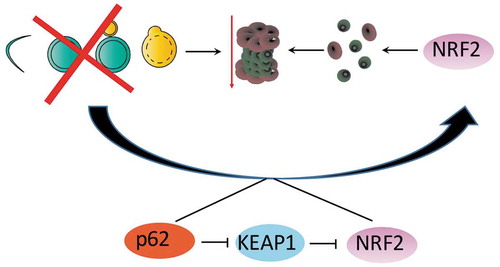
NRF2 is tightly regulated via post-translational mechanisms. While a number of proteins have been shown to regulate the transcription factor, the most well studied interactions involve the E3-ubiquitin ligase, KEAP1. Under normal conditions, NRF2 has a relatively short half-life and it is constitutively ubiquitinated by KEAP1 and targeted for degradation via the proteasome. NRF2 was first linked to autophagy when a novel interaction between the autophagy cargo adaptor protein, SQSTM1 (otherwise known as p62), and KEAP1 was identified [Citation82]. Specifically, p62 can bind to KEAP1 and compete for NRF2 binding. When autophagy is blocked, p62 accumulates and sequesters KEAP1 causing an increase in NRF2 protein levels and subsequent transcriptional activity.
We also showed that in the ATG7−/- clones generated from originally autophagy dependent cells, NRF2 was robustly upregulated via a p62 dependent mechanism [Citation53](). However, our work was the first to show that cells that have circumvented autophagy inhibition become exquisitely sensitive to loss of NRF2. Indeed, we found that the ATG7−/- cells generate more proteasome subunits in an NRF2 dependent manner and propose that this is due to an attempt to compensate for the decreased proteasomal activity (). Accordingly, knock down of NRF2 exacerbated the increased sensitivity to proteasome inhibition in the clones that circumvented autophagy inhibition. These results suggest that NRF2 expression may play a role in dictating autophagy dependence. In future clinical trials, it may be beneficial to assess NRF2 expression levels or activity in initial tumor biopsies as well as subsequent biopsies, particularly after a patient becomes refractory to autophagy inhibition. Tumor types like melanoma where biopsies are often more feasible may be the best place to run such a clinical trial. We hypothesize that NRF2 expression may be especially high after tumors become refractory to CQ/HCQ. Indeed, overexpression of either exogenous NRF2, or endogenous NRF2 – mediated by knockdown of KEAP1, in autophagy dependent WT cells, caused resistance to CQ induced apoptosis [Citation53].
Figure 2. An animation showing the mechanism by which autophagy dependent cancer cells upregulate NRF2 to generate more functional proteasomes in order to compensate for decreased proteasomal degradation after autophagy inhibition.
Further support for such ideas comes from studies by Strohecker et al who performed an interesting experiment with ATG7flox/flox; BRAFV600E/+ animals where intransal ingestion of CRE recombinase caused a co-deletion of ATG7 and oncogenic BRAF V600E activation in the lung. After crossing the mice with NRF2−/- animals they found that both ATG7−/- and NRF2−/- mice had an increase in life span compared to BRAFV600E/+ controls [Citation25]. However, the co-deletion of ATG7 and NRF2 did not have any additional affect. These data combined with our results suggest that the order of events may be important. We hypothesize that when cancer cells circumvent autophagy and therefore upregulate NRF2 as a survival mechanism, only then do they become much more dependent on that increased NRF2 signaling, and thus basal NRF2 before the selection may not be as critical. Studies to test this could be done where (1) autophagy inhibition is blocked after tumors are established and (2) with inducible knock down or knock out of NRF2 so that NRF2 expression can be modulated after loss of autophagy.
Concluding remarks
Although autophagy plays a complex role in tumor cell biology, many pre-clinical studies and early clinical trials suggest autophagy inhibition may be a beneficial therapeutic. As the field moves forward, it is important to gain a comprehensive understanding of how cancer cells respond to autophagy inhibition. Our data shows that while many cancer cells are very sensitive to autophagy inhibition, rare populations from these lines can circumvent autophagy inhibition (). In our studies we focused on protein homeostasis and identified NRF2 upregulation as a mechanism autophagy dependent cells can use to survive after knock out of the core autophagy gene, ATG7 (). Future studies are needed to understand how these cells are copying with loss of other critical pathways autophagy is involved such as mitochondrial homeostasis. An important finding from our work is that the process of circumventing autophagy also leads to new targetable susceptibilities. We showed that the ATG7−/- clones derived from autophagy dependent cell lines were more sensitive to pharmacological proteasome inhibition as well as genetic inhibition of NRF2. These studies may provide insight into the best combination therapies with CQ/HCQ to provide more robust clinical outcomes in patients.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–364.
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132.
- Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976.
- Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–750.
- HE C, LEVINE B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010;22:140–149.
- Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939.
- Li M, Hou Y, Wang J, et al. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J Biol Chem. 2011;286:7327–7338.
- Tsuboyama K, Koyama-HondA I, Sakamaki Y, et al. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016;354:1036–1041.
- Nguyen TN, Padman BS, Usher J, et al. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol. 2016;215:857–874.
- Tekirdag K, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: joint by a chaperone. J Biol Chem. 2018;293:5414–5424.
- Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012;69:1125–1136.
- Anding AL, Baehrecke EH. Cleaning house: selective autophagy of organelles. Dev Cell. 2017;41:10–22.
- Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019;9:1167–1181.
- Wang Y, Zhang N, Zhang L, et al. Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Mol Cell. 2016;63:34–48.
- Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42.
- Dou Z, Xu C, Donahue G, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527:105–109.
- Belaid A, Cerezo M, Chargui A, et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res. 2013;73:4311–4322.
- Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676.
- Takahashi Y, Coppola D, Matsushita N, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–1151.
- Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820.
- Yue Z, Jin S, Yang C, et al. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–15082.
- Liang C, Feng P, Ku B, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–698.
- Laddha SV, Ganesan S, Chan CS, et al. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol Cancer Res. 2014;12:485–490.
- Wirawan E, Lippens S, Berghe V, et al. Beclin1: a role in membrane dynamics and beyond. Autophagy. 2012;8:6–17.
- Strohecker AM, Guo JY, Karsli-Uzunbas G, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013;3:1272–1285.
- Yang A, Rajeshkumar NV, Wang X, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4:905–913.
- Rao S, Tortola L, Perlot T, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056.
- Rosenfeldt MT, O’prey J, Morton JP, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300.
- Karsli-Uzunbas G, Guo JY, Price S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4:914–927.
- Guo JY, Teng X, Laddha SV, et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016;30:1704–1717.
- Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–729.
- Xie X, Koh JY, Price S, et al. Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov. 2015;5:410–423.
- Guo JY, Chen HY, Mathew R, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470.
- Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64.
- Guo JY, Karsli-Uzunbas G, Mathew R, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27:1447–1461.
- Mgrditchian T, Arakelian T, Paggetti J, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci U S A. 2017;114:E9271–E9279.
- Baginska J, Viry E, Berchem G, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A. 2013;110:17450–17455.
- Devorkin L, Pavey N, Carleton G, et al. Autophagy regulation of metabolism is required for CD8(+) T cell anti-tumor immunity. Cell Rep. 2019;27:502–513 e5.
- Chen D, Xie J, Fiskesund R, et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat Commun. 2018;9:873.
- Cunha LD, Yang M, Carter R, et al. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell. 2018;175:429–441 e16.
- Sousa CM, Biancur DE, Wang X, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536:479–483.
- Poillet-Perez L, Xie X, Zhan L, et al. Autophagy maintains tumour growth through circulating arginine. Nature. 2018;563:569–573.
- Towers CG, Thorburn A. Therapeutic targeting of autophagy. EBioMedicine. 2016;14:15–23.
- Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528–542.
- Rojas-Puentes LL, Gonzalez-Pinedo M, Crismatt A, et al. Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat Oncol. 2013;8:209.
- Rangwala R, Leone R, Chang YC, et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1369–1379.
- Briceno E, Reyes S, Sotelo J. Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg Focus. 2003;14:e3.
- Perera RM, Stoykova S, Nicolay BN, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–365.
- Ojha R, Leli NM, Onorati A, et al. ER Translocation of the MAPK pathway drives therapy resistance in BRAF-mutant melanoma. Cancer Discov. 2019;9:396–415.
- Levy JM, Thompson JC, Griesinger AM, et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014;4:773–780.
- Morgan MJ, Gamez G, Menke C, et al. Regulation of autophagy and chloroquine sensitivity by oncogenic RAS in vitro is context-dependent. Autophagy. 2014;10:1814–1826.
- Maycotte P, Gearheart CM, Barnard R, et al. STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res. 2014;74:2579–2590.
- Towers CG, Fitzwalter BE, Regan D, et al. Cancer cells upregulate NRF2 signaling to adapt to autophagy inhibition. Dev Cell. 2019;50:690–703.e6.
- Eng CH, Wang Z, Tkach D, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci U S A. 2016;113:182–187.
- Blomen VA, Majek P, Jae LT, et al. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015;350:1092–1096.
- Hart T, Chandrashekhar M, Aregger M, et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015;163:1515–1526.
- Wang T, Birsoy K, Hughes NW, et al. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101.
- Cadwell K, Debnath J. Beyond self-eating: the control of nonautophagic functions and signaling pathways by autophagy-related proteins. J Cell Biol. 2018;217:813–822.
- Joo JH, Wang B, Frankel E, et al. The noncanonical role of ULK/ATG1 in ER-to-golgi trafficking is essential for cellular homeostasis. Mol Cell. 2016;62:491–506.
- Wang B, Maxwell BA, Joo JH, et al. ULK1 and ULK2 regulate stress granule disassembly through phosphorylation and activation of VCP/p97. Mol Cell. 2019;16;74:742–757.e8.
- Wang B, Kundu M. Canonical and noncanonical functions of ULK/Atg1. Curr Opin Cell Biol. 2017;45:47–54.
- Yang A, herter-sprie G, Zhang H, et al. Autophagy sustains pancreatic cancer growth through both cell autonomous and non-autonomous mechanisms. Cancer Discov. 2018;8:276–287.
- Lock R, Roy S, Kenific CM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22:165–178.
- Barnard RA, Wittenburg LA, Amaravadi RK, et al. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy. 2014;10:1415–1425.
- Pandey UB, Nie Z, Batlevi Y, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863.
- Ding WX, Ni HM, Gao W, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524.
- Milani M, Rzymski T, Mellor HR, et al. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with bortezomib. Cancer Res. 2009;69:4415–4423.
- Hui B, Shi YH, Ding ZB, et al. Proteasome inhibitor interacts synergistically with autophagy inhibitor to suppress proliferation and induce apoptosis in hepatocellular carcinoma. Cancer. 2012;118:5560–5571.
- Jia L, Gopinathan G, Sukumar JT, et al. Blocking autophagy prevents bortezomib-induced NF-kappaB activation by reducing I-kappaBalpha degradation in lymphoma cells. PLoS One. 2012;7:e32584.
- Kawaguchi T, Miyazawa K, Moriya S, et al. Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: crosstalk among proteasome, autophagy-lysosome and ER stress. Int J Oncol. 2011;38:643–654.
- Yao F, Wang G, Wei W, et al. An autophagy inhibitor enhances the inhibition of cell proliferation induced by a proteasome inhibitor in MCF-7 cells. Mol Med Rep. 2012;5:84–88.
- Marshall RS, Li F, Gemperline DC, et al. Autophagic degradation of the 26S proteasome is mediated by the dual ATG8/ubiquitin receptor RPN10 in arabidopsis. Mol Cell. 2015;58:1053–1066.
- DIKIC I. Proteasomal and autophagic degradation systems. Annu Rev Biochem. 2017;86:193–224.
- Vogl DT, Stadtmauer EA, Tan KS, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy. 2014;10:1380–1390.
- Wild AC, Moinova HR, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999;274:33627–33636.
- Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015;88:179–188.
- Holmstrom KM, Baird L, Zhang Y, et al. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open. 2013;2:761–770.
- Kovac S, Angelova PR, Holmstrom KM, et al. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta. 2015;1850:794–801.
- Kapeta S, Chondrogianni N, Gonos ES. Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J Biol Chem. 2010;285:8171–8184.
- Arlt A, Bauer I, Schafmayer C, et al. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene. 2009;28:3983–3996.
- Kwak MK, Wakabayashi N, Itoh K, et al. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem. 2003;278:8135–8145.
- Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223.