
ABSTRACT
Liver cancer stem cells (CSCs) are involved in tumorigenesis, progression, drug resistance and recurrence of hepatocellular carcinoma (HCC). However, the underlying mechanism for the propagation of liver cancer stem cells was unclear. Herein, we observed miR-206 expression was reduced in both chemoresistant HCCs and recurrent HCCs from patients. A dramatically decrease of miR-206 was detected in cluster of differentiation 133 (CD133) or epithelial cell adhesion molecule (EpCAM)–positive liver CSCs and in CSC-enriched hepatoma spheres. Functional studies revealed that a forced expression of miR-206 inhibited liver CSCs expansion by suppressing the dedifferentiation of hepatoma cells and attenuating the self-renewal of liver CSCs. Mechanistically, bioinformatic and luciferase reporter analysis identified epidermal growth factor receptor (EGFR) as a direct target of miR-206. Moreover, miR-206 downregulated the expression of EGFR in liver CSCs. There was a significant inverse correlation between miR-206 and EGFR mRNA expression in HCC samples. Special EGFR inhibitor Gefitinib abolished the discrepancy in liver CSC proportion and the self-renewal capacity between miR-206 overexpression hepatoma cells and control cells, which further confirmed that EGFR was required in miR-206-inhibited liver CSCs expansion. Conclusion: miR-206 could suppress HCC cell dedifferentiation and liver CSCs expansion by targeting EGFR signaling.
Introduction
Hepatocellular carcinoma (HCC) is the most common lethal malignancies in liver cancer [Citation1,Citation2]. It is the sixth most common death tumor and the second highest cause of cancer-related death in the world [Citation3]. There have about 850000 new cases of liver cancer recorded in the world a year and China accounts for about half of the new cases [Citation4]. The incidence of liver cancer is roughly equal with its mortality. It means most of the liver cancer patients died of liver cancer [Citation5]. Most HCC patients were diagnosed at late stages and lost the best operation time [Citation6]. In the past decade, 5-year survival of HCC patients remained at 15% to 40% and about 62%-82% patients will recurrence within two years [Citation7]. Liver cancer patients are not sensitive to conventional radiation and chemotherapy [Citation8]. Sorafenib was the most commonly used targeted drug in patients with liver cancer [Citation9]. While lots of HCC patients are also not benefited from sorafenib treatment. So, it is urgent to explore the underling mechanism of initiation and development of liver cancer, then find the new treatment strategies to prolong the survival time for HCC patients.
Several studies have demonstrated a distinct subpopulation of cells called cancer stem cells (CSCs) or tumor initiating cells (T-ICs) that exhibited extended self-renewal potential, tumor initiating ability and resistance to chemotherapy [Citation10]. The substantial heterogeneity and hierarchical organization in liver cancer support the theory of liver cancer stem cells (LCSCs) [Citation11]. Numerous studies reported that liver cancer stem cells could be identified by CD24, CD90, EpCAM and other biomarkers [Citation12,Citation13]. CD24 can drive self-renewal and tumor initiation of liver CSCs through STAT3-mediated Nanog regulation [Citation12]. It was accepted that chemo-resistance and recurrence of HCC was closely associated with the existence of liver CSCs [Citation14]. However, how liver CSCs generation and expansion remains unclear.
microRNAs (miRNAs) are small evolutionarily conserved, 20 to 22 nt,, non-coding RNA molecules that inhibit translation or induce mRNA degradation in general by binding to the 3ʹUTR of target mRNAs [Citation15]. miRNAs play an important regulatory role in various biological processes [Citation16]. They worked as tumor suppressor gene or oncogene and regulated the initiation and progression of tumors [Citation17,Citation18]. For example, miR-429 promotes liver tumor-initiating cell properties by targeting retinoblastoma protein (Rb) binding protein 4 [Citation19]. It was also reported that miR-206 suppresses the proliferation and invasion of lung cancer cells and gastric cancer cells [Citation20,Citation21], suggesting that this miRNA was involved in tumor progression. However, whether miR-206 was involved in liver CSCs regulation remains unclear.
In the present study, we found that the expression of miR-206 was dramatically downregulated in CD133 or EpCAM–positive liver CSCs and in CSC-enriched hepatoma spheres. Next, by gain-of-function analyzes in liver CSCs, we demonstrated that miR-206 inhibited liver CSCs expansion. Furthermore, EGFR was a direct target of miR-206 in liver CSCs. EGFR inhibitor Gefitinib abolished the discrepancy in liver CSC proportion and the self-renewal capacity between miR-206 overexpression HCC cells and control cells. We also found that miR-206 could affect the drug resistance of HCC cells to sorafenib and cisplatin. Taken together, our study showed that miR-206 was a novel cancer stem cell marker that plays a key role in liver CSCs expansion and drug resistance of HCC.
Materials and methods
Patients and samples
HCC patients’ tissue samples were collected from the Eastern Hepatobiliary Surgery Hospital (Shanghai, China). Patient informed consent was also obtained and the procedure of human sample collection was approved by the Ethics Committee of Eastern Hepatobiliary Surgery Hospital.
Cell lines and cell culture
Normal hepatocyte cell line HL7702 (L02), and liver cancer cell lines Huh7, HepG2, Hep3B, CSQT-2, PLC and HCCLM3 were purchased form Chinese Academy of Sciences, Shanghai, China. The HCC cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 2 mM L-glutamine, and 25 µg/ml of gentamicin and maintained at 37°C in 5% CO2 incubator. The culture cells were digested with 0.5% trypsin and moved to a new six-well plate twice a week.
The lenti–vector overexpressing miR-206 and its control virus were produced as described previously [Citation22]. Huh7 and HCCLM3 cells were infected with miR-206 or its control virus and the stable infectants were screened by puromycin.
Spheroid formation assay
Huh7 or HCCLM3 miR-206 and their control cells were cultured in a 6-well or 96-well Ultra-Low Attachment Microplates (Corning, USA) in serum-free DMEM/F12 (Invitrogen, USA), supplemented with B27 (1:50, Invitrogen), 20 ng/ml EGF (Peprotech), 10 ng/ml bFGF (Invitrogen), and 4 μg/ml insulin (Sigma). Spheres were photographed and counted 7 days after seeding (primary spheres).
Limiting dilution assay
Various numbers of Huh7 or HCCLM3 miR-206 and their control cells (64, 32, 26, 8, 4, 2) were seeded into 96-well ultra-low attachment culture plates for one week. CSC proportions were analyzed using Poisson distribution statistics and the L-Calc Version 1.1 software program (Stem Cell Technologies, Inc., Vancouver, Canada) as described [Citation23].
Real-time PCR
For detection of mature miR-206, total RNA was subjected to reverse transcription using a TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems). qRT-PCR analysis of miR-206 expression was carried out using TaqMan MicroRNA assay kits (Applied Biosystems). Results were normalized to U6 snRNA using the comparative threshold cycle (Ct) method.
The total cells RNA was extracted by using Trizol reagent (Invitrogen, 15,596–018). Total cDNAs were synthesized by ThermoScript TM RT-PCR system (Invitrogen, 11,146–057). The total mRNA amount presented in the cells was measured by RT-PCR using the ABI PRISM 7300 sequence detector (Applied Biosystems). The EGFR primer sequences were forward: 5ʹ CCCTCCTGAGCTCTCTGAGT 3ʹ, reverse: 5ʹ TTCCAGACAAGCCACTCACC 3ʹ.
Western blotting assay
The HCC cells were lysed with cell lysis buffer (Beyotime) followed by supersonic splitting. The total protein was quantified using the BCA Protein Quantification kit. A total of 20 μg of protein were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred onto nitrocellulose membranes. The membranes were blocked with 10% nonfat milk and incubated with the primary antibodies (EGFR, PARP and GAPDH) overnight. The protein band, specifically bound to the primary antibody, was detected using an IRDye 800CW-conjugated secondary antibody and LI-COR imaging system (LI-COR Biosciences, Lincoln, NE, USA). The EGFR, PARP and GAPDH antibodies were purchased from Abcam Company in USA.
Flow-cytometric analysis
Hepatoma cells were incubated with the primary anti–CD133 or anti-EpCAM for 30 min at room temperature. Flow-cytometric analysis was performed using a MoFlo XDP from Beckman Coulter according to the manufacturer’s instructions.
Huh7 or HCCLM3 miR-206 and their control cells were incubated with the primary anti-CD133 or anti- EpCAM for 30 min at room temperature. Flow cytometric analysis was performed using a MoFlo XDP cell sorter from Beckman Coulter according to the manufacturer’s instructions.
Apoptosis analysis
Huh7 or HCCLM3 miR-206 and their control cells were cisplatin (4 μg/ml) or sorafenib (10 μM) for 24 hours. Then the cells were measured by flow cytometry using a FITC Annexin V Apoptosis Detection Kit (BD Biosciences 51-66211E). Briefly, 1 × 106 cells were harvested and washed twice with cold cell staining buffer, resuspended in 100 μl Annexin V binding buffer, then incubated with 5 μl of FITC Annexin V and 5 μl of PI viability staining solution for 15 minutes at room temperature in the dark. The cell suspension was then incubated with 200 μl of Annexin V binding buffer followed by flow cytometry analysis.
Luciferase reporter assay
HCC cells were transfected with EGFR WT or EGFR mutant 3ʹUTR plasmids. Luciferase activity was measured using a Synergy 2 Multidetection Microplate Reader (BioTek Instruments, Inc.). Data were normalized for transfection efficiency by dividing firefly luciferase activity by Renillaluciferase activity.
Statistical analysis
GraphPad Prism (GraphPad Software, Inc. La Jolla, USA) was used for all statistical analyses. Statistical analysis was carried out using t test or Bonferroni Multiple Comparisons Test: *p < 0.05. A p value of less than 0.05 was considered significant.
Results
miR-206 expression was downregulated in liver CSCs
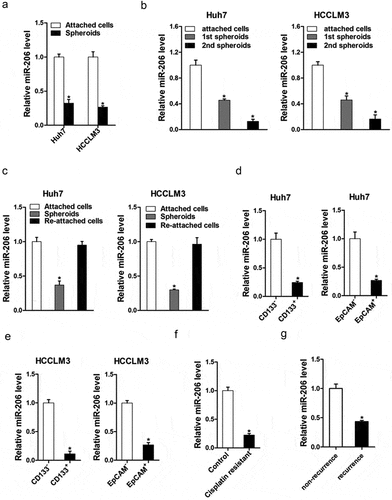
It was reported that miR-206 is involved in inhibiting proliferation and invasion in different cancer cell types [Citation24–Citation26]. So, we doubted whether miR-206 was also participated in the regulation of cancer stem cells. As self-renewal and chemo-resistance were distinct characteristics of CSCs, we investigated the expression of miR-206 in liver CSCs. As expected, miR-206 expression was downregulated in the self-renewing spheroids compared with the attached cells ()). Consistently, in serial passages of HCCLM3 or Huh7 spheroids, miR-206 expression was gradually decreased ()). Intriguingly, miR-206 levels could be partially restored during reattachment in parallel with the differentiation ()). Cluster of differentiation 133 (CD133) and epithelial cell adhesion molecule–positive (EpCAM) are well-accepted liver CSCs marker [Citation13,Citation27]. As expected, CD133+ and EpCAM+ liver CSCs sorted from trypsinized spheres of hepatoma cells displayed lower miR-206 level (). Considering the close association of liver CSCs with HCC recurrence and chemoresistance, cisplatin-resistant HCC xenografts were established as described. In comparison with control tumors, miR-206 expression was markedly decreased in the cisplatin-resistant, indicating that miR-206 expression was associated with chemoresistance ()). More importantly, miR-206 expression was dramatically decreased in recurrent HCC compared with the primary lesion ()), which further suggested that miR-206 expression was decreasing in liver CSCs.
Figure 1. miR-206 expression was downregulated in liver CSCs
miR-206 suppressed liver CSCs expansion
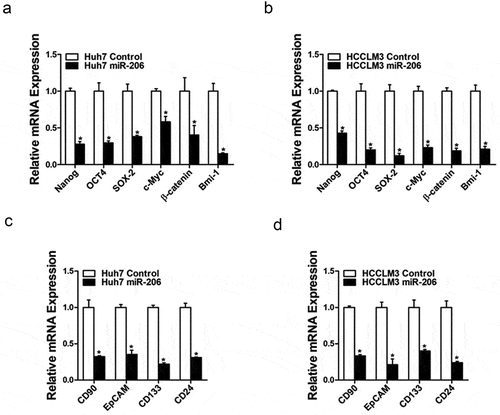
To explore the role of miR-206 in liver CSCs regulation, miR-206 expression was checked in series of HCC cell lines and miR-206 stable overexpressing infectants of HCC cells were used (). Flow-cytometric analysis revealed a decreased proportion of liver CSCs in miR-206 stably transfected HCC cells (). Consistently, HCC cells overexpressing miR-206 formed much fewer spheroids than control cells ()). An in vitro and in vivo limiting dilution assay illustrated that miR-206 overexpression dramatically decreased the CSC population and tumorigenicity capacity in HCC cells (). Moreover, miR-206 overexpression downregulated the expression of stemness-associated genes and liver CSC markers in HCC cells (), which further supported that miR-206 could inhibit liver CSCs expansion.
Figure 2. miR-206 was required for the expansion of liver CSCs
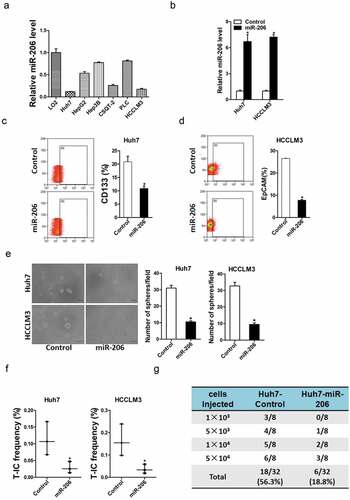
Figure 3. miR-206 downregulated stemness–like genes
miR-206 affected the drug resistance of HCC cells to sorafenib and cisplatin
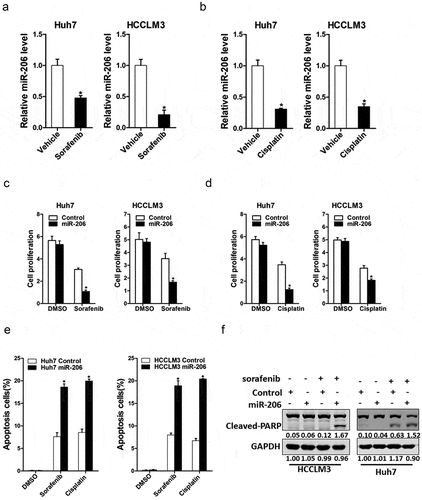
We next explored the role of miR-206 in chemoresistance of HCC to sorafenib and cisplatin. As expected, we found that miR-206 expression was markedly reduced in cisplatin-resistant or sorafenib-resistant hepatoma cells (); suggesting miR-206 was involved in drug resistance. Furthermore, miR-206 overexpression dramatically increased the sensitivity of hepatoma cells to the same dosages of sorafenib or cisplatin (). In addition, the population of apoptotic cells was also significantly increased in HCC cells with miR-206 overexpression when exposed to sorafenib or cisplatin ()). poly ADP-ribose polymerase (PARP) is a DNA repair enzyme. PARP is the cleavage substrate of caspase and plays an important role in DNA damage repair and apoptosis. Cleaved-PARP is an important indicator of apoptosis and is also generally considered to be an indicator of Caspase 3 activation [Citation28]. Moreover, western blot analyses showed that protein expression of cleaved-PARP in HCC cells with miR-206 overexpression was significantly increased when exposed to sorafenib or cisplatin at different dosages when compared with normal HCC cells ()). Collectively, these results showed that drug sensitivity of HCC to sorafenib and cisplatin was significantly increased when miR-206 was overexpressing, suggesting a possible role of miR-206 in the treatment of HCC drug resistance.
Figure 4. The effect of miR-206 on drug resistance of HCC to sorafenib and cisplatin
EGFR was a direct target of miR-206 in HCC cells
Next, we attempted to identify the target genes of miR-206 that may be involved in liver CSCs expansion. It was reported that miR-206 regulated EGFR in other tumors [Citation29]. So, we checked whether EGFR was also involved in miR-206 regulation in liver CSCs. As expected, EGFR protein level was downregulated in miR-206 overexpression liver CSCs ()). Consistently, EGFR mRNA expression was also downregulated in miR-206 overexpression liver CSCs ()). Bioinformatics analysis suggested that EGFR mRNA harbored a putative miR-206 binding site in its 3ʹ-UTR. To further explore whether miR-206 directly regulates EGFR expression via interaction with its mRNA 3ʹ-UTR, we transfected wild-type or mutant EGFR 3ʹ-UTR reporter in miR-206 overexpression hepatoma cells. The luciferase activity of wild-type reporter was significantly inhibited in the presence of miR-206 ()). However, miR-206-mediated repression of the reporter expression was compromised by mutation of the miR-206 binding site in the EGFR 3ʹ-UTR. ()), which further suggesting EGFR was a direct target of miR-206.
Figure 5. EGFR was a direct target of miR-206 in liver CSCs
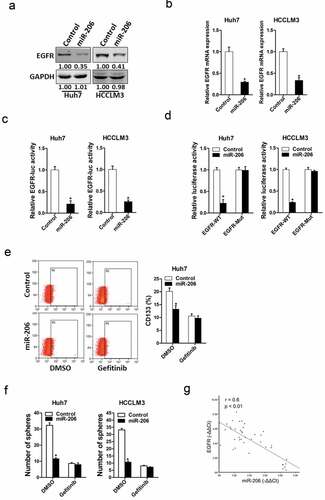
To investigate the role of EGFR in miR-206-mediated expansion of liver CSCs, the special EGFR inhibitor Gefitinib was used [Citation30]. As expected, Gefitinib abolished the difference in liver CSC proportion between miR-206 overexpression hepatoma cells and control cells ()). Consistently, Gefitinib entirely depleted the discrepancy of self-renewal capacity between miR-206 overexpression hepatoma cells and control cells ()). Moreover, the correlation between miR-206 expression and the levels of EGFR was observed in human HCC tissues ()). Taken together, these data suggest distinct regulation of EGFR by miR-206 in liver CSCs.
Discussion
Hepatocellular carcinoma (HCC) is one of most common and deadly malignant tumors in the world [Citation31]. Approximatively 70% HCC patients were diagnosed at late stage or had distant metastasis, their prognosis remains poor [Citation32]. The rate of recurrence after surgical was very high and only few HCC patients are benefited the treatment of TACE or sorafenib [Citation9]. The presence of CSCs in solid tumors has been confirmed, and these cells have the ability of self-renewal and differentiation, a high tumorigenic potential, and resistance to chemotherapeutics [Citation33]. The existence of liver CSCs is also considered as the origin of the chemoresistance and recurrence of patients with HCC. It is therefore important to explore the molecular mechanism underlying liver CSC regulation to develop novel therapeutic strategies targeting CSCs. In this study, our results showed that miR-206 plays a pivotal role in liver CSC expansion and may serve as a therapeutic target in personalized treatment of HCC.
miRNAs have important roles in the initiation and progression of numerous human cancers [Citation34], and might be proved to be a novel marker for the diagnosis and treatment of cancers. It was also reported that miRNAs were involved in CSCs regulation [Citation19]. Previous studies indicated that miR-206 worked as a tumor suppressor gene in numerous cancers [Citation35]. However, the potential role of miR-206 in liver CSCs has not been reported. In this study, we for first demonstrated that miR-206 was downregulated in a subpopulation of HCC cells with stem-like characteristics and was essential for maintaining self-renewal and oncogenic potential of this type of cancer cell.
Numerous studies have confirmed the presence of CSCs in solid tumors, and these cells are involved in self-renewal and differentiation, high tumorigenicity, and resistance to chemo treatments. The existence of liver CSCs is also considered to be the origin of chemoresistance and HCC recurrence [Citation25,Citation36]. In the current study, liver CSCs were enriched by establishing chemoresistant HCC xenograft tumors, and expression of miR-206 in these chemoresistant xenografts was dramatically down-regulated. More intriguingly, reduced miR-206 expression was found in recurrent HCC patients than non-recurrence HCC patients. Considering the importance of CSCs in tumor recurrence and chemoresistance, we investigated the influence of miR-206 on liver CSCs. Spheroid culture of cancer cells is a routine approach to enrich CSCs. We noted that miR-206 expression was barely expressed in hepatoma spheroids. To date, CD133 and EpCAM have been accepted as the predominant biomarkers of liver CSCs, which also include CD24, CD90, CD44, CD13, aldehyde dehydrogenase and OV6 [Citation37]. Our data showed that miR-206 levels decreased in CD133+ or EpCAM+ liver CSCs. Moreover, miR-206 overexpression in hepatoma cells suppressed the self-renewal capacity of liver CSCs, and downregulated stemness-associated genes and liver CSC markers. We also observed that miR-206 overexpression HCC cells are more sensitivity to sorafenib and cisplatin treatment. It means that miR-206-high expression HCC patients may benefit from sorafenib treatment.
Numerous studies showed that EGFR had important functions in most solid cancers, including ovarian cancer, NSCLC, breast cancer, colon cancer, lung and liver cancer [Citation38,Citation39]. Over-expression of EGFR was always found in aggressive and invasive cancers. Several EGFR-targeting strategies to interfere with EGFR-mediated malignant behaviors have shown prospective clinical benefits for several solid tumors [Citation40]. Treatment with gefitinib could down-regulate EGFR-TK activity, suppress tumor cell growth, promote apoptosis and cell cycle arrest, and reduce HCC-related angiogenesis both in vivo and in vitro [Citation41]. It was reported that miR-206 directly targeted EGFR in ovarian carcinoma. However, whether EGFR was also regulated by miR-206 in liver CSCs was unknown. In the present study, we found that EGFR was a direct target of miR-206. Overexpressing miR-206 in hepatoma cells downregulated EGFR expression through binding to its 3ʹUTR. In additional, special EGFR inhibitor gefitinib could abolish the discrepancy of the self-renewal ability between miR-206 overexpression HCC cells and their control cells, which further confirm EGFR was the downstream of miR-206 in regulating liver CSCs expansion. The combination of miR-206 overexpression virus and gefitinib showed better inhibitory effect on liver CSCs. Numerous studies showed that MUC1; MAP4K3; VEGF and Cyclin D1 was also directly targeted by miR-206 in other tumors. So, these above molecular might also be affected by miR-206 in liver CSCs. We will research these problems in future.
Here, we showed that miR-206 was downregulated in liver CSCs, which in turn suppressed the expansion of liver CSCs via directly regulating EGFR. miR-206 overexpression dramatically decreased the CSCs population and tumorigenicity capacity in HCC cells. It means miR-206 overexpression virus might be used to treat HCC patients by killing CSCs. The findings of the present study not only shed a new light on the mechanism of liver CSCs but suggest a novel prognostic marker and a potential therapeutic target against HCC.
Disclosure Statement
All authors declare no competing interests.
Additional information
Funding
References
- El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–1127.
- Laursen L. A preventable cancer. Nature. 2014;516(7529):S2–3.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29.
- Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–1255.
- El-Serag HB. Hepatocellular carcinoma: an epidemiologic view. J Clin Gastroenterol. 2002;35:S72–78.
- Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90.
- Worns MA, Galle PR. HCC therapies–lessons learned. Nat Rev Gastroenterol Hepatol. 2014;11(7):447–452.
- Shah C, Mramba LK, Bishnoi R, et al. Survival differences among patients with hepatocellular carcinoma based on the stage of disease and therapy received: pre and post sorafenib era. J Gastrointest Oncol. 2017;8:789–798.
- Han T, Xiang DM, Sun W, et al. PTPN11/Shp2 overexpression enhances liver cancer progression and predicts poor prognosis of patients. J Hepatol. 2015;63:651–660.
- Miyajima A, Tanaka M, Itoh T. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell. 2014;14(5):561–574.
- Li XF, Chen C, Xiang DM, et al. Chronic inflammation-elicited liver progenitor cell conversion to liver cancer stem cell with clinical significance. Hepatology. 2017;66(6):1934–1951.
- Lee TK, Castilho A, Cheung VC, et al. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011;9(1):50–63.
- Yamashita T, Ji J, Budhu A, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136(3):1012–1024.
- Zhou G, Latchoumanin O, Bagdesar M, et al. Aptamer-based therapeutic approaches to target cancer stem cells. Theranostics. 2017;7(16):3948–3961.
- DeSano JT, Xu L. MicroRNA regulation of cancer stem cells and therapeutic implications. Aaps J. 2009;11(4):682–692.
- Bayoumi AS, Sayed A, Broskova Z, et al. Crosstalk between long noncoding RNAs and microRNAs in health and disease. Int J Mol Sci. 2016;17(3):356. .
- Nucera S, Giustacchini A, Boccalatte F, et al. miRNA-126 orchestrates an oncogenic program in B cell precursor acute lymphoblastic leukemia. Cancer Cell. 2016;29(6):905–921.
- Wu J, Yuan P, Mao Q, et al. RETRACTED: miR-613 inhibits proliferation and invasion of breast cancer cell via VEGFA. Biochem Biophys Res Commun. 2016;478(1):274–278.
- Li L, Tang J, Zhang B, et al. Epigenetic modification of MiR-429 promotes liver tumour-initiating cell properties by targeting Rb binding protein 4. Gut. 2015;64(1):156–167.
- Pan JY, Sun CC, Bi ZY, et al. miR-206/133b cluster: a weapon against lung cancer? Mol Ther Nucleic Acids. 2017;8:442–449.
- Zheng Z, Yan D, Chen X, et al. MicroRNA-206: effective inhibition of gastric cancer progression through the c-Met pathway. PLoS One. 2015;10(7):e0128751.
- Xiang DM, Sun W, Ning BF, et al. The HLF/IL-6/STAT3 feedforward circuit drives hepatic stellate cell activation to promote liver fibrosis. Gut. 2018;67(9):1704–1715.
- Xiang D, Cheng Z, Liu H, et al. Shp2 promotes liver cancer stem cell expansion by augmenting beta-catenin signaling and predicts chemotherapeutic response of patients. Hepatology. 2017;65(5):1566–1580.
- Deng M, Qin Y, Chen X, et al. MiR-206 inhibits proliferation, migration, and invasion of gastric cancer cells by targeting the MUC1 gene. Onco Targets Ther. 2019;12:849–859.
- Heinemann FG, Tolkach Y, Deng M, et al. Serum miR-122-5p and miR-206 expression: non-invasive prognostic biomarkers for renal cell carcinoma. Clin Epigenetics. 2018;10(1):11.
- Liu F, Yin R, Chen X, et al. Over-expression of miR-206 decreases the Euthyrox-resistance by targeting MAP4K3 in papillary thyroid carcinoma. Biomed Pharmacother. 2019;114:108605.
- Ma S, Lee TK, Zheng BJ, et al. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27(12):1749–1758.
- Hassan S, Esch A, Liby T, et al. Pathway-enriched gene signature associated with 53Bp1 response to PARP inhibition in triple-negative breast cancer. Mol Cancer Ther. 2017;16(12):2892–2901.
- Choi BH, Ryu DY, Ryoo IG, et al. NFE2L2/NRF2 silencing-inducible miR-206 targets c-MET/EGFR and suppresses BCRP/ABCG2 in cancer cells. Oncotarget. 2017;8(63):107188–107205.
- Zou B, Lee VHF, Yan H. Prediction of sensitivity to gefitinib/erlotinib for EGFR mutations in NSCLC based on structural interaction fingerprints and multilinear principal component analysis. BMC Bioinformatics. 2018;19:88.
- Nishida N, Kudo M. Oncogenic signal and tumor microenvironment in hepatocellular carcinoma. Oncology. 2017;93(Suppl 1):160–164.
- Park YK, Song SK, Kim BW, et al. Prognostic significance of microvascular invasion in tumor stage for hepatocellular carcinoma. World J Surg Oncol. 2017;15:225.
- Xiao Y, Lin M, Jiang X, et al. The recent advances on liver cancer stem cells: biomarkers, separation, and therapy. Anal Cell Pathol (Amst). 2017;2017:5108653.
- Lim LP, Lau NC, Garrett-Engele P, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433(7027):769–773.
- Pan BL, Tong ZW, Wu L, et al. Effects of microRNA-206 on osteosarcoma cell proliferation, apoptosis, migration and invasion by targeting ANXA2 through the AKT signaling pathway. Cell Physiol Biochem. 2018;45(4):1410–1422. .
- Xiang DM, Sun W, Zhou T, et al. Oncofetal HLF transactivates c-Jun to promote hepatocellular carcinoma development and sorafenib resistance. Gut. 2019;68(10):1858–1871.
- Yang W, Wang C, Lin Y, et al. OV6(+) tumor-initiating cells contribute to tumor progression and invasion in human hepatocellular carcinoma. J Hepatol. 2012;57(3):613–620.
- Herbst RS, Langer CJ. Epidermal growth factor receptors as a target for cancer treatment: the emerging role of IMC-C225 in the treatment of lung and head and neck cancers. Semin Oncol. 2002;29(1):27–36.
- Fujino S, Enokibori T, Tezuka N, et al. A comparison of epidermal growth factor receptor levels and other prognostic parameters in non-small cell lung cancer. Eur J Cancer. 1996;32A(12):2070–2074.
- Ethier SP. Signal transduction pathways: the molecular basis for targeted therapies. Semin Radiat Oncol. 2002;12(3):3–10.
- Baselga J, Arteaga CL. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J Clin Oncol. 2005;23(11):2445–2459.