
ABSTRACT
Excessive migration and proliferation of vascular smooth muscle cells (VSMCs) are critical cellular events that lead to intimal hyperplasia in atherosclerosis and restenosis. In this study, we investigated the protective effects of ursodeoxycholic acid (UDCA) on intimal hyperplasia and VSMC proliferation and migration, and the underlying mechanisms by which these events occur. A rat unilateral carotid artery was ligated to induce vascular injury and the microRNA (miRNA) expression profiles were determined using miRNA microarray analysis. We observed that UDCA significantly reduced the degree of intimal hyperplasia and induced miR-21 dysregulation. Restoration of miR-21 by agomir-miR-21 reversed the protective effects of UDCA on intimal hyperplasia and proliferation in vivo. In vitro, UDCA suppressed PDGF-BB-induced VSMC proliferation, invasion and migration in a dose-dependent manner, whereas the suppressive effect of UDCA was abrogated by overexpression of miR-21 in PDGF-BB-incubated VSMCs. Furthermore, we identified that miR-21 in VSMCs targeted the phosphatase and tensin homolog (PTEN), a tumor suppressor gene, negatively modulated the AKT/mTOR pathway. More importantly, we observed that that UDCA suppressed AKT/mTOR signaling pathway in the carotid artery injury model, whereas this pathway was reactivated by overexpression of miR-21. Taken together, our findings indicated that UDCA inhibited intimal hyperplasia and VSMCs excessive migration and proliferation via blocking miR-21/PTEN/AKT/mTOR signaling pathway, which suggests that UDCA may be a promising candidate for the therapy of atherosclerosis.
Introduction
The abnormal proliferation and migration of vascular smooth muscle cells (VSMCs) contribute to the development of intimal hyperplasia in many vascular pathologies, including atherosclerosis and restenosis after angioplasty [Citation1,Citation2]. In normal blood vessel, VSMCs reside within the media and remain quiescent. After stimulation with atherogenic factors, VSMCs proliferate and migrate from the media to the intima, which ultimately results in intimal hyperplasia and vascular stenosis [Citation3]. Thus, therapeutic strategies against VSMC proliferation and migration may be beneficial for treating VSMC-related pathological conditions. In response to vascular injury, activated VSMCs, platelets and inflammatory cells release the growth factors, especially platelet derived growth factor (PDGF), thereby cause a change of VSMCs from a contractile phenotype to a synthetic phenotype [Citation4,Citation5]. PDGF-BB is considered to be the most potent stimulants for VSMC proliferation and migration [Citation6]; therefore, PDGF-BB was used in this study to induce these VSMC reactions.
Ursodeoxycholic acid (UDCA) is a naturally occurring hydrophilic bile acid that has been extensively used for the treatment of certain cholestatic liver diseases including chronic viral hepatitis and primary biliary cirrhosis [Citation7,Citation8]. UDCA has cytoprotective activity, such as anti-apoptotic and anti–inflammatory effects, but these effects have previously been described only in hepatocytes. Recently, Taurin-conjugated ursodeoxycholic acid (TUDCA), a taurine-conjugate derivative of UDCA, has been reported to attenuate neointimal hyperplasia by inhibiting proliferation and inducing apoptosis in the VSMCs of rats subjected to carotid artery balloon injury [Citation9]. Although both UDCA and TUDCA, two naturally occurring bile acids, have been already approved for clinical use for treatment of several cholestatic diseases, there are still some distinct effects of them since the difference in their hydrophility and affinity to cell receptors [Citation10,Citation11]. As compared to TUDCA, UDCA exerts anti-atherogenic effect on disturbed flow-induced atherosclerosis by repressing inflammatory response and endoplasmic reticulum stress [Citation12]; however, whether UDCA might act similarly of TUDCA on intimal hyperplasia and the underlying mechanisms by which these events occur remains unclear.
MicroRNAs (miRNAs) are endogenous small non-coding RNA molecules of 22–25 nucleotides that modulates post-transcriptional regulation of target genes through repressing translation or promoting RNA degradation [Citation13,Citation14]. It has been widely reported that miRNAs are involved in a variety of biological and pathological processes, including cellular apoptosis, proliferation, migration and carcinogenesis [Citation13,Citation15,Citation16]. One recent study has demonstrated that the expression of at least 20–30% of human protein-coding genes is regulated by miRNAs [Citation17]. Another study has revealed that miRNAs are highly expressed in the cardiovascular system and some miRNAs are dysregulated in diseased vessels [Citation18]. miRNAs have been documented to function as essential regulators of atherosclerosis by regulating crucial factors or key pathways [Citation19]. A recent study reported that UDCA improves rat liver regeneration by modulating miR-21 levels after partial hepatectomy [Citation20]. Castro et al. have also confirmed that UDCA modulated the disease severity in human nonalcoholic fatty liver disease via inhibiting miR-34a/SIRT1/p53 signaling pathway [Citation21]. Inspired by these studies, we speculated that UDCA might exert anti–intimal hyperplasia effect on atherosclerosis through regulating miRNA expression.
In the present study, we established a rat model of intimal hyperplasia induced by vascular injury to the rat carotid artery and evaluated how UDCA protects against intimal hyperplasia.
Materials and methods
Animals
Male Sprague-Dawley rats were used in our experiments. All experimental protocols approved by the Experimental Animal Care and Use Committee of the Shanghai University of Traditional Chinese Medicine and conformed to the Guide for the Care and Use of Laboratory Animal published by the US National Institutes of Health (NIH publication, 8th edition, 2011). The rats were housed on 12:12-h light–dark cycle in a temperature controlled (24 ± 2°C) and humidity-controlled room, with free access to standard chow and tap water.
Animal model of vascular injury
The unilateral carotid artery ligation was used to induce vascular injury in 8-week-old rats as previous reports [Citation22,Citation23]. In short, rats were anaesthetized with sodium pentobarbital (50 mg/kg, i.p.). The adequacy of anesthesia was determined by the absence of corneal reflexes and paw withdrawal response to a noxious pinch and surgical manipulation. A midline incision was made in the ventral surface of the neck. Bilateral carotid arteries were isolated. The left common carotid arteries were ligated with a 6.0 silk suture that was proximal to the carotid bifurcation for 8 weeks, and the uninjured right carotid arteries served as control.
Experimental design
Experiment 1: Effect of UDCA on animal model of vascular injury. Rats were randomly divided into four groups: injury group (n = 10 each group), where the rats subjected to unilateral carotid artery ligation. Injury + UDCA group, where the rats were injected intraperitoneally with UDCA (100 mg/kg/d body weight) at 3 d after vascular injury. Injury + vehicle group, where the rats were injected intraperitoneally with 10% cyclodextrin buffer in saline daily for 6 weeks. Sham group, where the rats underwent vascular injury, and the uninjured right carotid arteries were harvested and served as control. This dose of UDCA was selected according to previous studies [Citation24] and our preliminary studies. At the end of experiments, the rats were sacrificed by overdose of pentobarbital sodium (150 mg/kg, iv), and carotid arteries were collected for hematoxylin and eosin (HE) or immunohistochemical staining and molecular biological analyse [Citation25,Citation26].
Experiment 2: miR-21 agomir (agomir-miR-21) experiments. The rats were randomly divided into two groups: Injury + UDCA + agomir-miR-21/agomir-NC group (n = 10 each group), the vascular injury rats were injected intraperitoneally with UDCA (100 mg/kg/d body weight), and then injected with agomir-miR-21/agomir-NC (8 mg/kg) via tail vein in 0.2 ml saline. Agomir-miR-21/agomir-NC were designed and synthesized by RiboBio (Guangzhou, China). The carotid arteries were snap-frozen and stored at −80°C until use.
MiR-21 agomiR and antagomiR were synthesized by Ribobio Co. (Guangzhou,China). The sequences of miR-21 agomiR are as follows: 5′-CAACAGCAGUC GAUGGGCUGUC-3′. The antagomiR is a single stranded RNA analogue complementary to the mature miR-21, which was chemically modified and cholesterol conjugated from a hydoxyprolinol linked cholesterol solid support and 2′-Ome phosphoramidites.
Cell culture
VSMCs were harvested from the media of normal thoracic aorta of Sprague–Dawley rats (180 ± 30 g, Laboratory Animal Central of the Academy of Military Medical Sciences, Beijing, China), and were cultured in DMEM (Gibco BRL, Grand Island, NY) with 10% fetal bovine serum (Gibco BRL, Grand Island, NY), 100 U/ml penicillin, and 100 mg/ml streptomycin (Sigma, St. Louis, MO) at 37°C in a 5% CO2 humidified incubator. Cells in the 2 to 6 passages were used, and cells at 80–90% confluence were growth arrested by incubating them in serum-deprived DMEM for 24 h before stimulation.
Immunohistochemistry staining
The paraffin-embedded vascular sections were rehydrated and immersed in 3% hydrogen peroxide for 15 min to quench the endogenous peroxidase. The sections were blocked in 10% goat serum for 1 h at room temperature and then incubated overnight at 4°C with primary antibody: PCNA (Proteintech Group, Wuhan, China). Subsequently, the sections were incubated with secondary antibodies (anti-rabbit IgG antibodies). Finally, the immunoreactivity was visualized by staining with diaminobenzidine (DAB) for 5 min, covered with a cover-slip, and analyzed under a light microscope.
miRNA microarray analysis
miRNA microarray analysis was used to compared the miRNA expression profiles between the uninjured carotid arteries from 2 rats of sham group and injured carotid arteries from 2 rats of injury group. Total RNAs were extracted using TRIzol reagent (Molecular Research Center, Inc., Cincinnati, OH, USA), and the miRNA fraction was further purified by a mirVana miRNA isolation kit (Ambion, Austin, TX) according to the manufacturer’s recommendation. The isolated miRNAs were labeled with Hy3 using the miRCURY array labeling kit (Exiqon, Vedbaek, Denmark) and hybridized with miRCURY locked nucleic acid (LNA) microRNA arrays (v8.0; Exiqon). Microarray images were taken with a Genepix 4000B scanner (Axon Instruments, Foster City, CA, USA) and analyzed with Genepix Pro 6.0 software (Axon Instruments).
Quantitative real-time RT-PCR
Total RNA from carotid arteries tissues was isolated with TRIzol (Invitrogen, CA) according to manufacturer’s instructions. cDNA synthesis was performed with 500 ng of total RNA using a RT First Strand Kit (SA Biosciences, Frederick, MD). cDNA was used as a template for reverse transcription polymerase chain reaction analysis (RT-PCR) amplification (Mastercycler Gradient, Eppendorf, Germany) to observe the miRNAs expression by using the miRNAs-specific primers for miR-495, miR-221 and miR-21 (Ribobio, Guangzhou, China). The primers used to amplify U6 were 5ʹ-CTC GCT TCG GCA GCA CA-3ʹ (forward) and 5ʹ-AA CGC TTC ACG AAT TTG CGT-3ʹ (reverse). All reactions were performed in triplicate. The relative expression of miRNAs was normalized to U6. Data were analyzed using the 2−ΔΔCt method.
Cell counting Kit-8 assay
The proliferation of VSMCs was detected using the Cell Counting Kit-8 (CCK-8) assay according to the manufacturer’s instructions. The VSMCs (5 × 104 cells/well) were seeded in 96-well plate with 100 μl DMEM medium supplemented with 10% FBS. After 48 h incubation, CCK-8 reagent (10 μl) was added to each well and continuously cultured for 1 h in 5% CO2 (Thermo). The absorbance rate at 450 nm were measured by Microplate Reader (Bio-Rad, USA). All experiments were repeated three times in triplicate.
Immunofluorescence staining
After fixed with 4% paraformaldehyde for 30 min, the stimulated VSMCs were permeabilized with 0.1% Triton X-100 in PBS for 15 min. Cells were blocked with 5% BSA for 1 h at room temperature, and then incubated with indicated primary antibody (PCNA, 1:200) at 4°C overnight. After three washes with PBS, the cells were incubated with secondary antibodies (anti-rabbit IgG antibodies). Images were photographed using a fluorescence microscope (DX51; Olympus, Tokyo, Japan).
VSMC migration analysis
VSMC migration was evaluated by wound healing assay [Citation27]. VSMCs were seeded into six-well plates and the near-confluent VSMCs in six-well plates were wounded by scraping with a standard 1 ml pipette tip to make a gap across the diameter of the wells. The migration status was determined by measuring the movement of cells into a scraped area created by a micropipette pipette tip. Images were taken at different points of time following wounding. The wound area was measured and the percentage of closure of denuded area was calculated by Image J software (NIH, Bethesda, MD, USA).
VSMC invasion analysis
Cell invasion assays were performed by 24-well transwell chambers with 8.0 μm pore size polycarbonate membrane (Corning Incorporated, Corning, NY, USA). The VSMCs were seeded on the top side of the membrane precoated with Matrigel (BD, Franklin Lakes, NJ, USA). After 48 h of incubation, non-invading cells on the upper surface were carefully removed and invading cells on the lower surface of the membrane were fixed with methanol and stained with crystal violet (Sigma-Aldrich). The cell numbers were counted from six random fields in each well at 200 ×. Experiments were performed in triplicate.
Western blot analysis
Total protein from VSMCs and rat carotid arteries tissues was isolated using RIPA buffer with protease inhibitor Cocktail (Pierce, Rockford, IL, USA). Protein content was measured with a BCA Protein Assay Kit (Cwbiotech, Beijing, China). The equal amounts of proteins were resolved by 10% SDS-PAGE (Sigma Aldrich, St. Louis, MO) and then transferred onto polyvinylidene difluoride (PVDF) membranes (BD Pharmingen, San Diego, CA). After blocking with 5% nonfat milk at room temperature for 1 h, the PVDF membranes were incubated primary antibodies against PCNA (Proteintech Group, Wuhan, China), PTEN, p-AKT, AKT, p-mTOR, mTOR and β-actin (Santa Cruz, CA, USA) at 4°C overnight. All primary antibodies diluted 1:1000. The goat anti-mouse IgG horseradish peroxidase secondary antibody (diluted 1:5000) was purchased from Santa Cruz Biotechnology, Inc. Subsequently, the protein bands were scanned on the X-ray film using the enhanced chemiluminescence detection system (PerkinElmer Life and Analytical Sciences, Boston, MA). The alpha Imager software (Alpha Innotech Corporation, San Leandro, CA) was used to measure relative intensity of each band on western blots. The measurements were conducted independently for at least three times with similar results.
Luciferase reporter assay
The potential binding site between PTEN and miR-21 was searched in TargetScan (http://www.targetscan.org). The miR-21 mimics/inhibitor and corresponding NC were designed and synthesized by RiboBio (Guangzhou, China). The fragment of the 3ʹ-UTR of PTEN [wild-type (wt) or mutant (mut), respectively] was amplified and cloned into the pMIR-REPORT luciferase vector (Ambion, Thermo Fisher Scientific, USA). Site-directed mutagenesis of the PTEN 3ʹ-UTR at the putative miR-21 binding site was performed by a QuikChange Kit (Qiagen). Subsequently, VSMCs at a density of 2 × 105 per well were seeded into 24-well plates and co-transfected with 0.8 μg of pMIR-PTEN-3ʹ-UTR or pMIR-PTEN-mut-3ʹ-UTR, 50 nM miR-21 mimic/inhibitor or corresponding NC using Lipofectamine 2000 reagent (Invitrogen). The relative firefly luciferase activity normalized with Renilla luciferase was measured 48 h after transfection by using the Dual-Light luminescent reporter gene assay (Applied Biosystems). All experiments were repeated three times in triplicate. The sequences of miR-21 mimics and miR-21 inhibitor were as follows: miR-21 mimics 5′-UAGCUUAUCAGACUGAUGUUGA-3ʹ; miR-21 inhibitor 5′-UCAACAUCAGUCUGAUAAGCUA-3′.
Statistical analysis
All statistical analyses were performed using SPSS 14.0 software (Chicago, IL). Each experiment was repeated at least 3 times. Numerical data are presented as the mean ± SEM. A one-way analysis of variance (ANOVA) followed by post-hoc tests were used to verify statistically differences among the groups. P value of < 0.05 was considered significant and < 0.01 was considered very significant.
Results
UDCA attenuates intimal hyperplasia in the carotid artery injury model
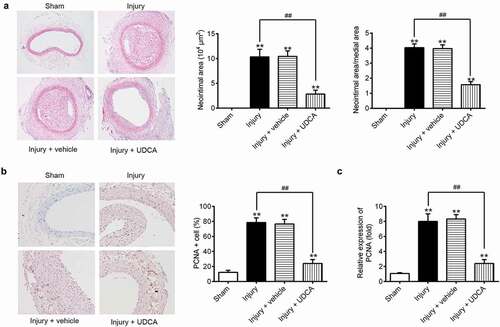
To evaluate the effects of UDCA on the rat model of vascular injury, the neointimal area was was inflicted. As shown in ), the intimal hyperplasia was observed in injury group and injury + vehicle group compared that in the sham group (P < 0.01). Injection of UDCA remarkably reduced neointimal area and the ratio of neointimal area to media area in injured carotid arteries (); P < 0.01). Furthermore, immunohistochemistry (IHC) staining showed that UDCA mitigated the protein expression of PCNA, a cellular proliferation marker, in injured carotid arteries compared with that in injury group (,); P < 0.01). These results indicated that UDCA exhibited protective effects against the formation of neointima hyperplasia.
Figure 1. The effects of UDCA on rat model of vascular injury. (a) Hematoxylin and eosin (HE) staining was used to evaluate the neointimal area in the vascular injury rat model. (b) PCNA protein expression was detected by immunohistochemistry staining in injured carotid arteries. (c) The mRNA level of PCNA was determined using qPCR analysis in injured carotid arteries. Data are presented as means ± SD of three individual experiments. (**P < 0.01 vs. sham group, ##P < 0.01 vs. injury group).
UDCA regulates miRNA expression in injured carotid arteries
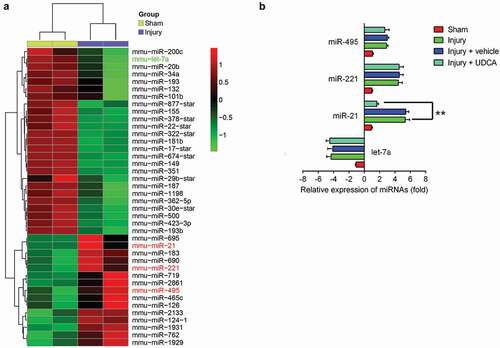
Recent studies reported that miRNA expressions, especially those of miR-132 [Citation28], miR-34 c [Citation29], miR-221 and miR-222 [Citation30], were dysregulated in a rat carotid artery injury model, which attenuated neointimal hyperplasia. Previous studies revealed that various types of drugs, such as Tongxinluo [Citation31] and Emodin [Citation32], inhibited neointimal hyperplasia in animal model of vascular injury by regulating miRNAs expression. Inspired by these studies, we hypothesized whether UDCA improved neointimal hyperplasia by regulating microRNA. First, we conducted a microarray analysis to measure the miRNA expression profiles in rat carotid arteries tissues. As shown in ), compared with sham group, a large set of miRNAs were ectopic expressions in injured carotid arteries. Interestingly, miR-21, miR-221, miR-let-7a, and miR-495 screened from our microarray analyses have been previously reported to modulate VSMC proliferation and migration and increase neointima hyperplasia in an animal model of vascular injury [Citation30,Citation33–Citation36]. To further explore whether UDCA inhibited neointimal hyperplasia via modulating these miRNAs expression, we used qRT-PCR to determine the miR-495, miR-221, miR-21 and miR-495 level in the rat carotid arterial injury model after UDCA treatment. We observed that UDCA markedly inhibited miR-21 expression in injury + UDCA group compared with that in injury group (); P < 0.01); whereas, the levels of miR-495, miR-221, and miR-495 level did not change. These data suggested that UDCA might attenuate intimal hyperplasia in the carotid artery injury model through modulating miR-21 expression.
Figure 2. UDCA modulates miRNA expression in injured carotid arteries. (a) The microarray analysis was performed to measure the miRNA expression profiles in injured carotid arteries (n = 2). Red or green color separately indicates high or low expression in the heatmap. (b) The relative expression of selected microRNAs including miR-495, miR-221, miR-21 and miR-495 were measured using qPCR analysis in a rat carotid arterial injury model after treatment with UDCA (n = 5). Data are presented as means ± SD of three individual experiments. (**P < 0.01 vs. injury group).
Agomir-miR-21 attenuates the effects of UDCA on neointimal hyperplasia
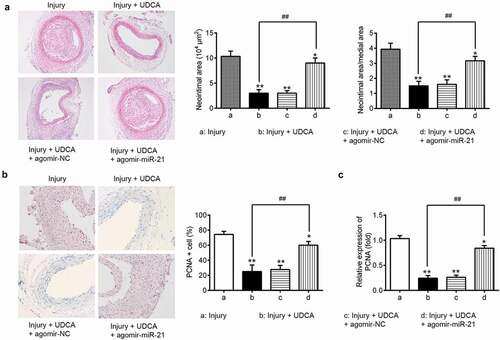
To further investigate whether UDCA improves intimal hyperplasia via regulating miR-21, the vascular injury rats were injected with agomir-miR-21 after UDCA treatment (injury + UDCA + agomir-miR-21 group), and levels of neointimal hyperplasia and PCNA protein expression were evaluated using HE and immunohistochemical staining. As shown in ), compared with that in injury group, UDCA treatment dramatically decreased the neointimal area and ratio of neointimal area to media area in injury + UDCA group (P < 0.01); however, miR-21 restoration using agomir-miR-21 significantly increased the neointimal area and ratio of neointimal area to media area (); P < 0.01). Moreover, IHC staining showed that UDCA inhibited increases in PCNA levels in the injury group; whereas, agomir-miR-21 reversed the reduction of PCNA resulting from UDCA treatment (,); P < 0.01). These results indicated that UDCA exerts the inhibitory effect on neointimal hyperplasia in injured carotid arteries by repressing miR-21 expression.
Figure 3. Agomir-miR-21 attenuates the protective effects of UDCA on neointimal hyperplasia. The vascular injury rats were injected with agomir-miR-21 after injection with UDCA. (a) HE staining was used to evaluate the neointimal area in the vascular injury rat model (n = 5). (b) PCNA protein expression was detected by immunohistochemistry staining in injured carotid arteries (n = 5). (c) The mRNA level was determined using qPCR analysis in injured carotid arteries. Data are presented as means ± SD of three individual experiments. (*P < 0.05, **P < 0.01 vs. sham group, ##P < 0.01 vs. injury group).
UDCA inhibits PDGF-BB-induced VSMC proliferation and metastasis
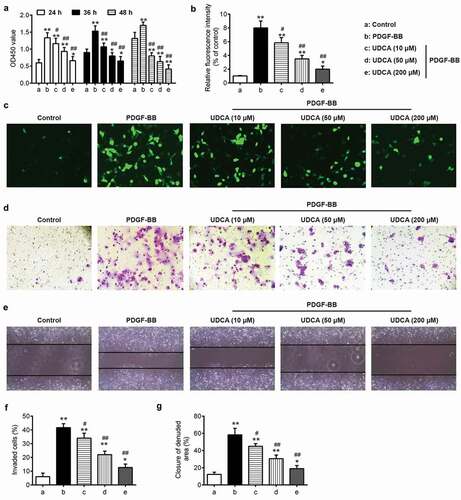
To test whether UDCA affects PDGF-BB-induced VSMC proliferation and metastasis, VSMCs were pretreated with 10, 50, or 200 μM UDCA for 6 h followed by stimulation with PDGF-BB for 24 h. The results showed that treatment of VSMCs with PDGF-BB markedly increased VSMC proliferation; however, UDCA treatment inhibited this PDGF BB–induced proliferation in a dose-dependent manner, as evidenced by CCK-8 assay (); P < 0.01). PCNA, a cellular proliferation marker, is considered to be involved in VSMC proliferation and migration [Citation37,Citation38]. Pretreatment with UDCA concentration-relatedly mitigated the protein levels of upregulated PCNA in PDGF-BB-incubated VSMCs () and C; P < 0.01). In addition, UDCA treatment dose-dependently suppressed PDGF-BB-induced invasion and migration of VSMCs, as evidenced by cell invasion and wound healing assay (); P < 0.01). These data suggested that UDCA has a suppressive effect on proliferation and metastasis of VSMCs incubated with PDGF-BB-.
Figure 4. The effect of UDCA on PDGF-BB-induced VSMC proliferation and invasion. VSMCs were pretreated with different doses of UDCA (10 μM, 50 μM, 200 μM) for 6 h followed by stimulation with PDGF-BB for 24 h. (a) The Cell Counting Kit-8 (CCK-8) assay was used to measure the cell proliferation. (b) Bar graph showing the relative protein expressions of PCNA. (c) The PCNA level was detected using immunofluorescence staining. (d) Cell invasion assay was performed to determine cell invasion. (e) Cell migration was measured by wound healing assay. (f) Bar graph showing PDGF-BB-induced invasion of VSMCs. (g) Bar graph showing PDGF-BB-induced migration of VSMCs. Data are presented as means ± SD of three individual experiments. (*P < 0.05, **P < 0.01 vs. control group; #P < 0.05, ##P < 0.01 vs. PDGF-BB group).
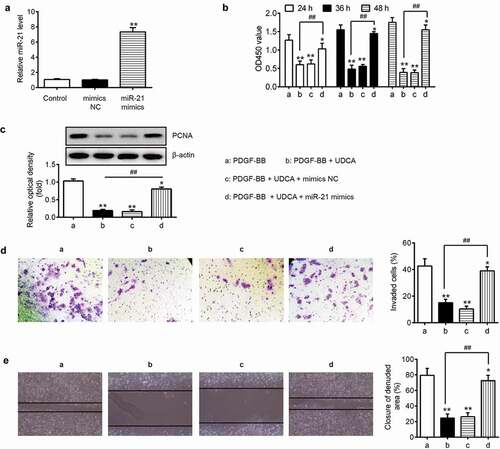
Overexpression of miR-21 ameliorates the suppressive effects of UDCA on PDGF-BB-induced VSMC proliferation and metastasis
To investigate whether miR-21 regulated the inhibitory effects of UDCA on PDGF-BB-induced VSMC proliferation and metastasis, VSMCs were treated with 50 μM of UDCA for 6 h followed by stimulation with PDGF-BB for 24 h after transfection with miR-21 mimics or mimics NC. As shown in ), miR-21 was significantly upregulated in VSMCs after transfection with miR-21 mimics compared with that in VSMC transfected mimics NC (P < 0.01). In addition, our results showed that UDCA inhibited PDGF BB–induced VSMC proliferation, while the suppressive effect of UDCA was partly alleviated by miR-21 mimics (); P < 0.01). Western blot analysis further confirmed that the suppressive effect of UDCA on PCNA level was rescued by overexpression of miR-21 in DGF-BB-treated VSMC cells (); P < 0.01), and the cell invasion and wound healing assay further confirmed that the overexpression of miR-21 abrogated the inhibitory effects of UDCA on PDGF BB – induced VSMC proliferation and migration (; P < 0.01). These data indicated that miR-21 attenuates the inhibitory effects of UDCA on PDGF-BB-induced VSMC proliferation and metastasis.
Figure 5. Overexpression of miR-21 weakens the suppressive effects of UDCA on PDGF-BB-induced VSMC proliferation and invasion. After transfection with miR-21 mimics or mimics NC, VSMCs were treated with 50 μM of UDCA for 6 h followed by stimulation with PDGF-BB for 24 h. (a) Overexpression efficiency of miR-21 level was measured using qPCR analysis. (**P < 0.01 vs. control group). (b) The Cell Counting Kit-8 (CCK-8) assay was used to measure the cell proliferation. (c) The PCNA level was detected using Western blot analysis. (d) Cell transwell assay was used to determine cell invasion. (e) Cell migration was determined by wound healing assay. Data are presented as means ± SD of three individual experiments. (*P < 0.05, **P < 0.01 vs. PDGF-BB group; ##P < 0.01 vs. PDGF-BB + UDCA group).
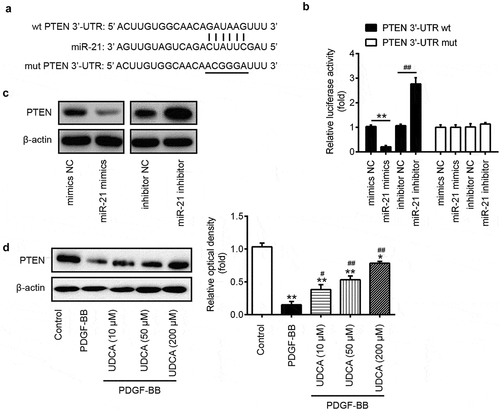
PTEN is a target gene of miR-21 in VSMCs
To investigate the molecular mechanism by which miR-21 regulates the effects of UDCA on PDGF-BB-induced VSMC development, we used the TargetScan to predict target genes for miR-21; PTEN was observed to be one of the best candidates ()). To verify this bioinformatic predication, we constructed luciferase reporter plasmids containing the wt or mut 3ʹ-UTR segments of PTEN ()). The reporters were co-transfected with either miR-21 mimics/inhibitor or corresponding NC to VSMCs, and the luciferase activity was measured. These experiments showed that miR-21 mimics significantly decreased the relative luciferase activity compared with mimic NC, while miR-21 inhibitor resulted in an opposite effect in the presence of the wild-type 3ʹ-UTR (P < 0.01; )). Expectedly, miR-21 mimics/inhibitor failed to modulate the relative luciferase activity of vector containing mut PTEN-3ʹ-UTR in the miR-21-binding site ()). Western blot analysis further confirmed that overexpression or inhibition of miR-21 obviously ether downregulated or upregulated, respectively, the PTEN protein level in VSMCs transfected with either miR-21 mimics/inhibitor or corresponding NC ()). To further determine whether the PTEN level is modulated by UDCA in PDGF-BB-incubated VSMCs, the VSMCs were pretreated with 10 μM, 50 μM, 200 μM UDCA for 6 h followed by stimulation with PDGF-BB for 24 h, and the PTEN level was measured using Western blot analysis. We observed that treatment of VSMCs with PDGF-BB dramatically reduced the PTEN, whereas UDCA treatment increased PDGF-BB-induced PTEN downregulation in VSMCs in a dose-dependent manner (P < 0.01; )). These results demonstrated that miR-21 inhibited PTEN expression by directly targeting its 3ʹ-UTR in PDGF-BB-incubated VSMCs.
Figure 6. PTEN is a target gene of miR-21 in VSMCs. (a) The PTEN 3ʹ-UTR region containing the wild (wt) or mutant (mut) binding site for miR-21. (b) The VSMCs were co-transfected with either pMIR-PTEN-3ʹ-UTR or pMIR-PTEN-mut-3ʹ-UTR, and miR-21 mimic/inhibitor or corresponding NC and the relative luciferase activity were measured (**P < 0.01 vs. mimic NC group; ##P < 0.01 vs. inhibitor NC group). (c) The VSMCs transfected with the miR-21 mimic/inhibitor or corresponding NC, and the PTEN expression was detected by Western blot analysis. β-actin was used as an internal control for protein loading. (d) The VSMCs were pretreated with different doses of UDCA (10 μM, 50 μM, 200 μM) for 6 h followed by stimulation with PDGF-BB for 24 h, and then PTEN level was measured using Western blot analysis. Data are presented as means ± SD of three individual experiments. (**P < 0.01 vs. control group; #P < 0.05, ##P < 0.01 vs. PDGF-BB group).
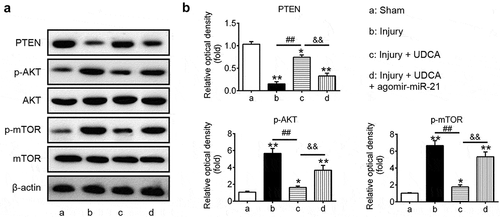
UDCA suppresses miR-21/PTEN/AKT/mTOR signaling pathway in the carotid artery injury model
AKT/mTOR signaling is a key pathway in cellular proliferation, cell survival, tumor growth and neointimal hyperplasia [Citation39–Citation42]. PTEN, a tumor suppressor gene, negatively modulated the PI3 K/AKT pathway, which plays an important role in various diseases through modulating cell proliferation, apoptosis, invasion and metabolism [Citation43]. To explore whether UDCA modulated the PTEN/AKT/mTOR signaling pathway via inhibiting miR-21 in injured carotid arteries, we assessed the levels of PTEN, p-AKT, total AKT, p-mTOR and total mTOR in UDCA-treated carotid arteries after administration with agomir-miR-21. Our results showed that the PTEN was significantly downregulated and p-AKT and p-mTOR were obviously upregulated in injure group compared with those in sham group (P < 0.01; ), while injection of UDCA remarkably resulted in PTEN upregulation and p-AKT and p-mTOR downregulation in injure + UDCA group (P < 0.01; ). Conversely, restoration of miR-21 by agomir-miR-21 reversed the UDCA-induced effects on PTEN, p-AKT and p-mTOR in injure + UDCA + agomir-miR-21 group (P < 0.01; ). Collectively, our results suggested that UDCA attenuated intimal hyperplasia through blocking miR-21/PTEN/AKT/mTOR signaling pathway in injured carotid arteries.
Figure 7. UDCA suppresses miR-21/PTEN/AKT/mTOR signaling pathway in the carotid artery injury model. (a) The vascular injury rats were administrated with agomir-miR-21 after injection with UDCA, and the PTEN, p-AKT, total AKT, p-mTOR and total mTOR were detected using Western blot analysis in injured carotid arteries. β-actin was used as an internal control for protein loading. (b) Bar graph showing the PTEN, p-AKT and p-mTOR protein expression in injured carotid arteries. The expression of PTEN was normalized to β-actin, and p-AKT and p-mTOR was normalized to total AKT and total mTOR, respectively. Data are presented as means ± SD of three individual experiments. (*P < 0.05, **P < 0.01 vs. sham group; ##P < 0.01 vs. injury group; &&P < 0.01 vs. injury + UDCA group).
Discussion
Dysregulated VSMCs in arterial walls function as a critical contributing factor in atherosclerosis, hypertension and vascular restenosis [Citation44]. In this study, our results demonstrated that UDCA ameliorated intimal hyperplasia and the excessive proliferation and invasion of VSMCs in the rat carotid artery injury model. In addition, our results identified that UDCA suppressed the formation of hyperplastic neointima in injured carotid arteries via inhibiting miR-21 expression. More importantly, our findings indicated that UDCA exerted the inhibitory effects on intimal hyperplasia both in vitro and in vivo via blocking miR-21/PTEN/AKT/mTOR signaling pathway.
UDCA, a tertiary bile acid, is the most widely used therapeutic agent for the treatment of hepatopathies, including primary biliary cirrhosis [Citation45]. It has also been reported that UDCA might be beneficial in atherosclerotic vascular disease. For example, Von Haehling et al. have demonstrated that UDCA improves endothelial function, as evaluated by flow mediated vasodilation in patients with chronic heart failure [Citation46]. UDCA was also identified to inhibit inflammatory response and endoplasmic reticulum stress in disturbed flow-induced atherosclerosis [Citation12]. In this study, we established a rat model of vascular injury used by unilateral carotid artery ligation and investigated the effects of UDCA on the formation of hyperplastic neointima. Our results showed that UDCA dramatically decreased neointimal area and the ratio of neointimal area to media area in injured carotid arteries. In addition, injection of UDCA resulted in the downregulation of PCNA, a cellular proliferation marker, downregulation. These results indicated that UDCA attenuate intimal hyperplasia and cell proliferation in injured carotid arteries.
miRNAs act as critical regulators of gene expression that exert their activity by regulating target mRNA stability or translation efficiency [Citation47]. A recent study has demonstrated that UDCA improves rat liver regeneration after partial hepatectomy by regulating miRNAs expression [Citation20]. Moreover, UDCA has been reported to regulate the severity of human nonalcoholic fatty liver disease by inhibiting miR-34a/SIRT1/p53 signaling pathway [Citation21]; therefore, we hypothesized that UDCA exerts the suppressive effect on intimal hyperplasia by modulating miRNAs expression. In this study, miRNA microarray analysis showed that a large set of miRNAs was differently expressed in injured carotid arteries using microarray assay. Among these aberrant miRNAs, miR-495, miR-221 and miR-21 were upregulated, and let-7a was downregulated in rat carotid arteries tissues, indicating the reliability of our microarray. A previous study reported that inhibition of miR-495 has beneficial effects on lesion formation, intimal hyperplasia and plasma cholesterol levels in experimental restenosis model [Citation36]. Liu et al. found that knockdown of miR-221 in rat carotid arteries suppressed VSMC proliferation in vivo and neointimal lesion formation after angioplasty [Citation30]. Cao et al. proved that upregulation of let-7a inhibits VSMCs proliferation in vitro and in vein graft intimal hyperplasia in rats model [Citation48]. To validate whether UDCA inhibits intimal hyperplasia by modulating these miRNAs, miR-21, miR-221, miR-let-7a and miR-495 were assessed in rat carotid arteries tissues. We observed that UDCA significantly inhibited the upregulation of injury-induced miR-21; whereas miR-495, miR-221 and miR-495 levels did not change in rat carotid arteries tissues. Moreover, our results revealed that restoration of miR-21 by agomir-miR-21 reversed the protective effects of UDCA in vivo on hyperplastic neointima. These data indicated that UDCA repressed intimal hyperplasia in the carotid artery injury model via inhibiting miR-21 expression.
In response to injured stimuli, VSMCs changed from contractile phenotype to highly proliferative and migratory synthetic phenotype [Citation45]. This phenotypic change increased VSMC proliferation and migration, which is one of the critical cellular events in the development of atherosclerosis [Citation45,Citation49]. To investigate the UDCA effect in VSMCs, we established the VSMC model and induced proliferation and invasion using PDGF-BB. We observed that UDCA treatment ameliorated PDGF-BB-induced proliferation, invasion and migration of VSMCs in a dose-dependent manner. Conversely, miR-21 overexpression reversed the inhibitory effects of UDCA on PDGF-BB-induced VSMC proliferation and metastasis. These results of our investigation of the effects of PDGF-BB on VSMCs suggested that UDCA exerts the suppressive effects on PDGF-BB-induced VSMC proliferation and metastasis by blocking miR-21 level; however, the molecular mechanism by which UDCA inhibits proliferation and metastasis in vitro and in vivo are not completely understood.
PTEN, a tumor suppressor, has been shown to modulate diverse cellular processes including apoptosis, proliferation and inflammation [Citation50–Citation53]. Datta et al. [Citation54] have demonstrated that the AKT/mTOR signaling pathway is an important oncogenic pathway to promote cell growth and survival, which is negatively modulated by PTEN [Citation55]. A recent study mTOR has identified mTOR as a regulator of VSMC proliferation and migration through its downstream target P70S6 K signaling [Citation42]. It is well reported that miR-21 inhibited the PTEN by directly targeting its 3ʹ-UTR in various types of cancer including breast cancer, hepatocellular cancer, lung cancer and gastric cancer [Citation56–Citation59]. Another recent study has demonstrated that miR-21 is upregulated in the vascular wall after balloon injury and mediates cell proliferation and apoptosis by downregulating PTEN and Bcl-2 [Citation59]. The results of our current study further confirmed that miR-21 suppresses the PTEN by directly targeting its 3ʹ-UTR in VSMCs. More importantly, our data validated that UDCA suppressed PTEN/AKT/mTOR signaling pathway in vivo by repressing miR-21 level. Taken together, our findings indicated that UDCA suppressed intimal hyperplasia in vivo by blocking miR-21/PTEN/AKT/mTOR signaling pathway.
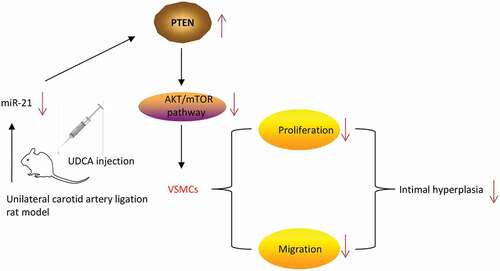
In conclusion, our results demonstrated that UDCA inhibits intimal hyperplasia by blocking miR-21/PTEN/AKT/mTOR signaling pathway (). Our findings suggest that UDCA may serve as a therapeutic agent for treatment of atherosclerosis.
Figure 8. Schematic diagram of the signaling pathway in which Ursodeoxycholic acid inhibits intimal hyperplasia in rat model. UDCA-treatment attenuated intimal hyperplasia and induced ectopic expression of miRNAs in injured carotid arteries. More importantly, UDCA inhibited intimal hyperplasia, VSMCs excessive migration and proliferation via blocking miR-21/PTEN/AKT/mTOR signaling pathway.
Authors contribution
Mengfan Li was responsiblefor the main conceive of the study and the draft of the manuscript. Rong Huang and Yi Huang helped to design the study and performed the statistical analysis. Guang Zeng and Yongzhi Jin helped to revise the manuscript and participated in its design. All authors have read and approved the final manuscript.
Disclosure Statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this Article
References
- Orr AW, Hastings NE, Blackman BR, et al. Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J Vasc Res. 2010;47:168–180.
- Yu H, Payne Thomas J, Mohanty Dillip K. Effects of slow, sustained, and rate-tunable nitric oxide donors on human aortic smooth muscle cells proliferation. Chem Biol Drug Des. 2011;78:527–534.
- Nicoletti A, Li Z, Lacolley P, et al. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. 2018;95:194–204.
- Heusch G, Libby P, Gersh B, et al. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet. 2014;383:1933–1943.
- Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2018;95:156–164.
- Shawky NM, Segar L. Sulforaphane inhibits platelet-derived growth factor-induced vascular smooth muscle cell proliferation by targeting mTOR/p70S6kinase signaling independent of Nrf2 activation. Pharmacol Res. 2017;119:251–264.
- Lindor K. Ursodeoxycholic acid for the treatment of primary biliary cirrhosis. N Engl J Med. 2007;357:1524–1529.
- Beuers U, Boyer JL, Paumgartner G. Ursodeoxycholic acid in cholestasis: potential mechanisms of action and therapeutic applications. Hepatology. 2003;28:1449–1453.
- Kim SY, Kwon Y-W, Jung IL, et al. Tauroursodeoxycholate (TUDCA) inhibits neointimal hyperplasia by suppression of ERK via PKC a -mediated MKP-1 induction. Cardiovasc Res. 2018;92:307–316.
- Malisova L, Kovacova Z, Koc M, et al. Ursodeoxycholic acid but not tauroursodeoxycholic acid inhibits proliferation and differentiation of human subcutaneous adipocytes. PloS One. 2013;8:e82086.
- Cabrera D, Arab JP, Arrese M. UDCA, NorUDCA, and TUDCA in liver diseases: a review of their mechanisms of action and clinical applications. Handb Exp Pharmacol. 2019;256:237–264.
- Chung J, An SH, Kang SW, et al. Ursodeoxycholic acid (UDCA) exerts anti-atherogenic effects by inhibiting RAGE signaling in diabetic atherosclerosis. Plos One. 2016;11:e0147839.
- Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–714.
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233.
- Bartel DP. MicroRNAs. Cell. 2004;116:281–297.
- Sage C, Agami R. Immense Promises for Tiny Molecules: uncovering miRNA Functions. Cell Cycle. 2006;5:1415–1421.
- Krichevsky AM. MicroRNA Profiling: from dark matter to white matter, or identifying new players in neurobiology. Sci World J. 2007;7:155–166.
- Jamaluddin MS, Weakley SM, Zhang L, et al. miRNAs: roles and clinical applications in vascular disease. Expert Rev Mol Diagn. 2011;11:79–89.
- Menghini R, Stöhr R, Federici M. MicroRNAs in vascular aging and atherosclerosis. Ageing Res Rev. 2014;17:68–78.
- Castro RE, Ferreira DMS, Zhang X, et al. Identification of microRNAs during rat liver regeneration after partial hepatectomy and modulation by ursodeoxycholic acid. Am J Physiol Gastrointest Liver Physiol. 2010;299:G887–G97.
- Castro RE, Ferreira DMS, Afonso MB, et al. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J Hepatol. 2013;58:119–125.
- Sun H-J, Zhao M-X, Ren X-S, et al. Salusin-β promotes vascular smooth muscle cell migration and intimal hyperplasia after vascular injury via ROS/NFκB/MMP-9 pathway. Antioxid Redox Signal. 2016;24:1045–1057.
- Menendez-Castro C, Cordasic N, Schmid M, et al. Intrauterine growth restriction promotes vascular remodelling following carotid artery ligation in rats. Clin Sci. 2012;123:437.
- Chung Y-R, Choi JA, Koh J-Y, et al. Ursodeoxycholic acid attenuates endoplasmic reticulum stress-related retinal pericyte loss in streptozotocin-induced diabetic mice. J Diabetes Res. 2017;2017:1-10.
- Wang B, Zhang M, Takayama T, et al. BET bromodomain blockade mitigates intimal hyperplasia in rat carotid arteries. EBioMedicine. 2015;2:1650–1661.
- Li Y, Zheng H, Yun Y, et al. De-agglomeration of IZM-2 zeolite crystals by post-synthetic treatment. Microporous Mesoporous Mater. 2016;225:185–191.
- Chien Y-C, Sheu M-J, Wu C-H, et al. A Chinese herbal formula “Gan-Lu-Yin” suppresses vascular smooth muscle cell migration by inhibiting matrix metalloproteinase-2/9 through the PI3K/AKT and ERK signaling pathways. BMC Complement Altern Med. 2012;12:137.
- Choe N, Kwon J-S, Kim J-R, et al. The microRNA miR-132 targets Lrrfip1 to block vascular smooth muscle cell proliferation and neointimal hyperplasia. Atherosclerosis. 2013;229:348–355.
- Choe N, Kwon J-S, Kim YS, et al. The microRNA miR-34c inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by targeting stem cell factor. Cell Signal. 2015;27:1056–1065.
- Liu X, Cheng Y, Zhang S, et al. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009;104:476.
- Zhang R-N, Zheng B, Li L-M, et al. Tongxinluo inhibits vascular inflammation and neointimal hyperplasia through blockade of the positive feedback loop between miR-155 and TNF-α. Am J Physiol Heart Circ Physiol. 2014;307:H552–H62.
- Hua J-Y, He Y-Z, Xu Y, et al. Emodin prevents intima thickness via Wnt4/Dvl-1/β-catenin signaling pathway mediated by miR-126 in balloon-injured carotid artery rats. Exp Mol Med. 2015;47:e170.
- Ji R, Cheng Y, Yue J, et al. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res. 2007;100:1579.
- Wang M, Li W, Chang G-Q, et al. MicroRNA-21 regulates vascular smooth muscle cell function via targeting tropomyosin 1 in arteriosclerosis obliterans of lower extremities. Arterioscler Thromb Vasc Biol. 2011;31:2044–2053.
- Li J, Liu B, Shao Y, et al. Upregulation of let-7a inhibits vascular smooth muscle cell proliferation in vitro and in vein graft intimal hyperplasia in rats. J Surg Res. 2014;192:223–233.
- Welten SMJ, de Jong RCM, Wezel A, et al. Inhibition of 14q32 microRNA miR-495 reduces lesion formation, intimal hyperplasia and plasma cholesterol levels in experimental restenosis. Atherosclerosis. 2017;261:26–36.
- Han J-H, Lee S-G, Jung S-H, et al. Sesamin inhibits PDGF-Mediated proliferation of vascular smooth muscle cells by upregulating p21 and p27. J Agric Food Chem. 2015;63:7317–7325.
- Liu J, Xiu J, Cao J, et al. Berberine cooperates with adrenal androgen dehydroepiandrosterone sulfate to attenuate PDGF-induced proliferation of vascular smooth muscle cell A7r5 through Skp2 signaling pathway. Mol Cell Biochem. 2011;355:127–134.
- Bai X, Jiang Y. Key factors in mTOR regulation. Cell Mol Life Sci. 2010;67:239–253.
- Arachchige Don AS, Tsang CK, Kazdoba TM, et al. Targeting mTOR as a novel therapeutic strategy for traumatic CNS injuries. Drug Discov Today. 2012;17:861–868.
- Xue M, Yao S, Hu M, et al. HIV-1 Nef and KSHV oncogene K1 synergistically promote angiogenesis by inducing cellular miR-718 to regulate the PTEN/AKT/mTOR signaling pathway. Nucleic Acids Res. 2014;42:9862–9879.
- Ha JM, Yun SJ, Kim YW, et al. Platelet-derived growth factor regulates vascular smooth muscle phenotype via mammalian target of rapamycin complex 1. Biochem Biophys Res Commun. 2015;464:57–62.
- Nakanishi A, Wada Y, Kitagishi Y, et al. Link between PI3K/AKT/PTEN pathway and NOX proteinin diseases. Aging Dis. 2014;5:203–211.
- Lim Y, Kwon J-S, Kim D-W, et al. Obovatol from Magnolia obovata inhibits vascular smooth muscle cell proliferation and intimal hyperplasia by inducing p21Cip1. Atherosclerosis. 2010;210:372–380.
- Roma Marcelo G, Toledo Flavia D, Boaglio Andrea C, et al. Ursodeoxycholic acid in cholestasis: linking action mechanisms to therapeutic applications. Clin Sci. 2011;121:523.
- Tousoulis D, Papageorgiou N, Stefanadis C. Ursodeoxycholic acid in patients with chronic heart failure. J Am Coll Cardiol. 2012;60:1579.
- Kataoka M, Wang D-Z. Non-coding RNAs including miRNAs and lncRNAs in cardiovascular biology and disease. Cells. 2014;3:883–898.
- Cao H, Hu X, Zhang Q, et al. Upregulation of let-7a inhibits vascular smooth muscle cell proliferation in vitro and in vein graft intimal hyperplasia in rats. J Surg Res. 2014;192:223–233.
- Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg. 2007;45:A25–A32.
- Hopkins BD, Hodakoski C, Barrows D, et al. PTEN function: the long and the short of it. Trends Biochem Sci. 2014;39:183–190.
- Singh AP, Arora S, Bhardwaj A, et al. CXCL12/CXCR4 protein signaling axis induces sonic hedgehog expression in pancreatic cancer cells via extracellular regulated Kinase- and Akt Kinase-mediated activation of nuclear factor κB: implications for bidirectional tumor-stromal interactionS. J Biol Chem. 2012;287:39115–39124.
- Iliopoulos D, Jaeger SA, Hirsch HA, et al. STAT3 Activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 2010;39:493–506.
- Kansal S, Bhatnagar A, Agnihotri N. Fish oil suppresses cell growth and metastatic potential by regulating PTEN and NF-κB signaling in colorectal cancer. Plos One. 2014;9:e84627.
- Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–296.
- Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100:387–390.
- Meng F, Henson R, Wehbe–Janek H, et al. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658.
- Yang Z, Fang S, Di Y, et al. Modulation of NF-κB/miR-21/PTEN pathway sensitizes non-small cell lung cancer to cisplatin. PLoS ONE. 2015;10:e0121547.
- Li LQ, Li XL, Wang L, et al. Matrine inhibits breast cancer growth via miR-21/PTEN/Akt pathway in MCF-7 cells. Cell Physiol Biochem. 2012;30:631–641.
- Yang S-M, Huang C, Li X-F, et al. miR-21 confers cisplatin resistance in gastric cancer cells by regulating PTEN. Toxicology. 2013;306:162–168.