
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The phosphoinositide-3-kinase like kinases (PIKK) such as ATM and ATR play a key role in initiating the cellular DNA damage response (DDR). One key ATM target is the cyclin-dependent kinase inhibitor p27Kip1 that promotes G1 arrest. ATM activates p27Kip1-induced arrest in part through phosphorylation of p27Kip1 at Serine 140. Here we show that this site is dephosphorylated by the type 2C serine/threonine phosphatase, WIP1 (Wildtype p53-Induced Phosphatase-1), encoded by the PPM1D gene. WIP1 has been shown to dephosphorylate numerous ATM target sites in DDR proteins, and its overexpression and/or mutation has often been associated with oncogenesis. We demonstrate that wildtype, but not phosphatase-dead WIP1, efficiently dephosphorylates p27Kip1 Ser140 both in vitro and in cells and that this dephosphorylation is sensitive to the WIP1-specific inhibitor GSK 2830371. Increased expression of wildtype WIP1 reduces stability of p27Kip1 while increased expression of similar amounts of phosphatase-dead WIP1 has no effect on p27Kip1 protein stability. Overexpression of wildtype p27Kip1 reduces cell proliferation and colony forming capability relative to the S140A (constitutively non-phosphorylated) form of p27. Thus, WIP1 plays a significant role in homeostatic modulation of p27Kip1 activity following activation by ATM.
Introduction
To preserve genomic integrity, systems of DNA damage detection, response, and repair have evolved. Key components of the DNA damage response (DDR) system include a set of kinases that recognize damage and transduce signals that activate the many cellular responses to DNA damage [Citation1–Citation3]. The phosphatidylinositol 3-kinase-like kinase (PIKK) family, typified by the intensively studied ATM, ATR, and DNA-PK, play a central role in the DDR [Citation1,Citation2]. These kinases are activated almost immediately following DNA damage and transduce their damage response signal through phosphorylation of DDR target proteins [Citation4]. PIKK targets include other kinases and signal transducers that amplify the damage signal, as well as downstream repair proteins and cell cycle regulatory molecules such as p53. p53 is a transcription factor that is directly phosphorylated by PIKKs such as ATM and ATR, resulting in p53 activation [Citation2,Citation5]. For example, ATM/ATR directly phosphorylate p53 at Serine 15, which contributes to increased p53 stability and activation as a transcription factor [Citation6,Citation7]. Activated p53 can initiate transcription of genes that enforce cell cycle arrest in some contexts or apoptosis in other contexts [Citation8,Citation9]. Induction of cell cycle arrest prevents propagation of damaged DNA templates as mutations whereas apoptosis protects the organism from the ill effects of irreparably damaged cells.
Among the many genes induced by p53 following DNA damage are genes with a homeostatic role. In those cases where DNA damage is repaired, mechanisms to return the damage-activated cell to an unstressed state are necessary [Citation10]. Among these p53-induced homeostatic genes are MDM2 and PPM1D. MDM2 encodes an E3-ubiquitin ligase that actively promotes degradation of p53 following successful repair, returning p53 protein to prestress levels [Citation11]. In addition, p53 activates the gene PPM1D (protein phosphatase, Mg2+/Mn2+ dependent 1D) that encodes the protein WIP1 (wildtype p53-induced phosphatase 1) [Citation12,Citation13]. WIP1 has been shown to dephosphorylate many of the same targets that PIKKs target, reversing their effects and returning them to a less activated state [Citation13]. For example, WIP1 dephosphorylates p53 at Serine 15 and MDM2 at Serine 395, facilitating MDM2 degradation of p53 [Citation14,Citation15]. Thus, once damage repair is completed, WIP1 assists the cell in its return to a prestress homeostatic state [Citation13]. Because the PPM1D gene product antagonizes p53, a potent tumor suppressor, it is not surprising that amplification or stabilizing mutations of PPM1D have been associated with human tumors and preneoplastic lesions [Citation13,Citation16–Citation20].
Among other cell cycle regulatory proteins activated by PIKKs, the cyclin-dependent kinase inhibitor p27Kip1 has recently been demonstrated to be a target of ATM [Citation21]. Encoded by the CDKN1B gene, activated p27Kip1 inhibits G1 progression and S phase entry and maintains cells in a non-proliferative state [Citation22,Citation23]. Emerging evidence indicates that it does play a significant role in maintaining genomic stability as well as regulating cell cycle progression [Citation24–Citation26]. An early screen of ATM/ATR phosphorylation targets revealed that ATM phosphorylates p27Kip1 at Serine 140 in response to DNA damage [Citation27]. More recently, Cassimere et al. [Citation21] confirmed ATM phosphorylation of p27Kip1 at Serine 140 and that this phosphorylation is important for stabilization and enforcement of the p27Kip1-mediated G1 checkpoint in response to DNA damage.
In this study we confirm ATM phosphorylation of p27Kip1 at Serine 140 and also demonstrate that WIP1 dephosphorylates p27Kip1 at this site in both in vitro and in cell culture assays. Following radiation damage, p27Kip1 Serine 140 phosphorylation increases rapidly but is gradually lost in a WIP1-dependent manner. p27Kip1 dephosphorylation by WIP1 is also associated with a reduction in p27Kip1 protein levels. We demonstrate that inability to phosphorylate p27Kip1 at Serine 140 is associated with enhanced cellular proliferation and colony formation, illustrating the relevance of this single phosphorylation site in mediating p27Kip1 growth suppression effects.
Results
Phosphopeptide screens show WIP1 preferences for ATM/ATR target sites in vitro
WIP1 is a serine/threonine phosphatase that has been shown to have an affinity for dephosphorylation of protein target sites with pSQ, pTQ or pTxY amino acid motifs [Citation13]. pS/pTQ motifs in particular represent sites that are often phosphorylated by DNA damage responsive kinases ATM and ATR. In fact, virtually all of the currently identified pS/pTQ sites on proteins dephosphorylated by WIP1 are known to be phosphorylated by ATM or ATR [Citation13,Citation28]. We sought a high-throughput method to identify potential WIP1 target proteins and the specific sites of dephosphorylation. Since WIP1 often dephosphorylates known sites of ATM/ATR phosphorylation, we selected 86 candidate phosphopeptides likely to be targeted by WIP1 from a large screen of ATM/ATR phosphorylation targets identified by Matsuoka et al. [Citation27] and from studies of individual ATM/ATR targets. In addition, we selected 78 novel pS/pTQ and 6 pTxY target sites of interest in proteins with DDR or cell cycle regulatory functions. We also assayed 123 non-pS/pTQ and non-TxY sites in proteins of interest related to DDR or cell cycle control as negative controls. We assayed a total of 293 phosphopeptides representing potential WIP1 target sites (Table S1).
Utilizing an in vitro phosphatase assay for each candidate phosphopeptide (see Methods), we were able to determine the degree to which these phosphopeptides could be dephosphorylated by recombinant WIP1 (). The complete list of peptides, sequences and in vitro phosphatase assay results are shown in Table S1. We selected a cutoff value of 1000 pmol of free phosphate released in one hour to identify positive dephosphorylation by WIP1. Of the 164 putative pS/pTQ sites tested, 131, roughly 80%, were found to be efficiently dephosphorylated by WIP1, indicating a strong selection for glutamine (Q) preceded by pS or pT. Of the 6 candidate TxY phosphopeptides, 4 (67%) were positive in our assay. Only 12 (10%) of the remaining 123 non-S/TQ sites were positive WIP1 target sites by our criteria. Analysis of amino acid sequences in the assayed 11 amino acid pS/pTQ phospho-peptides (pS or pT is always at amino acid position 6) showed a strong enrichment for aspartic acid (D) and glutamic acid (E) surrounding the phosphorylation site (Figure S1). Basic amino acids arginine, histidine and lysine (R,H, and K respectively) were selected against in our positive WIP1 targets (Figure S1). This corroborates WIP1 target site preferences presented previously by Appella and colleagues [Citation29].
Figure 1. WIP1 dephosphorylates p27Kip1 in vitro. (a) WIP1 target screening by in vitro phosphatase assay. Here, 264 synthetic 11 amino acid phosphopeptides (China Peptides Co.) were incubated with purified active WIP1 enzyme in the presence of Mg2+. The release of free phosphate from each of the phosphopeptides was quantified to determine phosphatase activity. Arrow indicates the location of p27Kip1 (CDKN1B) S140 phosphopeptide in relation to all others in the screen. Samples are duplicated for the assay. The green dotted line indicates the threshold value of 1000 pmol free phosphate released in the assay. We considered all phosphopeptides above that value to be strong candidate WIP1 target sites. The list of candidate phosphopeptides and WIP1 activity in the presence of each is indicated in Table S1. (b) Further validation of WIP1 dephosphorylation of p27Kip1 at serine 140. The 11 amino acid phosphopeptide including the central S140 residue was incubated with WIP1 and WIP1 D314A (phosphatase-dead mutant). WIP1-specific inhibitor GSK2830371 was used at 3 µM concentration. WIP1 enzyme without Mg2+ is used as a control. p53 pS15 and UNG2 pT31 are the positive [Citation14] and negative [Citation30] controls for WIP1 activity, respectively. Samples were assayed in duplicate. (c) WIP1 dephosphorylates intact p27Kip1 in vitro after p27Kip1 purification from HEK293 cells. After overexpression of wild-type Myc-p27Kip1 in HEK293 cells, the cells were irradiated with 10 Gy IR (0.5 h recovery) and then lysed in lysis buffer. Myc-p27Kip1, precipitated with anti-Myc antibody and purified from the HEK293 cell lysates, was incubated with purified recombinant WIP1 to assess phosphatase activity. The proteins were resolved on SDS-PAGE and followed by immunoblot analysis using anti-WIP1, anti-p27Kip1 protein, and anti-p27Kip1 pS140 antibodies.
![Figure 1. WIP1 dephosphorylates p27Kip1 in vitro. (a) WIP1 target screening by in vitro phosphatase assay. Here, 264 synthetic 11 amino acid phosphopeptides (China Peptides Co.) were incubated with purified active WIP1 enzyme in the presence of Mg2+. The release of free phosphate from each of the phosphopeptides was quantified to determine phosphatase activity. Arrow indicates the location of p27Kip1 (CDKN1B) S140 phosphopeptide in relation to all others in the screen. Samples are duplicated for the assay. The green dotted line indicates the threshold value of 1000 pmol free phosphate released in the assay. We considered all phosphopeptides above that value to be strong candidate WIP1 target sites. The list of candidate phosphopeptides and WIP1 activity in the presence of each is indicated in Table S1. (b) Further validation of WIP1 dephosphorylation of p27Kip1 at serine 140. The 11 amino acid phosphopeptide including the central S140 residue was incubated with WIP1 and WIP1 D314A (phosphatase-dead mutant). WIP1-specific inhibitor GSK2830371 was used at 3 µM concentration. WIP1 enzyme without Mg2+ is used as a control. p53 pS15 and UNG2 pT31 are the positive [Citation14] and negative [Citation30] controls for WIP1 activity, respectively. Samples were assayed in duplicate. (c) WIP1 dephosphorylates intact p27Kip1 in vitro after p27Kip1 purification from HEK293 cells. After overexpression of wild-type Myc-p27Kip1 in HEK293 cells, the cells were irradiated with 10 Gy IR (0.5 h recovery) and then lysed in lysis buffer. Myc-p27Kip1, precipitated with anti-Myc antibody and purified from the HEK293 cell lysates, was incubated with purified recombinant WIP1 to assess phosphatase activity. The proteins were resolved on SDS-PAGE and followed by immunoblot analysis using anti-WIP1, anti-p27Kip1 protein, and anti-p27Kip1 pS140 antibodies.](/cms/asset/27f1360b-f897-477d-8169-c1890a2326a2/kccy_a_1717025_f0001_c.jpg)
Since cell cycle arrest occurs early in the DNA damage response, we sought to identify WIP1 target proteins involved in cell cycle arrest. We tested 41 possible phosphorylation sites involved in cell cycle arrest. These sites spanned CDC25A, CDC25B, CDC25C, multiple cyclins, CDK4 and 9, as well as the CDK inhibitors p21Cip1 and p27Kip1. Only 5 pS/pTQ sites and 2 pS/pT sites were identified as WIP1 targets (Table S1). Of particular interest was p27Kip1, which had recently been validated as an ATM target protein. p27Kip1 has been shown to be phosphorylated by ATM at serine 140 in response to DNA damaging agents [Citation21]. We identified three additional pS/pTQ sites on p27Kip1 aside from Serine 140. We tested these four sites in our in vitro phosphatase assay along with 15 known p27Kip1 phosphorylation sites associated with other kinases. All four pS/pTQ sites were strongly positive in the in vitro phosphatase assay along with one CK2 kinase site at Serine 83 (Figure S2A). All other known phosphorylation sites were negative according to our standard criteria (<1000 pmol free phosphate released in one hour). Interestingly, all four pS/pTQ sites show significant evolutionary conservation (Figure S2B). To our knowledge, there is no evidence that p27Kip1 sites other than Serine 140 are phosphorylated by ATM. So for this reason, and the availability of phospho-specific antibodies for p27Kip1 Serine 140, we focused on this phosphorylation site.
WIP1 dephosphorylates p27Kip1 at Serine 140 in vitro
To validate that p27Kip1 Serine 140 is indeed a WIP1 target we performed a more rigorous in vitro phosphatase assay. The p27Kip1 S140 phosphopeptide was incubated with recombinant purified WIP1 in the presence or absence of magnesium, which is necessary for WIP1 phosphatase activity. In parallel experiments a negative control UNG2 T31 phosphopeptide [Citation30] and a positive control p53 S15 phosphopeptide [Citation14] were incubated with purified WIP1 under all of the same conditions as the p27Kip1 S140 phosphopeptide. As expected, in the presence of magnesium and purified WIP1, the p27Kip1 S140 and p53 S15 positive control phosphopeptides showed robust release of free phosphate in the in vitro phosphatase assay, whereas the negative control UNG2 T31 negative control phosphopeptide showed little or no phosphate release (). In the absence of magnesium there is no discernible dephosphorylation of the p27Kip1 S140 or control phosphopeptides (). If the phosphopeptides are incubated with the catalytically inactive D314A WIP1 (pdWIP1) there is no dephosphorylation of the p27Kip1 S140 phosphopeptide the other phosphopeptides. To further demonstrate that the dephosphorylation of p27Kip1 S140 is specifically due to WIP1, we incubated the complete reaction of p27 phosphopeptide, recombinant WIP1 and magnesium with the WIP1 inhibitor GSK 2830371 [Citation31]. Once again there was no detectable dephosphorylation of p27Kip1 S140, confirming that the p27Kip1 S140 phosphopeptide is specifically dephosphorylated by WIP1 in this assay.
The in vitro phosphatase assay utilizes an 11 amino acid phosphopeptide. Therefore, we determined whether WIP1 could also dephosphorylate full-length p27Kip1 in vitro. We immunoprecipitated Myc-tagged p27Kip1 from transfected cells exposed to 10 Gy ionizing radiation (to generate Ser140-phosphorylated p27Kip1) and incubated the immunoprecipitate with recombinant WIP1 in the presence or absence of magnesium. The immunoprecipitates were then subjected to immunoblotting for the S140 phosphorylated form of p27Kip1, using a phospho-specific antibody for this site. There is a robust phospho-p27Kip1 (S140) band in the Myc IP only lane, as well as in the WIP1 lane without magnesium (). However, that band is absent when the immunoprecipitate is incubated with WIP1 and magnesium, indicating that WIP1 does indeed dephosphorylate full-length p27Kip1 at serine 140.
Confirmation that ATM phosphorylates p27Kip1 at Serine 140
We further sought to confirm p27 phosphorylation by ATM, as previously described by Cassimere et al. [Citation21]. His-tagged recombinant p27 (p27-His) was incubated with immunoprecipitated Flag-ATM along with 32P-labeled ATP in an in vitro kinase assay. Incubation of Flag-ATM with p27-His resulted in a 32P-labeled band that was not present in lanes with either Flag-ATM only or p27-His only (). In a parallel control assay, known ATM target p53 was also robustly phosphorylated by ATM. These experiments indicate that ATM is indeed phosphorylating p27 in vitro. Since there are multiple pS/pTQ sites in the p27 protein sequence, we examined whether serine 140 was specifically being phosphorylated by ATM. To address this question, phospho-p27 S140 immunoblot analysis following Flag-ATM incubation with p27-His was performed and shows that p27 is indeed phosphorylated at S140 by ATM ().
Figure 2. ATM phosphorylates p27Kip1. (a) In vitro kinase assays demonstrate ATM phosphorylation of p27Kip1. HEK293 cells were transfected with FLAG-tagged ATM and were irradiated with 15 Gy IR. Two hours after IR treatment, the HEK293 cells were lysed and activated ATM was immunoprecipitated from the lysate with anti-Flag antibody. The immunoprecipitated FLAG-ATM was then incubated with recombinant His-p27Kip1 or His-p53 protein, and γ-32P-ATP in the presence or absence of ATM inhibitor (KU-55933) for 1 hour at 30◦C. 32P-labeled proteins were then separated by SDS-PAGE followed by autoradiography. (b) Radiation-activated ATM phosphorylates p27Kip1 in vitro. HEK293 cells were transfected with FLAG-tagged ATM and were irradiated with 15 Gy IR (2 h recovery). Activated ATM was immunoprecipitated with anti-FLAG antibody and incubated with recombinant His-p27Kip1 in the presence or absence of p27Kip1 for 1 h at 30◦C. The proteins were resolved on an SDS-PAGE, followed by immunoblot analysis using anti-ATM, anti-p27, and anti-p27 pS140 antibodies. (c) Time-course of phosphorylation on p27Kip1 Serine 140 following irradiation. HEK293 cells were transfected with Myc-p27Kip1 and then treated with 10 Gy ionizing radiation (IR). Cells were recovered up to 3 hours from irradiation. After cells were lysed at each indicated time, total lysates were resolved on SDS-PAGE, followed by immunoblot analysis with indicated antibodies. CHK2 T68 and p53 S15 are targets of ATM phosphorylation and serve as markers for activation of the DNA damage response. GAPDH served as a loading control. (d) p27Kip1 S140 phosphorylation is impaired by ATM-specific inhibitor in HEK293 cells. HEK293 cells were transfected with Myc-p27Kip1, treated with 0, 10 or 20 µM of ATM inhibitor KU-55933 and then irradiated to 10 Gy ionizing radiation (IR). CHK2 T68 and p53 S15 are DNA damage-associated targets of ATM phosphorylation. After 0.5 h recovery, immunoblot analyses were performed with the indicated antibodies.
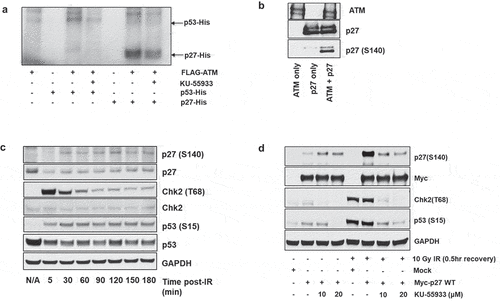
We next asked whether p27Kip1 is phosphorylated in response to DNA damage in cells. We treated Myc-p27 transfected HEK293 cells with 10 Gy ionizing radiation (IR) and allowed the cells to recover for the indicated time points. Whole cell lysates were prepared from each sample and subjected to immunoblotting for p27 phosphorylated at S140. The p27 (S140) band was detectable at 30 minutes post irradiation and was constant up to 180 minutes (). To determine whether ATM was specifically phosphorylating p27 in response to IR, Myc-p27 transfected HEK293 cells were pretreated with the ATM inhibitor KU-55933 prior to IR treatment. Immunoblot analysis of cell lysates show a robust increase in p27 (S140) in response to IR treatment, which is decreased back to background levels in samples pre-treated with KU-55933 (). This complements previous studies indicating that ATM phosphorylates p27Kip1 at serine 140 following DNA damage [Citation21,Citation27].
p27Kip1 is dephosphorylated by WIP1 in cells
To determine whether p27Kip1 phosphorylated at S140 is dephosphorylated by WIP1 in cells, we co-transfected a Flag-tagged WIP1 with Myc-p27 in HEK293 cells. Transfected cells were again treated with 10 Gy IR followed by immunoblotting for target proteins. In response to IR we again see p27Kip1 (S140) phosphorylation in the IR sample but decreased levels in the un-irradiated sample (). When the Flag-WIP1 co-transfected cells were exposed to IR, we lose the robust p27Kip1 (S140) phosphorylated band (). To verify that the effect we see is due to WIP1 phosphatase activity, we co-transfected the D314A phosphatase-dead mutant WIP1 construct (pdFlag-WIP1) instead of wildtype WIP1. pdFlag-WIP1 transfected cells show increased levels of phosphorylated p27Kip1 (S140) compared to the Flag-WIP1 samples and is comparable to the no Flag-WIP1 irradiated lane ().
Figure 3. WIP1 dephosphorylates p27Kip1 in vivo. (a) WIP1 dephosphorylates p27Kip1 S140 in HEK293 cells. HEK293 cells were transfected with Myc-p27 or p53 and Flag-WIP1 or Flag-WIP1-PD (phosphatase-dead) and irradiated with 10 Gy IR with 0.5 hours recovery. Immunoblot analysis was performed with indicated antibodies. (b) WIP1 inhibition in MCF-7 cells leads to a delay in p27Kip1 S140 phosphorylation levels returning to homeostatic levels after DNA damage treatment. MCF7 cells were pretreated with 3 uM of a WIP1-specific inhibitor (GSK 2830371A) and cells were irradiated with 10 Gy IR. Cells were recovered up to 3.5 h post IR treatment. For each indicated time point, cells were lysed and subjected to immunoblot analysis with the indicated antibodies. In addition to using antibodies to p27Kip1 and p27 S140, we immunoblotted with antibodies to two other known WIP1 target proteins and their targeted sites (CHK2 T68 and H2AX S139). To clarify relative p27 protein and S140 phosphorylation levels, the intensity of p27 S140 and p27 protein bands for each condition were quantified with Image J and the ratio between p27 S140 and total p27 was calculated and noted in the bar graph to the right. P values were determined by t test. (c) WIP1 inhibition leads to decreased p27 stability before and after DNA damage treatment. HEK293 cells were co-transfected p27 with WIP1-WT or WIP1-PD and incubated for 24 h at 37◦C. Cells were irradiated with 5 Gy IR and recovered up to 8 h. Cells were lysed at indicated time points and subjected to immunoblot analysis with the indicated antibodies. (d) Quantitation of WIP1 effects on p27Kip1 stability. The intensities of p27 protein bands for each condition from Panel C were quantified with Image J and graphed. Bars compare relative p27Kip1 protein levels in non-transfected and WIP1-transfected cells. **p < 0.01 (n = 6). P values were determined by t test.
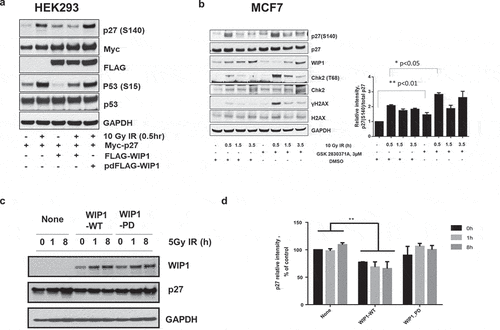
Since overexpression of WIP1 could diminish p27Kip1 (pS140) levels following DNA damage, we asked whether inactivation of WIP1 could enhance p27Kip1 phosphorylation. To answer this question we turned to MCF7 breast cancer cells. Some breast cancers have been shown to have amplified and overexpressed PPM1D/WIP1 and the MCF7 line exhibits this property [Citation32]. High expression of WIP1 by this line allows a more sensitive readout on whether WIP1 knockdown has effects on WIP1 target dephosphorylation. MCF7 cells were exposed to 10 Gy IR and harvested at multiple time points post-IR for immunoblot analysis. Prior to IR treatment p27Kip1 S140 phosphorylation levels relative to p27Kip1 protein levels were higher in cells treated with WIP1-specific inhibitor GSK2830371A (). An increase in phosphorylation at serine 140 was observed at 30, 90, and 210 minutes post IR treatment, but again prior WIP1 inhibitor treatment resulted in higher overall levels of p27Kip1 S140 phosphorylation relative to their counterparts not treated with the WIP1 inhibitor (). Positive control WIP1 target sites CHK2 T68 [Citation33] and H2AX S139 [Citation34] are also affected in a similar manner. These data indicate that p27Kip1 is phosphorylated at serine 140 in response to IR and that WIP1 phosphatase is responsible for reduced levels of p27Kip1 (S140) phosphorylation.
WIP1 overexpression is associated with reduced p27Kip1 stability
The phosphorylation of p27Kip1 at Serine 140 by ATM in response to DNA damage is associated with its stabilization and increased protein levels [Citation21]. We confirmed the earlier studies of Cassimere et al. [Citation21] by showing that IR-treated cells overexpressing wildtype or S140D versions of p27Kip1 retained high levels of stable p27, while those overexpressing the mutant S140A version of p27Kip1 showed reduced stability over time in the presence of cycloheximide (Figure S3). We then tested whether dephosphorylation at phosphoserine 140 on p27Kip1 by WIP1 is associated with p27Kip1 destabilization. HEK293 cells were co-transfected with p27Kip1 expression vectors along with either empty vector, WT WIP1, or phosphatase-dead WIP1 expression vectors and subjected to 5 Gy ionizing radiation 24 hours later. Treated cells were harvested before and after radiation and p27Kip1 protein levels were shown to be significantly reduced in wildtype WIP1 transfected cells but not in WIP1 empty vector or phosphatase-dead WIP1 expressing cells (). Thus, the reduction of p27Kip1 protein levels associated with increased WIP1 complements the report of enhanced p27Kip1 protein levels after its phosphorylation by ATM [Citation21].
Mutations in p27Kip1 Serine 140 affect cellular proliferation and colony formation
p27Kip1 is a regulator of cell cycle progression through its inhibition of cyclin-CDK complexes [Citation22,Citation23]. Moreover, ATM phosphorylation of p27Kip1 at Ser140 appears to enhance its cell cycle inhibitory functions [Citation21]. Thus, if WIP1 targets p27Kip1 and dephosphorylates it at Ser140, it might be expected that WIP1 reduces p27Kip1 anti-proliferative functions. To test p27Kip1 effects on proliferation in the presence and absence of WIP1 we performed a four day cell growth assay on HEK293 cells transfected with a wildtype p27Kip1 expression construct either with or without a WIP1 expression construct. As controls, HEK293 cells irradiated with 5 Gy ionizing radiation or untreated, non-transfected cells were monitored four days for cell growth. As shown in the cell growth curves in , overexpression of p27Kip1 inhibits cell growth to about the same extent as 5 Gy ionizing radiation and both groups of cells grow poorly, while untreated cells grow well. After four days, cells overexpressing both p27Kip1 and WIP1 grow at intermediate rates, suggesting that WIP1 modestly attenuates effects p27Kip1 anti-proliferative function. To determine how Ser140 phosphorylation status may affect cellular proliferation, HEK293 cells were transfected with wildtype Myc-p27, the phospho-dead mutant S140A or the phospho-mimetic S140D mutant. Proliferation of transfected cells was subsequently determined over a period of 4 days. While there was little difference in proliferation between mock transfected cells and cells expressing p27Kip1 S140A mutants, cells expressing wildtype p27Kip1 and S140D mutant p27Kip1 showed reduced cell growth at four days after cell seeding (). All three p27 vector-transduced cell types expressed high levels of Myc-p27 (, lower panel). The enhanced growth rate of the S140A mutant compared to the WT p27 expressing cells suggests that the inability to phosphorylate S140 may result in attenuated p27Kip1 activities in inhibiting cell proliferation.
Figure 4. The p27Kip1 S140A overexpressing cells have a growth advantage over p27Kip1 WT overexpressing cells. (a) To determine the effects of p27 S140 phosphorylation on cell growth, HEK293 cells were plated in 6 cm dishes and transfected with p27-WT and p27-WT plus WIP1-WT and on the next day non-transfected cells were irradiated with 5 Gy IR. Each cell was trypsinized and plated into 96 well cell culture. Growth of cells was monitored up to 4 days by cell proliferation assay using the CCK-8 kit. Left panel, Each time point was analyzed at N = 6.*** p < 0.001. Central panel, Bar graph of day 4 from left panel. Right panel, Expression of p27Kip1 and WIP1 protein in cells. Western blot analysis of cells from left panel were tested for p27Kip1 and WIP1 protein expression. After 24 h of transfection, cells in each condition were lysed with SDS-PAGE treatment buffer and subjected to immunoblot analysis with the indicated antibodies. GAPDH served as a loading control. (b) p27Kip1 S140 status affects cell growth. To determine the effect of phosphorylation of p27Kip1 S140 on cell growth, HEK293 cells containing stable inducible p27Kip1 WT, p27Kip1 S140D and p27Kip1 S140A expression constructs were plated 96 well plate and incubated with treatment of doxycycline (0.1 ug/ml) for 4 days. Relative cell numbers were determined by the CCK-8 kit to compare differences of cell growth in each cell line (upper panel). Expression of p27Kip1 in cells is shown in the lower panel. (c) Colony formation is reduced for cells transfected with WT p27Kip1 and p27Kip1 S140 vectors, but not p27Kip1 S140A vectors. In a clonogenic assay 1000 stable HEK293 cells containing inducible p27Kip1 WT, p27Kip1 S140D and p27Kip1 S140A expression system were plated in 6 well plates and grown for 2 weeks in the presence or absence of doxycycline. Colonies were stained with 0.5% crystal violet. (d) Bar graph quantitating the relative intensity of colonies formed in the clonogenic assay two weeks after plating. *, p < 0.05 and **, p < 0.01 (n = 3). P values were determined by t test.
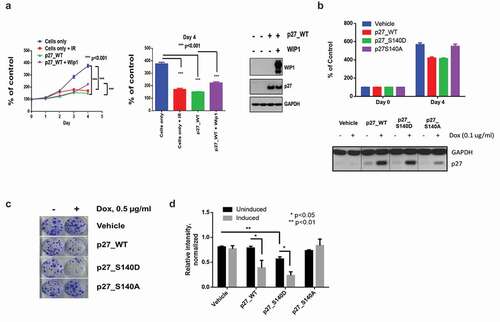
Since these results were based on the transient expression of p27Kip1, we examined whether a similar effect would be seen when p27Kip1 was constitutively expressed. Using a stable inducible expression system, we analyzed the proliferative potential of WT p27, S140A and S140D in a colony formation assay. HEK293 cells were left uninduced or induced with doxycycline for p27 expression and plated at low density in 6-well plates. Cells were allowed to grow for two weeks and stained with crystal violet to assay for surviving colonies. Vehicle control cells show no difference in colony forming potential with the addition of doxycycline (). Induction of WT p27 and S140D both show decreased numbers of surviving colonies, while S140A induced cells show similar colony numbers to uninduced cells (). Overall, this data corroborates the previous proliferation assay demonstrating that cells overexpressing WT p27 or S140D p27 have lower proliferation/survival rates than cells expressing S140A.
While the previous experiments clearly showed that WT p27Kip1 and the mutant S140D version showed enhanced inhibition of cell growth and colony formation over a cumulative period of several days, we also attempted to determine whether short term inhibition of G1 to S phase transition could be regulated by overexpression of WT p27Kip1 and its Ser140 mutant forms. HEK293 cells stably transfected with doxycline-inducible WT, S140A, or S140A p27Kip1 expression vectors were subjected to an aphidicolin-induced G1 block and induced or not induced with doxycycline for 24 hours. Cells were then released from aphidicolin and examined for progression out of G1 by cell cycle flow cytometry at 0, 2, 4, 6, and 8 hour intervals. As shown in Figure S4, by 4 hours post-release from the block, a higher proportion of cells overexpressing WT and S140D p27Kip1 remained in G1 compared to their uninduced WT or induced S140A counterparts. Despite this apparent enhanced short term inhibition of cell cycle progression by the WT and mutant S140D version of p27Kip1, in two experiments the differences between these mutants and the less inhibitory S140A version of p27Kip1 did not quite reach statistical significance. We noted in the flow cytometry profiles of Figure S4A that the numbers of cells in the sub-G1 area of the profiles showed extremely low cell numbers, regardless of p27Kip1 status, suggesting that p27Kip1 has its primary effects on cell growth inhibition by inhibiting cell cycle progression and not by inducing apoptosis.
Discussion
Among the PIKKs, ATM, ATR, and DNA-PK are central kinases that phosphorylate a vast array of other target proteins that enact many aspects of the DNA damage response and repair network [Citation1,Citation2,Citation4]. In addition to direct activation of DNA repair systems, mechanisms to prevent propagation of damaged DNA templates by halting DNA replication and cell division are implemented. One such cell cycle modulator, p53, is activated and stabilized in part through direct phosphorylation by ATM, ATR, and DNA-PK. In turn phosphorylated p53 transcriptionally upregulates important negative cell cycle regulators such as the cyclin-dependent kinase inhibitor p21Cip1 [Citation5,Citation9]. In addition to phosphorylation of p53, there is growing evidence that p27Kip1, a member of the CIP/KIP family of CDK cell cycle inhibitors, plays a role in the DNA damage response and may be directly regulated by ATM [Citation21,Citation35]. Recently, it has been shown by Cassimere et al. [Citation21] that p27Kip1, is a direct phosphorylation target of ATM. ATM phosphorylation of p27Kip1 takes place on Serine 140 in response to DNA damage and this phosphorylation is associated with enhanced p27Kip1 stability and an enhanced G1 arrest response [Citation21].
We have found that WIP1 plays a major role in reversal of the DNA damage response initiated by PIKKs such as ATM, ATR, and DNA-PK [Citation13]. In particular, ATM/ATR are known to phosphorylate S/TQ sites on their target proteins while WIP1 often dephosphorylates pS/pTQ sites initially phosphorylated by these kinases. In a broad screen to identify novel WIP1 targets, half of our 264 tested phosphopeptides contained pS/pTQ sites and most had exhibited prior activity as DNA damage-associated ATM/ATR target sites [Citation27]. Roughly 80% of our pS/pTQ phosphopeptides were efficiently dephosphorylated by WIP1 in vitro, while less than 10% of non-pS/pTQ phosphopeptides (derived from likely target proteins of other kinases) were dephosphorylated by WIP1. While a positive WIP1 in vitro phosphatase assay result is not a guarantee that WIP1 dephosphorylates the parental protein target site in vivo, we have historically found a very high correlation between positive in vitro phosphopeptide assay results and in vivo functionality as a WIP1 target site [Citation13]. Thus, we postulate that WIP1 may reverse the effects of PIKK phosphorylation at S/TQ sites in a near universal manner.
We had previously shown that p53 is inhibited by WIP1 dephosphorylation of p53 at Serine 15, an ATM target site [Citation14]. Here, we have subsequently investigated whether p27Kip1, a direct ATM target, is also dephosphorylated by WIP1 at Serine 140. Indeed, WIP1 efficiently dephosphorylated 27Kip1 at Serine 140 both in vitro and in cellular contexts. We also showed in in vitro phosphatase assays that the other three pS/pTQ sites at Ser56, Ser106 and Thr162 are dephosphorylated, along with one non-pS/pTQ site at Ser83 (Figure S2A). While it is not clear that the other three pS/pTQ sites are phosphorylated by ATM in vivo, all four pS/pTQ sites are evolutionarily conserved, suggesting that these may be damage-targeted phosphorylation sites (Figure S2B).
We confirmed the data of Cassimere et al. [Citation21] that ATM phosphorylates p27Kip1 at Serine 140 following radiation-induced DNA damage and that overexpressed WIP1 inhibited this phosphorylation event (). Moreover, specific inhibitors of WIP1 enhanced p27Kip1 Ser140 phosphorylation following DNA damage (). While Cassimere et al. [Citation21] detected enhanced stability of p27Kip1 protein following ATM phosphorylation, we were able to detect reduced stability of p27Kip1 following overexpression of wildtype WIP1, but not phosphatase-dead WIP1 in the presence and absence of DNA damage (). Thus, Serine 140 phosphorylation serves as a key control point for regulation of p27Kip1
protein stability and function.The functional consequences of p27Kip1 phosphorylation at Ser140 by ATM were shown to facilitate initiation of G1 arrest, allowing time for the prolonged maintenance of G1 arrest by activated p53-induced p21Cip1. Our own functional results are consistent with WIP1 dephosphorylation of p27Kip1 Ser140 significantly enhancing cell proliferation () and colony forming capabilities (). When we performed short term cell cycle progression experiments comparing wildtype and mutant forms of overexpressed p27Kip1, we were able to detect modestly increased cell cycle inhibition by the more inhibitory WT and mutant S140D p27Kip1 forms relative to their mutant S140A form (Figure S3). However, the differences were not statistically significant, perhaps due in part to the shorter frame of the experiment and insufficient sensitivity in detecting large functional changes resulting from modification of a single phosphorylation site. Since we also had preliminary evidence that other potential pS/pTQ sites on p27Kip1 were targeted by WIP1 (Figure S2A), it is possible that ATM/ATR may phosphorylate p27Kip1 on multiple sites to regulate the activities of this CDK inhibitor. Modification of all pST/Q sites potentially targeted by ATM/ATR (and dephosphorylated by WIP1) may be necessary to observe more robust short term effects on p27Kip1 stability and function. In sum, these experiments confirm and further elucidate the importance of the oncogenic phosphatase WIP1 in homeostatic regulation of proteins that not only initiate and maintain the DNA damage response but also regulate key cell cycle checkpoints.
Materials and methods
Plasmids and mutagenesis
The human WIP1-FLAG-CMV-Neo-Bam plasmid and WIP1-FLAG-CMV-Neo-Bam-D314A were a gift of Dr. Ettore Appella and have been previously characterized [Citation12]. The construct for bacterial expression of recombinant human WIP1 (WIP1- ΔExon6-His-pET-23a+) was previously obtained from Dr. H. Yamaguchi [Citation29], and the point mutant WIP1-D314A was generated using site directed mutagenesis [Citation36].
The Myc-tagged p27Kip1 vector was graciously donated by Dr. Tsz-Kwong Man at Texas Children’s Hospital (Houston, TX). pCMV-Myc-p27S140A and pCMV-Myc-p27S140D mutants were created using the Agilent Quik Change II Site-Directed Mutagenesis Kit (Santa Clara, CA) according to the manufacturer’s protocol. Primer sequences utilized for the site-directed mutagenesis are as follows: S140A forward 5ʹ-ctgatccgtcggacgcccagacggggttag-3ʹ, S140A reverse 5ʹ-ctaaccccgtctgggcgtccgacggatcag-3ʹ, S140D forward 5ʹ-actgatccgtcggacgaccagacggggttagc-3ʹ, and S140D reverse 5ʹ-gctaaccccgtctggtcgtccgacggatcagt-3ʹ.
In vitro phosphatase assay/screen
Phosphopeptides for individual phosphorylation sites were generated from either New England Peptides (Gardner, MA) or China Peptides (Shanghai, China). A complete list of all phosphopeptides used and their sequences can be found in Table S1.
Recombinant human WIP1-ΔEx6 and phosphatase-dead WIP1-D314A-ΔEx6 were purified from bacteria as previously described [Citation36,Citation37]. 50 ng of purified WIP1 was incubated with 100 µM phosphopeptide in PP2C buffer (50 mM Tris-HCl pH 7.5, 0.1 mM EGTA, and 0.02% 2-mercaptoethanol), 1 mg/mL Bovine Serum Albumin (BSA) (Sigma-Aldrich), and 30 mM MgCl2 for 1 hour at 25°C. Reactions were stopped by the addition of BIOMOL Green (Enzo Life Sciences) to the reaction. Samples were subsequently incubated at room temperature for 30 minutes and released phosphates were measured at an absorbance of 630 nm in a Victor2 1400 Multilabel 96-well Plate Reader (Perkin Elmer). The WIP1 inhibitor GSK2830371 was kindly provided by GlaxoSmithKline and has been previously described [Citation31,Citation38]. WIP1 activity toward full-length immunoprecipitated proteins were performed using 100 ng of recombinant human WIP1 along with 1mg/mL BSA, 50mM MgCl2, and 2µM GSK2830371 when necessary.
Cell lines and transfections
HEK 293, 293T, and MCF-7 cell lines were previously obtained from the American Type Culture Collection (ATCC, Manassas, VA). All cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco) with 10% fetal bovine serum (FBS) (Gibco), 1% Penicillin/Streptomycin (Gibco), and grown in a 37°C humidified incubator with 5% CO2. All transient transfections were performed with Lipofectamine 2000 (ThermoFisher) according to manufacturer instructions.
Lentivirus and stable cell generation
Lentiviral constructs inducible for WT p27Kip1, S140A p27Kip1 and S140D p27Kip1 were generated using PCR amplified p27Kip1 from the previously described pCMV-myc-p27 vectors. The forward primer used was 5ʹ-ggggacaagtttgtacaaaaaagcaggcttcaccatgtcaaacgtgcgagtgtctaa-3ʹ and the reverse primer sequence was 5ʹ-ggggaccactttgtacaagaaagctgggttttacgtttgacgtcttctga-3ʹ. The addition of the attB1 and attB2 sequences to the forward and reverse primers respectively allowed for the integration of the PCR products into the doxycycline-inducible destination vector, pHAGE_Ubc_Inducible_Dest_HA (gifted by Dr. Zhou Songyang) using Gateway BP and LR Clonase II (Life Technologies) according to the manufacturer’s instructions. Lentiviral particles were produced by transient transfection of 1 µg p27-WT, p27-S140A or p27-S140D lentiviral vectors along with packaging vectors pMD2.G (750 ng) and psPAX2 (250 ng) in 60% confluent HEK293T cells. The lentiviral supernatants were collected at 24 h and 48 h and pooled. Filtered supernatant was added to HEK293 cells for 24 hours and then removed. Cells stably expressing p27Kip1 were subsequently selected for with 500 µg/ml Geneticin (Gibco) for 2 weeks. Selected cells were maintained in growth media containing 100 µg/ml of Geneticin for further study.
Western blot analysis
Cells were lysed in lysis buffer (40 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.6% CHAPS, 0.5 mM EDTA, 0.2% NP-40, 1% Glycerol) and the supernatant collected. Proteins were resolved on a 10% Bis-Tris or a 4-12% Bis-Tris gradient gel (Invitrogen) and transferred onto PVDF or nitrocellulose membrane. Membranes were probed with the indicated antibodies and visualized with chemiluminescent detection reagent (Perkin Elmer).
Immunoprecipitation
HEK293 cells were transfected with pCMV-Myc-p27 and incubated for 24 hours at 37°C. Cells were treated with 10 Gy IR and recovered for 30 minutes prior to lysis in lysis buffer. The resulting supernatant was incubated with an anti-Myc antibody (BioLegend) overnight at 4°C. Protein A/G agarose beads (Santa Cruz) were subsequently added and samples were incubated an additional 2 h at 4°C followed by 2 washes in lysis buffer without phosphatase inhibitors and 2 washes in PP2C buffer. Washed beads were resuspended in PP2C buffer for use in downstream in vitro phosphatase assays.
In vitro kinase assay
pcDNA3.1(+)Flag-His-ATM wt was a gift from Michael Kastan (Addgene plasmid # 31985) [Citation7]. HEK293 cells that were transfected with this ATM vector and irradiated with 15 Gy IR 48 hours post transfection. Cells were lysed with lysis buffer (50 mM Trish pH7.5, 50 mM glycerophosphate, 150 mM NaCl, 10% glycerol, 1% Tween-20, 1 mM NaF, 1 mM NaVO4, 1 mM PMSF, 1 mM DTT and protease inhibitor cocktail) 30 minutes post-IR and incubated with FLAG-conjugated M2 protein beads (Sigma Aldrich, #M8823-5mL) for 2 hours at 4°C. Beads were washed twice in lysis buffer and twice in kinase buffer (10 mM HEPES pH 7.5, 50 mM glycerophosphate, 50 mM NaCl, 10 mM MgCl2, 10 mM MnCl2, 5 µM ATP and 1 mM DTT). 100 ng of recombinant p27 (Abcam) or recombinant p53 (Enzo Life Sciences) was incubated with 15 ul of ATM-IP, 2.5 µCi γ-32P-ATP (Perkin Elmer) in the presence or absence of 2.5 µM KU55933 ATM inhibitor (Sigma Aldrich) for 1h at room temperature. Reactions were subsequently run on a 4-12% Bis-Tris gel (Invitrogen) and analyzed by autoradiography.
Cell proliferation assay
HEK293 cells were plated in 6 cm dishes and transfected with p27-WT, p27-S140D and p27-S140A expression vectors. The next day, cells were irradiated with 5 Gy ionizing radiation. Irradiated cells were trypsinized and plated into 96 well cell culture plates at 3,000 cells/well. Cells were assayed daily for viability. Briefly, CCK-8 reagent was added to the appropriate wells for 2 hours, and absorbance at 450 nm was measured.
Clonogenic assay
HEK293 cells stably expressing inducible p27 WT, p27S140A and p27S140D were seeded at a density of 1,000 cells/well in 6 well plates in 3 ml of medium in the presence or absence of 1 µg/ml of doxycycline. Plates were incubated for 1–2 weeks and the resulting colonies were stained with 0.5% crystal violet. Wells were extensively washed with water to remove background staining. Resulting stained colonies were washed in 10% acetic acid to redissolve the crystal violet and absorbance was read at 630 nM for quantification.
Flow cytometry and aphidicolin block
HEK 293 cells with stably-expressing doxycycline inducible p27WT, p27S140A, and p27S140D constructs were seeded in 60mm tissue culture dishes at 5 × 105 cells/plate in 4mL of growth medium. The day after seeding, p27 expression was induced via 1g/mL doxycycline treatment in addition to 2µl of 1mg/mL Aphidicolin (Calbiochem, catalog# 178273-1MG) solubilized in DMSO for cell-cycle synchronization. After 24 hours, treatment media was removed, cells were washed carefully with PBS and placed back in normal growth media. Cells were then harvested at 0 hr, 2 hr, 4 hr, 6 hr, and 8 hr timepoints, washed carefully with PBS, and fixed in cold 70% ethanol at −20°C overnight.
To stain the cells for flow cytometry, fixed cells were centrifuged at 300 RCFs for 5 minutes and washed with PBS. The washed pellet was then resuspended in 50 µg/mL Propidium Iodide (PI) (Sigma, catalog #P4864-10ML) and 0.1 mg/mL RNAse A solution. Cells were stained for 30 minutes, and then analyzed for PI content on a BD FACSCanto II system for a total of 10,000 cells per sample. All flow experiments were conducted at the Baylor College of Medicine Flow Cytometry Core.
Supplemental Material
Download Zip (463.5 KB)Acknowledgments
We thank Avani Raythata, Sung-Hwan Moon, and Thuy-Ai Nguyen for technical support and helpful discussions. We also thank Tsz-Kwong Man for providing p27Kip1 expression constructs and helpful discussions. This project was supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the CPRIT Core Facility Support Award (CPRIT-RP180672), the NIH (P30 CA125123 and S10 RR024574) and the expert assistance of Joel M. Sederstrom. This work was also supported by the Cancer Prevention and Research Institute of Texas (CPRIT) under grant RP160451 and the National Cancer Institute under grant CA237291.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Smith J, Tho LM, Xu N, et al. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112.
- Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66:801–817.
- Hanawalt PC. Historical perspective on the DNA damage response. DNA Repair (Amst). 2015;36:2–7.
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210.
- Morgan SE, Kastan MB. p53 and ATM: cell cycle, cell death, and cancer. Adv Cancer Res. 1997;71:1–25.
- Banin S, Moyal L, Shieh S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677.
- Canman CE, Lim DS, Cimprich KA, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679.
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431.
- Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170:1062–1078.
- Wasylishen AR, Lozano G. Attenuating the p53 pathway in human cancers: many means to the same end. Cold Spring Harb Perspect Med. 2016; ;6(8).
- Moyer SM, Larsson CA, Lozano G. Mdm proteins: critical regulators of embry ogenesis and homeostasis. J Mol Cell Biol. 2017;9:16–25.
- Fiscella M, Zhang H, Fan S, et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci U S A. 1997;94:6048–6053.
- Lu X, Nguyen TA, Moon SH, et al. The type 2C phosphatase Wip1: an oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008;27:123–135.
- Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19:1162–1174.
- Lu X, Ma O, Nguyen TA, et al. The Wip1 Phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell. 2007;12:342–354.
- Bulavin DV, Demidov ON, Saito S, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet. 2002;31:210–215.
- Rauta J, Alarmo EL, Kauraniemi P, et al. The serine-threonine protein phosphatase PPM1D is frequently activated through amplification in aggressive primary breast tumours. Breast Cancer Res Treat. 2006;95:257–263.
- Coombs CC, Zehir A, Devlin SM, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell. 2017;21:374–82 e4.
- Hsu JI, Dayaram T, Tovy A, et al. PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell. 2018;23:700–13 e6.
- Yokoyama A, Kakiuchi N, Yoshizato T, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. 2019;565:312–317.
- Cassimere EK, Mauvais C, Denicourt C. p27Kip1 is required to mediate a G1 cell cycle arrest downstream of ATM following genotoxic stress. PLoS One. 2016;11:e0162806.
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267.
- Bencivenga D, Caldarelli I, Stampone E, et al. p27(Kip1) and human cancers: A reappraisal of a still enigmatic protein. Cancer Lett. 2017;403:354–365.
- Payne SR, Zhang S, Tsuchiya K, et al. p27kip1 deficiency impairs G2/M arrest in response to DNA damage, leading to an increase in genetic instability. Mol Cell Biol. 2008;28:258–268.
- Cuadrado M, Gutierrez-Martinez P, Swat A, et al. p27Kip1 stabilization is essential for the maintenance of cell cycle arrest in response to DNA damage. Cancer Res. 2009;69:8726–8732.
- Berton S, Cusan M, Segatto I, et al. Loss of p27(kip1) increases genomic instability and induces radio-resistance in luminal breast cancer cells. Sci Rep. 2017;7:595.
- Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166.
- Lowe J, Cha H, Lee MO, et al. Regulation of the Wip1 phosphatase and its effects on the stress response. Front Biosci (Landmark Ed). 2012;17:1480–1498.
- Yamaguchi H, Durell SR, Chatterjee DK, et al. The Wip1 phosphatase PPM1D dephosphorylates SQ/TQ motifs in checkpoint substrates phosphorylated by PI3K-like kinases. Biochemistry. 2007;46:12594–12603.
- Lu X, Bocangel D, Nannenga B, et al. The p53-induced oncogenic phosphatase PPM1D interacts with uracil DNA glycosylase and suppresses base excision repair. Mol Cell. 2004;15:621–634.
- Gilmartin AG, Faitg TH, Richter M, et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat Chem Biol. 2014;10:181–187.
- Li J, Yang Y, Peng Y, et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat Genet. 2002;31:133–134.
- Fujimoto H, Onishi N, Kato N, et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006;13:1170–1180.
- Moon SH, Lin L, Zhang X, et al. Wild-type p53-induced phosphatase 1 dephosphorylates histone variant gamma-H2AX and suppresses DNA double strand break repair. J Biol Chem. 2010;285:12935–12947.
- Denicourt C, Dowdy SF. Cip/Kip proteins: more than just CDKs inhibitors. Genes Dev. 2004;18:851–855.
- Nguyen TA, Slattery SD, Moon SH, et al. The oncogenic phosphatase WIP1 negatively regulates nucleotide excision repair. DNA Repair (Amst). 2010;9:813–823.
- Moon SH, Nguyen TA, Darlington Y, et al. Dephosphorylation of gamma-H2AX by WIP1: an important homeostatic regulatory event in DNA repair and cell cycle control. Cell Cycle. 2010;9:2092–2096.
- Richter M, Dayaram T, Gilmartin AG, et al. WIP1 phosphatase as a potential therapeutic target in neuroblastoma. PLoS One. 2015;10:e0115635.