
ABSTRACT
Cyclin A-cyclin dependent kinase (CDK) activity is regulated by cyclin A proteolysis and CDK inhibitors (CKIs) during M and G1 phases. Our previous work has shown that constitutive activation of cyclin A-CDK in mouse somatic cells, by ectopic expression of stabilized human cyclin A2 (lacking the destruction box: CycAΔ80) in triple CKI (p21, p27, and p107)-knocked-out mouse embryonic fibroblasts, induces rapid tetraploidization. However, effects of such cyclin A-CDK hyperactivation in human cells have been unknown. Here, we show hyperactivity of cyclin A-CDK induces G2/M-phase arrest in human cell lines with relatively low expression of p21 and p27. Moreover, adenovirus E1A protein promoted CycAΔ80-derived G2/M-phase arrest by increasing the amount of cyclin A and cyclin A-CDK2 complex. This response was suppressed by an addition of ATR or Chk1 inhibitor. The amount of repressive phosphorylation of CDK1 at tyrosine 15 (Y15) was decreased by Chk1 inhibitor treatment. Moreover, we observed that co-expressing CDK1AF mutant, which is resistant to the repressive phosphorylation at threonine 14 and Y15, or cdc25A, which dephosphorylates CDK1 at Y15, suppressed the G2/M-phase arrest by CycAΔ80 with E1A. These results suggest that G2/M-phase arrest in human cells by hyperactivity of cyclin A-CDK2 is caused by repression of CDK1 via the cell cycle checkpoint ATR-Chk1 pathway.
Introduction
Cyclin-cyclin-dependent kinase (CDK) complexes are the major regulators on cell cycle progression [Citation1,Citation2]. CDK activity is regulated by three mechanisms: cyclin degradation, tyrosine or threonine phosphorylation, and CDK inhibitor (CKI) binding. The anaphase-promoting complex (APC) is one of the E3 ubiquitin-protein ligases that initiates the proteasomal degradation of cyclin [Citation3]. Both cyclin A and cyclin B contain a motif, destruction box (D-box), that is required for ubiquitination by the APC [Citation4]. CDK activation is not only regulated by cyclin binding. Phosphorylation of CDK2 on tyrosine 15 by Wee1 inhibits CDK2 activity, which is antagonized by dephosphorylation by cdc25 [Citation5]. By contrast, phosphorylation of CDK2 on threonine 160 by CDK-activating kinase (CAK) activates CDK2 [Citation5]. Among CKIs in mammals, p21 and p27 are major ones and inhibit CDK activity via binding to cyclin-CDK complex, inducing G1-phase arrest [Citation6] and block of S-phase entry [Citation7].
Cyclin A associates with CDK1 and CDK2 [Citation8]. Cyclin A is required during S phase and G2 to M phases, and degraded after prometaphase, which is dependent on polyubiquitination by APC [Citation4]. Moreover, cyclin A-CDK complex is inactivated by CKIs binding during G1 phase [Citation9]. Cyclin A stabilization or overexpression delays chromosome alignment and anaphase onset, resulting in genome instability [Citation4,Citation10]. On the other hand, CDK2 activity induces cell cycle checkpoint mediated by ATR-Chk1 after DNA damage [Citation11]. Chk1 activation by CDK2 leads to phosphorylation of CDK1 on tyrosine 15 via cdc25A inactivation. Thus, CDK2 plays an important role in cell cycle checkpoint after DNA damage.
We have previously studied the effect of hyperactivity of cyclin A-CDK in mouse, by ectopically expressing a stabilized human cyclin A2 (lacking the D-box) in triple CKI (p21, p27, and p107)-knocked-out mouse embryonic fibroblasts (MEFs) [Citation12]. In this setting, the mutant cyclin A-CDK is active during the M and G1 phases when the wild-type cyclin A-CDK is inactivated by redundant proteolytic and CKI pathways, and therefore, the mutant cyclin A-CDK is constitutively active throughout the cell cycle (“hyperactive”). Our data have shown that the hyperactive cyclin A-CDK induces rapid tetraploidization in MEFs [Citation12]. However, effects of such cyclin A-CDK hyperactivation in human cells have been unknown. Here, we show hyperactivity of cyclin A2-CDK induces G2/M-phase arrest in human cells via activation of ATR-Chk1 pathway. This response may contribute to checking and eliminating abnormal cells in which cyclin A-CDK activity is deregulated.
Materials and methods
Cells and plasmids
All cells were obtained from RIKEN Bioresource Center Cell Bank. All cells used were grown in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) at 37°C with 5% CO2.
pCS2MT [Citation13]-based expression vector carrying myc-tagged human cyclin A2 (pCSMTCycA WT) has been described elsewhere [Citation12]. Myc-tagged CycAΔ80 expression vector was constructed by subcloning the human cyclin A2 coding region, from which the N-terminal 80 codons containing the D-box were truncated, into pCS2MT. An expression vector for adenovirus E1A (pE1A) was obtained from B. Clurman (Fred Hutchinson Cancer Research Center, Seattle, WA, USA). Expression vectors for HA-tagged human wild-type and dominant-negative CDK2 were obtained from D. Morgan (University of California, San Francisco, CA) and S. van den Heuvel (Utrecht University, Netherlands), respectively. pCS2HA [Citation13]-based expression vectors carrying HA-tagged human wild-type and AF mutant (resistant to the repressive phosphorylation, with alanine and phenylalanine substitutions at T14 and Y15, respectively) CDK1 were provided by M. Ohtsubo (Beppu University, Ooita, Japan). Human cdc25A expression vector was constructed by cloning the cDNA derived from WI-38 cells into pCSHA.
Antibodies and drugs
Anti-cyclin A polyclonal antibodies (Abs) [Citation14] were obtained from E. Firpo (Fred Hutchinson Cancer Research Center, Seattle, WA, USA). Anti-E1A (M58), anti-cyclin E (E-4), anti-p21 (C-19), anti-p27 (C-19) and anti-p107 (C-18) Abs were purchased from Santa Cruz Biotechnology. Others were as follows: anti-CDK1 from BD Biosciences, anti-Y15-phosphorylated (pY15) CDK1 from Abcam, anti-myc tag monoclonal and polyclonal Abs from MBL.
Nocodazole, caffeine and UCN-01 were purchased from SIGMA-Aldrich, and PV1019 and MK8776 were from Calbiocem and Selleckchem, respectively.
RNA interference experiments
Double-stranded small interfering RNA (siRNA) against adenovirus E1A gene was prepared by annealing the following RNAs which were designed according to siDirect (http://sidirect2.rnai.jp/): 5ʹ-AUAAUCUAACACAAACUCCUC-3ʹ and 5ʹ-GGAGUUUGUGUUAGAUUAUGU-3ʹ. The resulting siRNA was introduced into HEK293 cells using X-tremeGENE transfection reagent (Roche Diagnostics).
Transfection and flow cytometry
Those expression vectors described above were co-transfected into asynchronous human cell lines with 1 µg of green fluorescent protein (GFP)-expression vector, pEGFP-C1 (Clontech Laboratories, Inc., Mountain View, CA), using the modified Ca-phosphate method [Citation15]. Sixteen hours after the transfection, cells were refed and then cultured for an additional 24 h before being harvested. Flow cytometry was performed using FACSCalibur cytometer and CellOuest software (BD Biosciences), and data were gated for GFP-positive cells. Statistical analyses of cell cycle distribution were performed using MacCycle AV software (Phoenix Flow systems). Statistical significances of differences were analyzed by the Student t test.
Preparation of cell extracts and immunoprecipitation
Cells were rinsed once with Tris-buffered saline (pH 7.4) and lysed on ice in RIPA buffer (10 mM Tris-HCl, pH 7.4, 0.15 M NaCl, 0.5% Nonidet P-40, 50 mM NaF, 1 mM Na-vanadate, 10 mg/ml each of aprotinin, leupeptin and pepstatin). Immunoprecipitation and histone H1 kinase assay were performed as described [Citation16,Citation17] using the following Abs: anti-myc tag monoclonal PL-14 and polyclonal (MBL), anti-CDK2 polyclonal (Merck Millipore), anti-E1A monoclonal M58 (Santa Cruz Biotechnology).
Results
CycAΔ80 overexpression induces G2/M-phase arrest in the cell lines expressing low levels of p21 and p27
To examine the effect of hyperactivity of cyclin A2 in human cells, we constructed a stabilized mutant of cyclin A2 (CycAΔ80) [Citation18], which lacks its N-terminal domain containing the D-box. With the combination of a stabilized cyclin A mutant and lack of three CKIs (p21, p27 and p107), cyclin A-CDK is constitutively active throughout the cell cycle in MEFs, as we have previously reported [Citation12]. We transiently overexpressed CycAΔ80 and wild-type cyclin A (CycA WT) as myc epitope-tagged (mt) proteins in asynchronous human cell lines, U-2 OS, Saos-2, HT1080, WI-38, MDAH041 and HEK293, and analyzed the effect of CycAΔ80 overexpression on cell cycle by flow cytometry ()). Overexpression of CycA WT resulted in decreased G1-phase and increased S-phase populations, evidently in U-2 OS, MDAH041 and Saos-2 cells. This is consistent with previous reports that cyclin A accelerates G1/S transition [Citation9,Citation19]. CycAΔ80 overexpression had similar effects on the cell cycle distribution in U-2 OS, Saos-2, HT1080 and WI-38, while it led to an increase in G2/M population in MDAH041 and HEK293 ()).
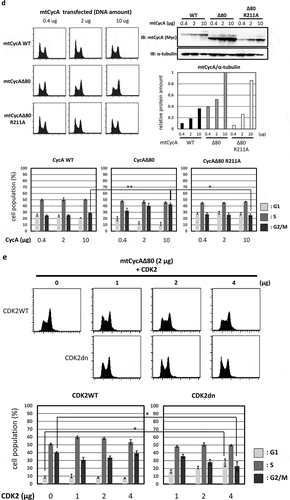
Figure 1. Effect of CycAΔ80 overexpression on cell cycle in human cells. (a) 10 µg each of myc-tagged cyclin A (mtCycA) WT or mtCycAΔ80 expression vector or empty vector (EV) was co-transfected with 1 µg of GFP expression vector in the indicated cell lines and cell cycle profiles of the GFP-positive cells were analyzed by flow cytometry (top). Arrows indicate the increase in G2/M populations. Cell cycle distributions of mtCycA WT- and mtCycAΔ80-transfected cells are shown in the bottom panel. **p < 0.01. (b) Extracts of the indicated cell lines were subjected to western analysis using antibodies against various CDK inhibitors and α-tubulin (internal control) (top). Protein amounts of p21, 27 and p107 relative to those of α-tubulin are shown (bottom). (c) Extracts from the indicated cells co-transfected with 10 µg of the mtCycA WT or Δ80 vector were immunoprecipitated with anti-myc tag, immunoblotted for mtCycA (top), and assayed for histone H1 kinase activity. H1 kinase activity (arbitrary units) relative to the amount of mtCycA proteins is shown in the bottom panel. (d) Indicated amounts of the expression vector for mtCycA WT, Δ80, or Δ80 R211A, which cannot bind to CDK, were co-transfected with 1 µg of the GFP vector in HEK293 cells and cell cycle profiles of the GFP-positive cells were analyzed by flow cytometry (top, left). Relative expression levels of exogenous CycA proteins are shown in the top right panel (maximum CycAΔ80 level = 1). Cell cycle distributions are shown in the bottom panel. *p < 0.05, **p < 0.01. (e) 2 µg of the mtCycAΔ80 vector was co-transfected with 1 µg of the GFP vector, and 4 µg of empty vector (0 µg CDK2) or indicated amounts of wild-type CDK2 (CDK2WT) or dominant-negative CDK2 mutant (CDK2dn) vector in HEK293 cells, and cell cycle profiles of the GFP-positive cells were analyzed by flow cytometry (top). Cell cycle distributions are shown in the bottom panel. *p < 0.05.
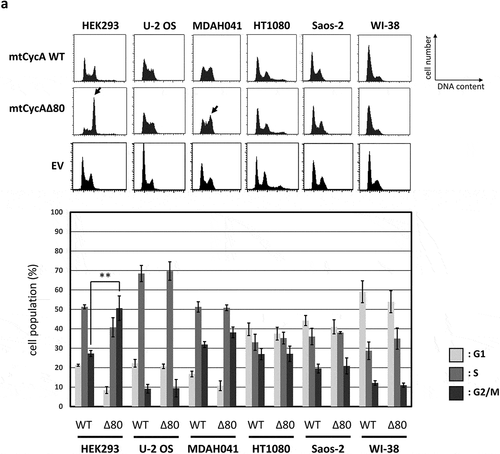
Figure 1. Continued.
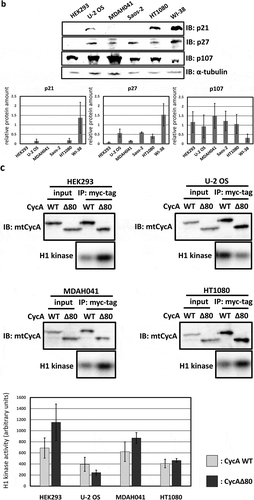
Figure 1. Continued.
Next, we assumed the expression level of CDK inhibitors (CKIs) to be a potential cause of different effects of CycAΔ80 overexpression in various human cell lines, and checked the expression levels of CKIs p21, p27 and p107 that could inhibit cyclin A-CDK activation in these cell lines by western blotting ()). In HEK293 cells, both p21 and p27 proteins are undetectable. In MDAH041 cells, p21 protein is undetectable and p27 protein level is relatively low in comparison with other cell lines. Meanwhile, p107 protein level is comparable among these cell lines, except for WI-38, whose p107 level is about three to five times lower than that in other cell lines. Therefore, relatively low expression levels of p21 and p27 correlate with the induction of G2/M-phase arrest by CycAΔ80. Moreover, we compared wild-type cyclin A-CDK activity with CycAΔ80-CDK activity in some of these cell lines. CycAΔ80-CDK activity was higher than CycA WT-CDK activity in HEK293 and MDAH041 where CycAΔ80 overexpression led to G2/M-phase arrest. In contrast, CycAΔ80-CDK activity was no higher than CycA WT-CDK activity in U-2 OS and HT1080 where CycAΔ80 overexpression had little effect on the cell cycle distribution ()).
In order to investigate whether the G2/M arrest by CycAΔ80 overexpression is dependent on CDK activity, we first overexpressed CycAΔ80 R211A mutant, which cannot bind to CDK [Citation20] (Figure S1), as well as wild-type cyclin A and CycAΔ80 in HEK293 cells and analyzed the effect of these CycA proteins by flow cytometry. CycAΔ80 R211A mutant overexpression had little effect as well as wild-type cyclin A, while CycAΔ80 overexpression led to an increase in G2/M population in a dose-dependent manner ()). The expression level of CycAΔ80 R211A is nearly comparable to that of CycAΔ80. This suggests that the G2/M arrest by CycAΔ80 is dependent on its binding to CDK. Next, we co-transfected CycAΔ80 with wild-type CDK2 (CDK2WT) or dominant-negative CDK2 mutant (CDK2dn). We observed that co-expression of CDK2dn but not CDK2WT suppressed the G2/M arrest by CycAΔ80 in a dose-dependent manner ()). Taken together, these results suggest that G2/M-phase arrest by CycAΔ80 overexpression depends on its associated CDK activity.
G2/M-phase arrest is promoted by co-expressing CycAΔ80 with E1A, which increases the amount of CycA-CDK2 complex
In human cancer cell line MDAH041, G2/M-phase arrest was only weakly induced by CycAΔ80 overexpression even though the expression levels of both p21 and p27 proteins are as low as those in HEK293. We assumed that the different effect of CycAΔ80 is caused by the expression of adenovirus-derived protein E1A in HEK293, and investigated the relationship between CycAΔ80 and E1A. First, we confirmed the interaction of CycA-CDK complexes and E1A in HEK293 cells by co-immunoprecipitation. E1A interacted with CycA and CDK2, but not with CDK1 ()). Next, we co-transfected HEK293 cells with CycAΔ80 and E1A-specific siRNA, and observed that the G2/M-phase arrest was suppressed by knocking-down E1A ()). Then, we investigated endogenous CDK2 activity and the amount of CycA-CDK2 complex in these cells by immunoprecipitation and histone H1 kinase activity assay. We observed that both CDK2 activity and the amount of CDK2-associated CycAΔ80 decreased by the introduction of E1A-specific siRNA ()). The amount of CycAΔ80 also decreased in the presence of E1A-specific siRNA, suggesting that E1A increases the expression of CycAΔ80.
Figure 2. G2/M-phase arrest is enhanced by co-expressing E1A. (a) Extracts from HEK293 cells were immunoprecipitated with control IgG and anti-E1A antibody, and immunoblotted for cyclin A (CycA), CDK1 and CDK2. (b) 10 µg of the mtCycAΔ80 vector was co-transfected with indicated concentrations of E1A-specific siRNA and 1 µg of the GFP vector in HEK293 cells, and cell cycle profiles of the GFP-positive cells were analyzed by flow cytometry (top). Cell cycle distributions are shown in the bottom panel. **p < 0.01. (c) Extracts from HEK293 cells co-transfected with the mtCycAΔ80 vector and E1A-specific siRNA were immunoprecipitated with anti-CDK2, immunoblotted for E1A and mtCycAΔ80, and assayed for histone H1 kinase activity (left panel). Relative E1A protein amount (0 nM siRNA = 1) in the cell extracts (input) is shown in the top middle panel. H1 kinase activity relative to the amount of CDK2 proteins (0 nM E1A siRNA = 1) is shown in the top right panel. Relative amounts of CDK2-associated mtCycAΔ80 and E1A (0 nM E1A siRNA = 1) are shown in the middle and right bottom panels, respectively. *p < 0.05. (d) Indicated amounts of the mtCycAΔ80 vector was co-transfected with 5 µg of E1A expression vector (pE1A) and 1 µg of the GFP vector in MDAH041 cells, and cell cycle profiles of the GFP-positive cells were analyzed by flow cytometry (top). Cell cycle distributions are shown in the bottom panel. *p < 0.05, **p < 0.01. (e) Extracts from MDAH041 cells co-transfected with 10 µg of the mtCycA WT or mtCycAΔ80 vector, with or without 5 µg of pE1A, were immunoprecipitated with anti-myc-tag and immunoblotted for CDK2 and E1A (left panel). Relative amounts of mtCycA-associated CDK2 (mtCycA WT only = 1) are shown in the right panel. **p < 0.01. (f) Extracts from MDAH041 cells co-transfected with 10 µg of the mtCycAΔ80 vector, with or without 5 µg of pE1A, were immunoprecipitated with anti-CDK2, immunoblotted for E1A, cyclin E, and mtCycAΔ80, and assayed for histone H1 kinase activity (left panel). H1 kinase activity relative to the amount of CDK2 proteins is shown in the middle top panel (E1A- = 1). Relative amounts of CDK2-associated mtCycAΔ80 and cyclin E (E1A- = 1) are shown in the bottom panels (middle and right, respectively).
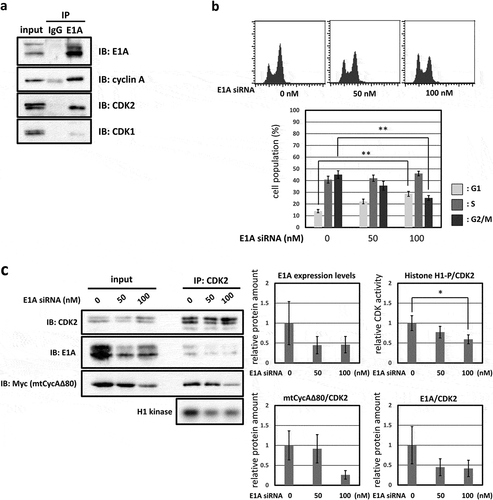
Figure 2. Continued.
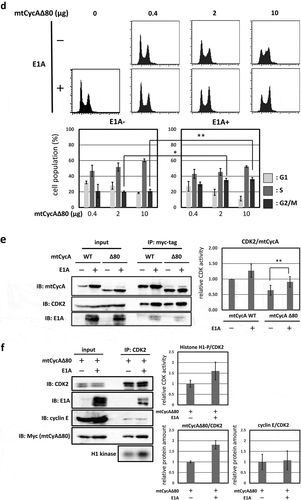
By contrast, we co-transfected CycAΔ80 with E1A in MDAH041 cells, and observed that the G2/M-phase arrest was promoted by co-expressing E1A ()). We also investigated the interaction of CycA and CDK2 in these cells by immunoprecipitation and observed that the amount of CycA-associated CDK2 increased with co-expression of E1A ()). This suggests that E1A facilitates the formation of CycA-CDK2 complex. Furthermore, to investigate whether endogenous CDK2 activity is increased by E1A co-transfection, we extracted CDK2 from MDAH041 cells co-transfected with CycAΔ80, with or without E1A, by immunoprecipitation and assayed for histone H1 kinase activity. We observed that CycAΔ80-associated CDK2 activity from the cells co-transfected with E1A was higher than without E1A ()). The amount of CycAΔ80 expression also increased with E1A co-expression as shown in ). Co-expression of E1A increased the amount of CycAΔ80, but not cyclin E, associated with CDK2 ()). These results suggest that endogenous CDK2 activity is enhanced by co-expressing E1A, which increases the amount of both CycA and CycA-CDK2 complex, and that the elevated CDK2 activity promotes G2/M-phase arrest.
G2/M-phase arrest is induced by CycAΔ80 overexpression via ATR-Chk1 pathway
A previous report has shown that CDK2 induces the G2/M-phase arrest via ATR-Chk1 pathway, which is p53-independent [Citation11]. We, therefore, asked if G2/M-phase arrest induced by overexpression of CycAΔ80 depends on ATR-Chk1 pathway. Thus, in p53-negative MDAH041 cells, we co-transfected CycAΔ80 with E1A and treated them with specific inhibitors of Chk1, Chk2 and ATR. We observed that the G2/M-phase arrest by CycAΔ80 was suppressed by caffeine (ATM/ATR inhibitor), MK8766 and UCN-01 (Chk1-specific inhibitors), but not by PV1019, a specific inhibitor of Chk2 ()). Moreover, to identify the period of Chk1 function in the G2/M-phase arrest, we treated MDAH041 cells co-expressing CycAΔ80 and E1A with nocodazole, which blocks cell cycle at prometaphase in mitosis, and MK8766. After the release from nocodazole block, a substantial population of cells expressing CycAΔ80 was still arrested at G2/M phase, while a majority of cells expressing CycA WT was released (), ②). A treatment with MK8766 8 h before the release from nocodazole block (), ③) suppresses G2/M-phase arrest by the CycAΔ80 overexpression more efficiently than a treatment after the release (), ④). These results suggest that the G2/M-phase arrest by CycAΔ80 overexpression is induced by DNA damage checkpoint via ATR-Chk1, which functions before prometaphase.
Figure 3. G2/M-phase arrest is induced by CycAΔ80 overexpression via ATR-Chk1 pathway. (a) MDAH041 cells co-transfected with 10 µg of the mtCycAΔ80 vector, 5 µg of pE1A and 1 µg of the GFP vector were treated with ATM/ATR, Chk1, and Chk2 inhibitors (final concentration: 4 mM caffeine, 1 μM MK8776, 50 nM UCN-01, 300 nM PV1019, respectively) for 24 h, and cell cycle profiles of the GFP-positive cells were analyzed by flow cytometry (top). Cell cycle distributions are shown in the bottom panel. *p < 0.05, **p < 0.01. (b) MDAH041 cells co-transfected with 10 µg of the mtCycAΔ80 vector, 5 µg of pE1A and 1 µg of the GFP vector were treated for 24 h with 0.1 µM nocodazole added 16 h after transfection (①), then released from nocodazole for 8 h (②) or treated with 1 µM MK8776 for 8 h, before (③) or after (④) the release from nocodazole, and cell cycle profiles of the GFP-positive cells were analyzed by flow cytometry (top). As for the nocodazole-released condition (②), the cell cycle profile of the cells co-transfected with the mtCycA WT vector was also shown. Cell cycle distributions are shown in the bottom panel. **p < 0.01. (c) Extracts from MDAH041 cells co-transfected with 10 µg of the mtCycAΔ80 vector, with or without 5 µg of pE1A, were immunoprecipitated with anti-myc-tag and immunoblotted for the CDK1 and CDK1 phosphorylated at Y15 (pY15) (left). Relative amounts of mtCycA-associated CDK1-pY15 (E1A – = 1) are shown in the right panel. (d) Extracts from MDAH041 cells co-transfected with 10 µg of the mtCycAΔ80 and 5 µg of p E1A, treated for 24 h with 0.1 µM nocodazole 16 h after transfection and treated with or without 1 µM MK8776 for 8 h before harvest, were immunoprecipitated with anti-myc-tag and immunoblotted for the CDK1 phosphorylated at Y15 (pY15) and CDK1 (left). Relative amounts of mtCycA-associated CDK1-pY15 (-Chk1 inhibitor = 1) are shown in the right panel. *p < 0.05. (e) MDAH041 cells co-transfected with 10 µg of the mtCycAΔ80 vector, 5 µg of pE1A, 1 µg of the GFP vector and 3 µg each of empty vector (EV), wild-type CDK1 (CDK1WT), unphosphorylatable mutant CDK1 (CDK1AF) or cdc25A expression vector were treated for 24 h with 0.1 µM nocodazole after transfection, released for 8 h, and cell cycle profiles of the GFP-positive cells were analyzed by flow cytometry (top). Cell cycle distributions are shown in the bottom panel. *p < 0.05, **p < 0.01.
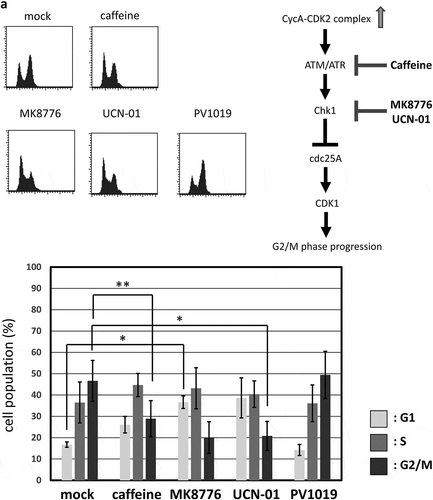
Figure 3. Continued.
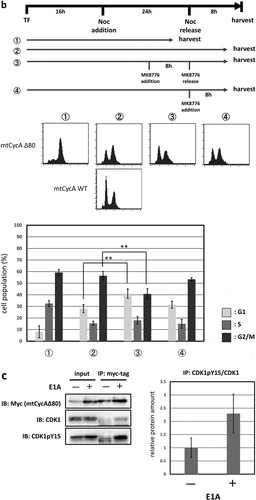
Figure 3. Continued.
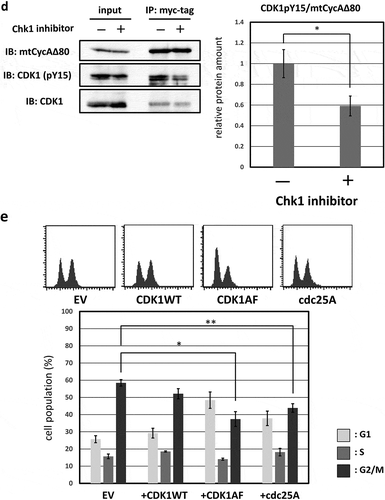
Furthermore, the previous report has shown that ATR-Chk1 activation by CDK2 suppresses dephosphorylation by cdc25A, which leads to CDK1 activation, at tyrosine 15 (Y15) [Citation11]. We, therefore, asked if hyperactivation of CycAΔ80-CDK increases phosphorylation of CDK1 at Y15 (CDK1-pY15), via ATR-Chk1 pathway. First, in MDAH041 cells, we co-transfected CycAΔ80 with or without E1A. We observed that amount of CDK1-pY15 associated with CycAΔ80 was increased by co-expression of E1A ()). Next, we co-transfected CycAΔ80 with E1A and treated them with or without MK8766 8 h before the release from nocodazole block in MDAH041 cells. We observed that amount of CDK1-pY15 associated with CycAΔ80 was decreased by MK8766 treatment ()). Moreover, we co-transfected CycAΔ80 with E1A and CDK1WT or CDK1AF mutant, which eliminates the repressive phosphorylation at T14 and Y15, in MDAH041 cells. Co-expressing CDK1AF suppressed the G2/M arrest by CycAΔ80, after the release from nocodazole block, more efficiently than co-expressing CDK1WT ()). We also co-transfected CycAΔ80 with E1A and cdc25A in MDAH041 cells, and observed that co-expressing of cdc25A similarly suppressed the G2/M arrest by CycAΔ80 ()). These results suggest that CycAΔ80 overexpression and activation of its associated CDK2 lead to ATR-Chk1 activation, which results in inactivation of CDK1 via cdc25A suppression and induces G2/M arrest.
Discussion
CycA-CDK is negatively regulated by degradation via ubiquitin-dependent proteolysis and inactivation via CKI (p21, p27, p107) binding. Failure of these negative regulations causes genetic instability. Our previous work has shown that a stabilized mutant cyclin A induces tetraploidization in p21−/- p27−/- p107−/-, but not in p21−/- p27−/-, MEFs [Citation12]. Here, we investigated the effect of hyperactivity of cyclin A2 in human cells and observed that CycAΔ80 overexpression induced G2/M-phase arrest in two human cell lines, HEK293 and MDAH041 ()). These human cell lines express low levels of p21 and p27 compared to other human cell lines in which CycAΔ80 overexpression did not induce G2/M-phase arrest. By contrast, in p21−/- p27−/- MEFs, CycAΔ80 overexpression did not induce G2/M-phase arrest. These results suggest that CycAΔ80 overexpression causes different effects between MEFs and human cell lines.
We probed the mechanisms that G2/M-phase arrest by CycAΔ80 overexpression in human cell lines. A previous report has shown that CDK2 regulates the G2/M-phase checkpoint by activating ATR-Chk1 pathway, which is p53-independent [Citation11]. We demonstrated that CycAΔ80 overexpression induced G2/M-phase arrest in human cells. In addition, we observed that CDK2 activity is enhanced by co-expressing E1A, which facilitates the formation of CycA-CDK2 complex and thereby promotes G2/M-phase arrest ()). Intriguingly, a previous report has shown that E1A inhibits 26S proteasome activity [Citation21]. We observed that the amount of exogenous CycA expression is fluctuated by E1A expression level. This result could be caused by an inhibition of 26S proteasome by E1A. However, the amount of CDK2 associated with CycAΔ80 was increased by co-expression of E1A ()), suggesting that E1A not only increases the CycA protein level but facilitates the formation of CycA-CDK2 complex. Our observation that E1A increased CycAΔ80, but not cyclin E, which is associated with CDK2 ()) suggests that E1A does not enhance cyclin E-CDK2 activity. Indeed, we observed that cyclin E and E1A co-expression does not induce G2/M-phase arrest (Figure S2).
These results correlate with the observations that CDK2 activity stimulates ATR-Chk1 pathway and induces G2/M-phase arrest in the previous report [Citation11]. We indeed observed that G2/M-phase arrest induced by CycAΔ80 overexpression was suppressed by the addition of ATR and Chk1 inhibitors ()). In addition, suppression of CycAΔ80-derived G2/M-phase arrest by Chk1 inhibitor was observed without co-expressing E1A (Figure S3). Moreover, the previous paper has shown that CDK1pY15 is increased via inhibition of cdc25A by Chk1 [Citation11]. Chk1 has been shown to inhibit interaction between cdc25A and cyclin-CDK complex [Citation22]. We also observed that CycAΔ80-induced G2/M-phase arrest was suppressed by co-expressing cdc25A or unphosphorylatable CDK1AF mutant ()). These results suggest that G2/M-phase arrest by CycAΔ80 overexpression in human cells is caused by inactivation of CDK1 via ATR-Chk1-mediated cdc25A suppression. On the other hand, another report has shown that CDK2 is not required for Chk1 activation and G2/M-phase checkpoint [Citation23]: the amount of p21 binding to CDK1 increases upon DNA damage in CDK2−/- cell line, which induces G2/M-phase checkpoint. Our present studies, however, demonstrate that aberrant activation of CDK2 induces G2/M-phase arrest via ATR-Chk1 pathway in the cell lines expressing undetectably low level of p21, which is different from the status of CDK2 activity and p21 expression in the cell lines described in that report.
In conclusion, we show that aberrant activation of CDK2 by CycAΔ80 overexpression induces G2/M-phase arrest via the cell cycle checkpoint ATR-Chk1 pathway. This response may contribute to checking and eliminating abnormal cells in which CycA-CDK activity is deregulated. It should be noted that this response depends on the stabilization of CycA (and concomitant activation of CycA-CDK) from metaphase to G1-phase, the period when CycA is disrupted in normal cell cycle, since CycAΔ80 induces G2/M-phase arrest stronger than wild-type CycA. Further analyses on pathway(s) downstream of aberrant stabilization of CycA during M/G1 phase would shed light on a physiological role of this response.
Supplemental Material
Download PDF (192.5 KB)Acknowledgments
We thank M. Ohtsubo, B. Clurman, D. Morgan, S. van den Heuvel, E. Firpo, and RIKEN Bioresource Center for materials; and N. Mori and D. Sato for technical assistance. This work was supported by Tokyo NODAI Doctoral Research Grant Program.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Nurse P. Universal control mechanism regulating onset of M-phase. Nature. 1990;344:503–508.
- Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294.
- King RW, Deshaies RJ, Peters JM, et al. How proteolysis drives the cell cycle. Science. 1996;274:1652–1659.
- Den EN, Pines J. Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J Cell Biol. 2001;153:121–135.
- Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134.
- Harper JW, Elledge SJ, Keyomarsi K, et al. Inhibition of cyclin-dependent kinases by p21. Mol Biol Cell. 1995;6:387–400.
- Polyak K, Lee MH, Erdjument-Bromage H, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66.
- Pagano M, Pepperkok R, Verde F, et al. Cyclin A is required at two points in the human cell cycle. Embo J. 1992;11:961–971.
- Resnitzky D, Hengst L, Reed SI. Cyclin A-associated kinase activity is rate limiting for entrance into S phase and is negatively regulated in G1 by p27Kip1. Mol Cell Biol. 2015;15:4347–4352.
- Kabeche L, Compton DA. Cyclin A regulates kinetochore microtubules to promote faithful chromosome segregation. Nature. 2013;502:110–113.
- Chung JH, Bunz F. Cdk2 is required for p53-independent G2/M checkpoint control. PLoS Genet. 2010;6:e1000863.
- Chibazakura T, McGrew SG, Cooper JA, et al. Regulation of cyclin-dependent kinase activity during mitotic exit and maintenance of genome stability by p21, p27, and p107. Proc Natl Acad Sci. 2004;101:4465–4470.
- Turner DL, Weintraub H. Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev. 1994;8:1434–1447.
- Marraccino RL, Firpo EJ, Roberts JM. Activation of the p34 CDC2 protein kinase at the start of S phase in the human cell cycle. Mol Biol Cell. 1992;3:389–401.
- Clurman BE, Sheaff RJ, Thress K, et al. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 1996;10:1979–1990.
- Ghiara JB, Richardson HE, Sugimoto K, et al. A cyclin B homolog in S. cerevisiae: chronic activation of the Cdc28 protein kinase by cyclin prevents exit from mitosis. Cell. 1991;65:163–174.
- Surana U, Amon A, Dowzer C, et al. Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. Embo J. 2018;12:1969–1978.
- Geley S, Kramer E, Gieffers C, et al. Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J Cell Biol. 2001;153:137–148.
- Chibazakura T, Kamachi K, Ohara M, et al. Cyclin A promotes S-phase entry via interaction with the replication licensing factor Mcm7. Mol Cell Biol. 2011;31:248–255.
- Kobayashi H, Stewart E, Poon R, et al. Identification of the domains in cyclin A required for binding to, and activation of, p34cdc2 and p32cdk2 protein kinase subunits. Mol Biol Cell. 1992;3:1279–1294.
- Turnell AS. Regulation of the 26S proteasome by adenovirus E1A. Embo J. 2002;19:4759–4773.
- Uto K, Inoue D, Shimuta K, et al. Chk1, but not Chk2, inhibits Cdc25 phosphatases by a novel common mechanism. Embo J. 2004;23:3386–3396.
- Bačević K, Lossaint G, Achour TN, et al. Cdk2 strengthens the intra-S checkpoint and counteracts cell cycle exit induced by DNA damage. Sci Rep. 2017;7:13429.