
ABSTRACT
Microvesicles (MVs) derived from human umbilical cord mesenchymal stem cells (hUC-MSCs-MVs) and miR-21 were demonstrated to ameliorate renal ischemia-reperfusion injury (IRI). Since hUC-MSC-MVs contained a substantial quantity of miR-21, we speculated that miR-21 might account for a part of the therapeutic effects of hUC-MSCs-MVs. The human tubule epithelial (HK-2) cells were cultured under low oxygen (LO) condition to mimic a cellular IRI model. A rat model of unilateral renal IRI was established. A co-culture model of HK-2 cells and MSC-MVs was utilized to examine the therapeutic role of MSC-MVs in HK-2 cell apoptosis and mechanism. The results showed that hUC-MSCs-MVs inhibited LO-induced HK-2 cell apoptosis through transferring miR-21 to HK-2 cells. Mechanistically, miR-21 directly targeted and negatively regulated programmed cell death protein 4 (PDCD4) in HK-2 cells. Moreover, PDCD4 overexpression in HK-2 cells abrogated the hUC-MSCs-MVs-inhibited HK-2 cell apoptosis under LO condition. Additionally, the beneficial effect of MSC-MVs on rat renal IRI was partly eliminated when miR-21 was knocked down in MSCs. Taken together, MSC-MVs inhibit tubular epithelial cell apoptosis and ameliorate renal IRI, at least partially, via delivery of miR-21.
Introduction
Renal ischemia-reperfusion injury (IRI) is common in several clinical situations including shock, sepsis or during transplantation [Citation1]. IRI is a major cause of intrinsic acute kidney injury (AKI), which is considered as a major contributor to end-stage kidney disease and leads to high mortality [Citation2]. Therefore, a potent therapeutic intervention for ischemic AKI is imperative.
In recent years, a promising and effective approach of therapeutic strategy for renal IRI is the use of mesenchymal stromal cells (MSCs). Intriguingly, microvesicles (MVs) derived from MSCs were found to mimic the efficacy of MSCs on the recovery from ischemic or toxic AKI [Citation3,Citation4], indicating that MVs are critical mediators. MVs are membranous structures delivering bioactive molecular content including proteins, mRNAs and miRNAs sequences to the recipient cells. Thus, MVs are described as a novel avenue for cell-to-cell communication and a crucial point of endocrine [Citation5]. Our previous study demonstrated that administration of human umbilical cord MSCs (hUC-MSCs)-derived MVs (hUC-MSCs-MVs) could ameliorate renal IRI in rats [Citation6–Citation9]. The mechanisms underlying the therapeutic effects of hUC-MSCs-MVs involve anti-inflammatory and anti-oxidative properties of MVs. However, the mechanism still remains incompletely understood.
microRNAs (miRNAs) are small noncoding RNAs with 19–23 nucleotides in lengths and play a significant role in various biological processes. Recent evidence indicated that miRNAs are implicated in IRI [Citation10]. Among these miRNAs, miR-21 has been shown to protect against renal IRI [Citation11]. Specifically, miR-21 has been considered as a strong anti-apoptotic factor at least in part by targeting pro-apoptotic genes including programmed cell death protein 4 (PDCD4) [Citation12]. PDCD4 has been demonstrated to be associated with IRI [Citation11]. In addition, tubular epithelial cell apoptosis contributes importantly to renal IRI [Citation13]. Recently, Song et al [Citation11] reported that IRI elevated miR-21 expression in the tubular epithelial cells, which prevented kidney injury by resisting apoptosis of epithelial cells.
Our preliminary study revealed that miR-21 was enriched in hUC-MSC-MVs. Thus, we speculated that miR-21 account for a part of the therapeutic effects of hUC-MSCs-MVs. In the present study, we explored the possible potential of hUC-MSCs-MVs to resist apoptosis of tubular epithelial cells under hypoxia condition and to clarify whether the possible mechanism was associated with hUC-MSCs-MVs delivering miR-21 to tubular epithelial cells. Furthermore, we investigated whether hUC-MSCs-MVs ameliorated renal IRI via transferring miR-21.
Materials and methods
Ethics statement
All research involving human participants was approved by the Research Ethics Committee of Zhengzhou University People’s Hospital, and written informed consent was obtained from each participant.
Isolation and characterization of hUC-MSCs
hUC-MSCs were isolated from umbilical cords within 4 h from post-partum, as described previously [Citation14]. Briefly, human umbilical cords were aseptically collected from full-term cesarean-section infants at Zhengzhou University People’s Hospital and were negative for hepatitis B virus (HBV), hepatitis C virus (HCV), human immunodeficiency virus (HIV), and cytomegalovirus. The umbilical cords were washed in phosphate‐buffered saline (PBS), pulverized into approximately 1–2 mm3 pieces, and then incubated in Dulbecco’s modified Eagle’s medium with low glucose (DMED-LG, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) in a humidified atmosphere containing 95% air and 5% CO2 at 37°C. The medium was changed every two days. Two weeks later, the adherent cells were subcultured. The cells at the 3rd to 5th passage were used in subsequent experiments. For identification, the immunophenotype (CD34, CD45, CD44, CD90, CD29, CD105; 1:100; BD Biosciences, San Jose, CA, USA) of hUC-MSCs was analyzed on a FACScan flow cytometer (BD Biosciences).
Isolation and characterization of hUC-MSCs-MVs
To isolate hUC-MSCs-MVs, hUC-MSCs were cultured in low-glucose DMEM deprived of FBS and supplemented with 0.5% bovine serum albumin (BSA) (Sigma-Aldrich). Following 48 h of incubation, the supernatants were centrifuged at 300 g for 10 min to remove cells, then at 2,000 g for 20 min to remove cellular debris. The supernatants were filtered through a 0.22 μm filter and centrifuged at 100,000 g to sediment the MVs for 1 h at 4°C. MVs were washed once with serum-free M199 medium (Sigma-Aldrich) containing 25 mM HEPES (pH = 7.4) and submitted to second ultracentrifugation in the same conditions. The supernatants were abandoned and the resultant sediment was washed in PBS, and resuspended in serum-free M199 and stored at −80°C until further use.
The protein concentration of the hUC-MSCs-MVs was quantified using the Bradford method (Bio-Rad, Hercules, USA). The morphologic characteristics of hUC-MSCs were observed under a transmission electron microscope (TEM). The phenotypic profile of hUC-MSCs-MVs (CD63 and CD9) was determined by western blot.
HK-2 cell culture
Human proximal tubule epithelial (HK-2) cells were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China) and maintained in Keratinocyte Serum Free Medium (K-SFM; Invitrogen) supplemented with gentamicin and amphotericin in a humidified atmosphere at 37°C with 5% CO2.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cells using TRIzol reagent (Invitrogen) and was reverse transcribed into cDNAs using the Reverse Transcription Kit (Takara). qRT-PCR was performed to amplify the cDNA templates using SYBR Premix Dimmer Eraser kit (TaKaRa) by the ABI7900 system (Applied Biosystem). The relative expression of miR-21 and PDCD4 was calculated by the 2−ΔΔCt method and normalized to the internal control GAPDH.
Cell transfection
The miR-21 inhibitor (miR-21I), inhibitor negative control (NCI), miR‐21 mimic, and mimic NC were synthesized and purchased from GenePharma (Shanghai, China). To overexpress PDCD4, the full-length PDCD4 cDNA fragment was cloned into pcDNA 3.1 (Invitrogen, USA), generating pcDNA3.1-PDCD4, with an empty pcDNA3.1 vector used as a control. hUC-MSCs and HK-2 cells were transfected with these plasmids or miRNAs using Lipofectamine® 3000 (Invitrogen) according to different experimental purposes. Following transfection for 48 h, hUC-MSCs and HK-2 cells were harvested for qRT-PCR analysis to examine the knockdown or overexpression efficiency.
Cell apoptosis assay
Cell apoptosis was qualified using an annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) cell apoptosis kit (Invitrogen, Thermo Fisher Scientific, Inc.). After treatments, HK-2 cells were digested with trypsin and washed twice with PBS. Then cells were incubated with 5 µL Annexin V‑FITC and 5 µL PI. Following 10 min of incubation at room temperature in the dark, the mixtures were analyzed using a flow cytometry system (FACScan®, BD Biosciences) equipped with FlowJo 7.6 software (Treestar, Inc., Ashland, OR, USA).
Western blot
Total protein was extracted from the cells in RIPA lysis buffer. The protein concentrations were determined by BCA assay. Then equal protein from cell lysates was loaded per lane and separated using 10% SDS-PAGE gel electrophoresis and then transferred to PVDF membranes. Subsequently, the membranes were blocked with 5% fat-free milk, and then incubated with primary antibodies against caspase-3, Bax, Bcl-2, and PDCD4 at a 1:1000 dilution (all purchased from Abcam, Cambridge, MA, USA) overnight at 4 °C. After washed with TBST three times, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Finally, the blots were analyzed using the ECL detection system (Beyotime, Shanghai, China). The band intensity was analyzed by Image-Pro Plus 6.0 software. GAPDH was used as an internal control.
Luciferase reporter assay
Dual-luciferase reporter assays were performed to verify the direct interactions between miR‐21 and the 3ʹ‐untranslated region (UTR) of PDCD4 mRNA. Briefly, 3ʹ-UTR of PDCD4 cDNA containing the predicted miR‐21 binding site was amplified by PCR and then cloned into a pGL3 vector. After the cells reached 80% confluence, the HK-2 cells were cotransfected with wild type (WT) or mutant (Mut) PDCD4 3ʹ-UTR luciferase reporter plasmids, together with control mimics or miR-21 mimics (GenePharma) by Lipofectamine 2000™ (Invitrogen). The luciferase activity was conducted after 48 h of transfection using the Dual-Luciferase Reporter Assay System (Promega, USA) following the manufacturer’s instructions. Firefly luciferase activity was normalized to Renilla luciferase activity.
Animal models of unilateral renal IRI and experimental groups
All animal experimental protocols were performed in accordance with the guidelines for the Care and Use of Laboratory Animals and approved by the Ethics Committee of Zhengzhou University People’s Hospital. Specific pathogen-free (SPF) Sprague-Dawley rats (male, weighing 180–200 g) were given free access to food and water before use in experiments. After 1 week of acclimatization, rats were randomly divided into the following 5 groups (n = 8/group): Sham, IRI, MVs, NCI-MVs, and miR-21I-MVs. A rat model of unilateral renal IRI was established. Briefly, the left renal pedicles were clamped for 45 min to reduce renal ischemia. The clamps were removed for reperfusion, and then the abdomen was closed in two layers with sutures. MVs-derived from MSCs, NCI-transfected MSCs, and miR-21I-transfected MSCs were named as MVs, NCI-MVs, and miR-21I-MVs, respectively. The rats in the IRI group were given an equal amount of vehicle. 100 μg MVs in 1 mL PBS or 1 mL PBS only was administered via tail vein immediately after reperfusion. For the sham group, rats were subjected to the same surgical procedure, except the renal pedicles were not clamped. Forty-eight hours and two weeks after reperfusion, rats were anesthetized by intraperitoneal injection of pentobarbital sodium (50 mg/kg; Sigma-Aldrich) and the renal tissues were collected for the following experiments.
TdT-mediated dUTP nick-end labeling (TUNEL) staining
Briefly, renal tissue was fixed in 4% paraformaldehyde and embedded in paraffin before being cut into 4-μm thick sections. Apoptotic cells in the renal tissues were detected by TUNEL staining using the In Situ Cell Death Detection kit (Roche Diagnostics, GmbH, Mannheim, Germany) according to the manufacturer’s protocols. The number of TUNEL-positive cells (brown) was counted from 5 fields and averaged.
Masson’s trichrome staining
Briefly, the 4-μm thick renal sections were stained with the Masson’s Trichrome Stain Kit (Solarbio, Beijing, China) to assess the level of renal fibrosis following the routine staining procedures. The percentage of Masson staining positive area was determined in 10 fields for each sample, and Image J was used for quantitative analysis.
Immunohistochemistry staining
The sections were incubated at room temperature for 15 min in 3% H2O2 to quench endogenous peroxidase activity. Sections were then treated with trypsinization to retrieve the antigen. Nonspecific binding was blocked by 5% BSA in PBS for 30 min. The sections were then incubated with primary antibodies against α-smooth muscle actin (α-SMA) (1:500; Abcam) and Ki67 (1:250; Abcam) followed by horseradish peroxide-conjugated secondary antibody using diaminobenzidine reagents as substrates and then counterstained with hematoxylin. Ki67- positive cells were counted as the number of positively-staining cells from 5 fields and averaged. The α-SMA stained area was quantitated as a percentage of the total area using Image J software.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 7 software (Graph Pad, San Diego, USA). The data are presented as the mean ± standard deviation (SD) from three independent experiments. The unpaired Student’s t-test was used to analyze differences between the two groups. One-way analysis of variance (ANOVA) was used to analyze differences among three or more groups. p < 0.05 was considered to be statistically significant.
Results
Identification of hUC-MSCs and hUC-MSCs-MVs
Flow cytometry analysis showed hUC-MSCs were positive for some surface-expressed molecules typically expressed by MSCs, such as CD44, CD90, CD29, CD105, and negative for CD34 and CD45 ()), indicating these cells belonged to high-purity MSCs. hUC-MSCs-MVs were then successfully isolated using ultracentrifugation. As shown in ), the yielded MVs were spherical or oval-shaped, and bilayered structures with the diameters of approximately 30–500 nm. Furthermore, western blot analysis showed that hUC-MSCs-MVs highly expressed MVs markers CD63 and CD9 ()). These data indicated that the obtained hUC-MSCs and hUC-MSCs-MVs confirmed to the well-established criteria.
Figure 1. Identification of hUC-MSCs and hUC-MSCs-MVs. (a) Analysis of the immunophenotype of hUC-MSCs by flow cytometry. hUC-MSCs were positive for CD44, CD90, CD29, and CD105, while negative for CD34, CD45. (b) Morphology of hUC-MSCs-MVs under a transmission electron microscope. Scale bar, 500 nm. (c) Western blot analysis on the expression of the MVs markers CD63 and CD9.
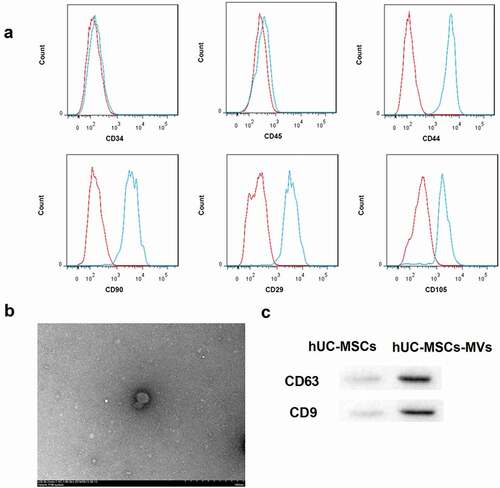
hUC-MSCs-MVs inhibited LO-induced HK-2 cell apoptosis through transferring miR-21
To investigate the role of hUC-MSCs-MVs in regulating HK-2 cell apoptosis under low oxygen (LO) condition, we co-cultured HK-2 cells with hUC-MSCs-MVs, followed by LO exposure. As expected, LO exposure led to a significant increase in the percentage of apoptotic cells in HK-2 cells ()). Consistently, the protein levels of pro-apoptotic caspase-3 and Bax were notably increased, whereas the levels of anti-apoptotic Bcl-2 were decreased in HK-2 cells after LO exposure ()). Importantly, hUC-MSCs-MVs could be absorbed by HK-2 cells ()). Furthermore, hUC-MSCs-MVs effectively rescued the LO-induced HK-2 cell apoptosis, as indicated by a decrease in cell apoptosis rate ()) and protein levels of caspase-3 and Bax, as well as an increase in protein levels of Bcl-2 ()).
Figure 2. hUC-MSCs-MVs inhibited LO-induced HK-2 cell apoptosis through transferring miR-21. (a) qRT-PCR analysis showed that miR-21 expression was downregulated in MVs derived from miR-21 inhibitor-transfected hUC-MSCs. (b) The MVs derived from FAM (green)-miR-21-transfected hUC-MSCs were labeled with Dil (red), and co-cultured with HK-2 cells (nuclei stained with DAPI, blue). The distribution and intensity of fluorescence were observed to analyze MVs uptake by HK-2 cells. Scale bar: 10 μm. (c-e) HK-2 cells were co-cultured with PBS, hUC-MSCs-MVs, MVs derived from inhibitor NC-transfected hUC-MSCs (inhibitor NC MVs), and MVs derived from miR-21 inhibitor-transfected hUC-MSCs (miR-21 inhibitor MVs), followed by exposure to low oxygen (LO; 1.5 h of hypoxia and then 24 h of reoxygenation). (c) miR-21 expression in HK-2 cells in each group was detected by qRT-PCR. (d) The apoptosis rate of HK-2 cells in each group was examined using flow cytometry after annexin V-FITC/propidium iodide (PI) staining. (e) The protein levels of apoptosis-related proteins (caspase-3, Bax, Bcl-2) in HK-2 cells were analyzed by western blot.
Simultaneously, miR-21 expression in HK-2 cells was significantly decreased by LO exposure and then elevated by hUC-MSCs-MVs ()), indicating that hUC-MSCs-MVs might transfer miR-21 to HK-2 cells. To elucidate whether hUC-MSCs-MVs rescued the LO-induced HK-2 cell apoptosis through transferring miR-21 to HK-2 cells, we performed rescue experiments. To this end, miR-21 inhibitor and inhibitor NC were transfected into hUC-MSCs, and MVs were isolated from hUC-MSCs. The yielded miR-21 inhibitor MVs and inhibitor NC MVs were then co-cultured with HK-2 cells under LO exposure. the qRT-PCR analysis showed that miR-21 expression was downregulated in miR-21 inhibitor MVs ()). Furthermore, both the red (Dil-labeled MVs) and green signals (FAM-miR-21 signals) were detected in the cytoplasm of HK-2 cells (nuclei stained with DAPI, blue) exposed to these Dil-labeled MVs under fluorescence microscopy, indicating that miR-21 could be transferred into HK-2 cells via MVs delivery ()). Of note, compared with inhibitor NC MVs, miR-21 inhibitor MVs significantly decreased the miR-21 expression ()) and facilitated HK-2 cell apoptosis under LO exposure (,)). Collectively, these data indicated that hUC-MSCs-MVs inhibited the LO-induced HK-2 cell apoptosis through transferring miR-21 to HK-2 cells.
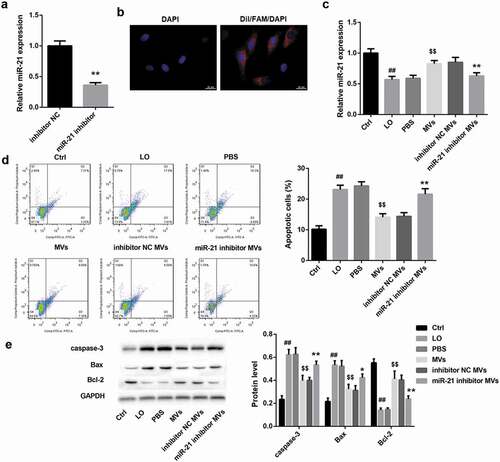
miR-21 significantly inhibited HK-2 cell apoptosis
To further clarify the role of miR-21 in regulating HK-2 cell apoptosis, we transfected HK-2 cells with miR-21 mimic, miR-21 inhibitor, and corresponding controls. Flow cytometry analysis revealed that miR-21 mimic significantly decreased the percentage of apoptotic cells in HK-2 cells ()). Furthermore, the protein levels of pro-apoptotic caspase-3 and Bax were notably decreased, whereas the levels of anti-apoptotic Bcl-2 were increased in HK-2 cells after miR-21 mimic transfection ()). In contrast, miR-21 inhibitor had the opposite effect (,)). These results indicated that miR-21 significantly inhibited HK-2 cell apoptosis.
Figure 3. miR-21 significantly inhibited HK-2 cell apoptosis. HK-2 cells were transfected with miR-21 mimic, miR-21 inhibitor, and corresponding negative controls, followed by exposure to low oxygen (LO; 1.5 h of hypoxia and then 24 h of reoxygenation). (a) The apoptosis rate of HK-2 cells in each group was examined using flow cytometry after annexin V-FITC/propidium iodide (PI) staining. (b) The protein levels of apoptosis-related proteins (caspase-3, Bax, Bcl-2) in HK-2 cells were analyzed by western blot.
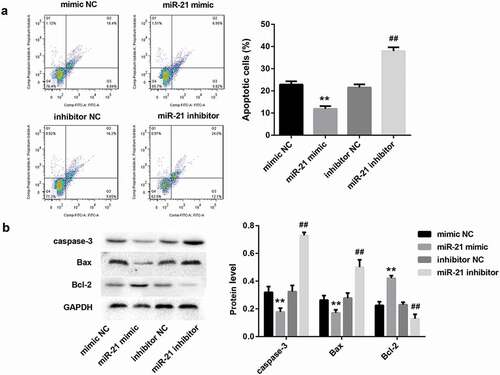
miR-21 directly targeted to PDCD4 3ʹ-UTR and negatively regulated PDCD4 expression
Our bioinformatics analysis showed that miR‐21 harbors putative target sequences at the 3ʹ-UTR of PDCD4 ()). Luciferase reporter assay showed that miR-21 mimic led to a marked decrease in luciferase activity in PDCD4-WT reporter compared with the mimic NC group, whereas had no obvious effect on the luciferase activity in PDCD4-Mut reporter, verifying the direct binding between miR-21 and PDCD4 ()). Furthermore, miR‐21 overexpression by miR-21 mimic in HK-2 cells markedly downregulated PDCD4 expression, both at mRNA ()) and protein levels ()). In contrast, miR-21 inhibitor notably increased mRNA ()) and protein levels ()) of PDCD4. These data indicated that miR-21 directly targeted to PDCD4 3ʹ-UTR and negatively regulated PDCD4 expression.
Figure 4. miR-21 directly targeted to PDCD4 3ʹ-UTR and negatively regulated PDCD4 expression(a). The putative miR-21 binding sites in PDCD4 (PDCD4-WT) or and the designed mutant sequence (PDCD4-Mut) were indicated. (b) Luciferase report assay delineated a reduction in luciferase activity of PDCD4-WT reporter after introduction of miR-21. The mRNA (c) and protein (d) levels of PDCD4 in HK-2 cells which were transfected with miR-21 mimic, miR-21 inhibitor, and corresponding negative controls.
hUC-MSCs-MVs inhibited LO-induced HK-2 cell apoptosis through inhibiting PDCD4 in HK-2 cells
The mRNA ()) and protein ()) levels of PDCD4 in HK-2 cells was significantly increased by LO exposure and then decreased by hUC-MSCs-MVs. Furthermore, miR-21 inhibitor MVs significantly increased mRNA ()) and protein ()) levels of PDCD4. These results suggested that hUC-MSCs-MVs might transfer miR-21 to HK-2 cells and then inhibited PDCD4 expression in HK-2 cells.
Figure 5. hUC-MSCs-MVs inhibited LO-induced HK-2 cell apoptosis through inhibiting PDCD4 in HK-2 cells. (a-b) HK-2 cells were co-cultured with PBS, hUC-MSCs-MVs, MVs derived from inhibitor NC-transfected hUC-MSCs (inhibitor NC MVs), and MVs derived from miR-21 inhibitor-transfected hUC-MSCs (miR-21 inhibitor MVs), followed by exposure to low oxygen (LO; 1.5 h of hypoxia and then 24 h of reoxygenation). (a) The PDCD4 mRNA in HK-2 cells in each group was detected by qRT-PCR. (b) The PDCD4 protein levels in HK-2 cells in each group were detected by western blot. (c) The apoptosis rate of HK-2 cells in each group was examined using flow cytometry after annexin V-FITC/propidium iodide (PI) staining. (d) The protein levels of apoptosis-related proteins (caspase-3, Bax, Bcl-2) in HK-2 cells was analyzed by western blot.
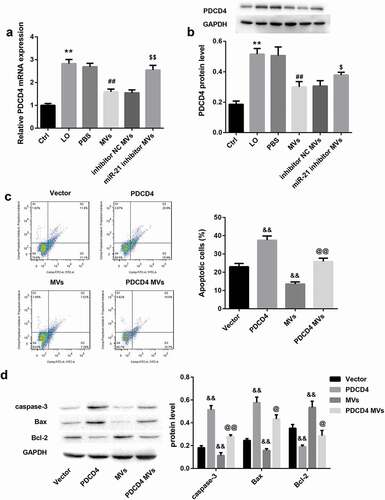
To elucidate whether inhibition of PDCD4 by miR-21 secreted by hUC-MSCs-MVs is involved in hUC-MSCs-MVs-inhibited HK-2 cell apoptosis under LO condition, we co-cultured PDCD4 overexpressing HK-2 cells and hUC-MSCs-MVs under LO exposure. PDCD4 overexpression in HK-2 cells alone significantly increased the percentage of apoptotic cells ()), increased the protein levels of caspase-3 and Bax, whereas decreased levels of Bcl-2 in HK-2 cells under LO exposure ()). These data indicated that PDCD4 overexpression facilitated HK-2 cell apoptosis under LO condition. In contrast, hUC-MSCs-MVs treatment notably inhibited HK-2 cell apoptosis (,)). Importantly, PDCD4 overexpression in HK-2 cells effectively abrogated the hUC-MSCs-MVs-inhibited HK-2 cell apoptosis (,)). Furthermore, hUC-MSCs-MVs treatment also abolished the PDCD4 overexpression-induced HK-2 cell apoptosis (,)). Taken together, these data indicated that hUC-MSCs-MVs inhibited the LO-induced HK-2 cell apoptosis through inhibiting PDCD4 expression in HK-2 cells via transferring miR-21 to HK-2 cells.
MSC-MVs ameliorated rat renal IRI via transferring miR-21
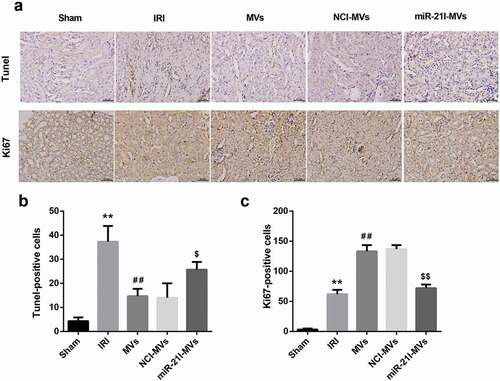
To confirm the in vivo role of MSC-MVs and miR-21 in renal IRI, we established a rat model of IRI and treated these IRI rats with vehicle, MVs, NCI-MVs, and miR-21I-MVs. TUNEL staining showed that the number of TUNEL-positive cells was increased following IRI, but was attenuated by MSC-MVs administration for 48 h. However, compared with the NCI-MVs group, the renal tissue cell apoptosis was enhanced in the miR-21I-MVs group (,)). Furthermore, Ki67 staining demonstrated that the number of Ki67-positive cells was increased at 48 h after IRI and further enhanced by MSC-MVs administration. However, administration of miR-21I-MVs attenuated the promoting effect of MSC-MVs on the number of Ki67-positive cells (,)).
Figure 6. Effects of MSC-MVs administration on the renal cell apoptosis and proliferation. (a) Representative images of TUNEL and Ki67 immunostaining in the injured kidneys at 48 h after reperfusion treated with MSC-MVs (Scale bar = 25 μm). (b) Quantification of TUNEL-positive cells in the kidney sections after reperfusion with MSC-MVs. (c) Quantification of Ki67-positive cells in the kidney sections after reperfusion with MSC-MVs. N = 8 in each group.
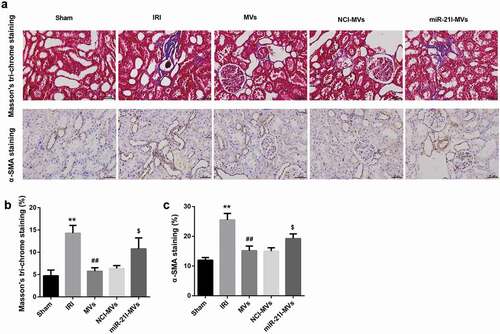
Moreover, Masson’s trichrome staining at 2-week post-IRI showed that MSC-MVs could somewhat ameliorate the degree of renal fibrosis induced by IRI. However, miR-21I-MVs administration attenuated the anti-fibrosis effect of MSC-MVs on renal tissues (,)). Furthermore, the number of α-SMA-positive cells, an indicator of epithelial-mesenchymal transition, was increased at 2 week after IRI but then decreased by MSC-MVs administration. However, miR-21I-MVs administration increased the number of α-SMA-positive cells when compared with the NCI-MVs group (,)).
Figure 7. Effects of MSC-MVs administration on the renal fibrosis. (a) Representative images of Masson’s trichrome staining and α-SMA immunostaining in the injured kidneys at 2 week after reperfusion treated with MSC-MVs (Scale bar = 25 μm). (b) Quantification of percentage of Masson staining positive area in the kidney sections after reperfusion with MSC-MVs. (c) Quantification of α-SMA-positive cells in the kidney sections after reperfusion with MSC-MVs. N = 8 in each group.
Discussion
MSCs, derived from various sources such as bone marrow or adipose, are now under extensive investigation as a potential therapy for AKI. Various preclinical studies indicated the beneficial effects of MSCs in alleviating renal injury and accelerating tissue repair by a paracrine or endocrine mechanism. Intriguingly, the efficacy of MSC-derived MVs for kidney repair following AKI is similar to that of cells [Citation15–Citation17], which indicates that MVs are critical mediators. MVs, which shuttle selected patterns of RNA, are regarded as vehicles for genetic information exchange between cells [Citation5]. MVs are small vesicles released by cells bearing the surface antigens characteristic of the original cells. They can also enter the target cells through specific receptor-ligand interactions and transfer miRNAs, mRNA, proteins and other bioactive materials [Citation3,Citation5]. Our previous study demonstrated that administration of hUC-MSCs-MVs could ameliorate renal IRI in rats, as evidenced by mitigated renal cell apoptosis, decreased collagen deposition, improved renal function, and abrogated renal fibrosis [Citation6–Citation9].
Although the mechanisms underlying the therapeutic effects of hUC-MSCs-MVs involve anti-inflammatory and anti-oxidative properties of MVs [Citation6,Citation7], the underlying mechanism still remains incompletely understood. In a previous report from our laboratory, hUC-MSC-MVs might induce synthesis of hepatocyte growth factor (HGF) via transferring HGF mRNA to damaged tubular cells, thereby facilitating tubular cell dedifferentiation and regeneration [Citation9]. This indicates that in the AKI context, hUC-MSC-MVs can regulate tubular cell viability by transferring certain RNA to damaged tubular cells. A recent study indicated that [Citation11] miR-21 protects against renal IRI-induced AKI by preventing epithelial cell apoptosis and inhibiting dendritic cell maturation. Tubular epithelial cell apoptosis contributes importantly to renal IRI [Citation13]. In the present study, we observed that hUC-MSCs-MVs elevated miR-21 expression and inhibited LO-induced cell apoptosis in HK-2 cells. However, miR-21 inhibition in hUC-MSCs decreased miR-21 expression in both hUC-MSCs-MVs and HK-2 cells, concomitantly facilitated HK-2 cell apoptosis. Further assays showed that miR-21 mimic led to a notable inhibition of HK-2 cell apoptosis, whereas miR-21 inhibitor promoted HK-2 cell apoptosis. These findings indicated that hUC-MSCs-MVs inhibited LO-induced HK-2 cell apoptosis through transferring miR-21 to HK-2 cells. Further in vivo assay showed that the beneficial effect of MSC-MVs on rat renal IRI was partly eliminated when miR-21 was knocked down in MSCs. In the present study, we confirmed the hypothesis that the therapeutic effects of MSC-MVs in renal IRI were also partly mediated by miR-21.
miRNAs have come into focus as powerful regulators of gene expression during IRI. miR-21, one of the most extensively studied miRNAs, is importantly implicated in several pathophysiological processes related to IRI, such as apoptosis and inflammation as well as angiogenesis [Citation13,Citation18,Citation19]. Existing evidence indicates that miR-21 exerts significant functional protection in renal IRI. XU et al. [Citation13] showed that preconditioning-induced upregulation of miR-21 contributes to the protection against subsequent renal IRI through the targeting of pro-apoptotic genes such as PDCD4 and interactions between miR-21 and hypoxia-inducible factor. Afterward, Hu et al [Citation20] further confirmed that miR-21 is endowed with anti-apoptotic properties by suppressing the expression of PDCD4 gene and active caspase 3/8 fragments and attenuated renal IRI. Consistent with this, our results confirmed that miR-21 directly targeted to PDCD4 3ʹ-UTR and negatively regulated PDCD4 expression in HK-2 cells. In addition, rescue experiments showed that PDCD4 overexpression in HK-2 cells effectively abrogated the hUC-MSCs-MVs-inhibited HK-2 cell apoptosis under hypoxia injury. Our results indicate the importance of miR-21 targeting PDCD4 in hUC-MSCs-MVs-mediated protection of renal IRI.
In summary, our findings demonstrated that hUC-MSCs-MVs inhibit HK-2 cell apoptosis under hypoxia injury and the delivery of miR-21 via hUC-MSCs-MVs and the subsequent suppression of PDCD4 may be a potential mechanism. This study provides a novel mechanism for the therapeutic effects of MSCs-MVs on renal IRI and AKI.
Disclosure statement
The authors declare no conflicts of interest.
Additional information
Funding
References
- Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. 2011;17(11):1391–1401.
- Malek M, Nematbakhsh M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J Renal Injury Prev. 2015;4(2):20–27.
- Bruno S, Grange C, Deregibus MC, et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol. 2009;20(5):1053–1067.
- Gatti S, Bruno S, Deregibus MC, et al. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol Dialysis Trans. 2011;26(5):1474–1483.
- Camussi G, Deregibus MC, Bruno S, et al. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010;78(9):838–848.
- Zou X, Zhang G, Cheng Z, et al. Microvesicles derived from human Wharton’s Jelly mesenchymal stromal cells ameliorate renal ischemia-reperfusion injury in rats by suppressing CX3CL1. Stem Cell Res Ther. 2014;5(2):40.
- Zhang G, Zou X, Miao S, et al. The anti-oxidative role of micro-vesicles derived from human Wharton-Jelly mesenchymal stromal cells through NOX2/gp91(phox) suppression in alleviating renal ischemia-reperfusion injury in rats. PloS One. 2014;9(3):e92129.
- Chen W, Yan Y, Song C, et al. Microvesicles derived from human Wharton’s Jelly mesenchymal stem cells ameliorate ischemia-reperfusion-induced renal fibrosis by releasing from G2/M cell cycle arrest. Biochem J. 2017;474(24):4207–4218.
- Ju GQ, Cheng J, Zhong L, et al. Microvesicles derived from human umbilical cord mesenchymal stem cells facilitate tubular epithelial cell dedifferentiation and growth via hepatocyte growth factor induction. PloS One. 2015;10(3):e0121534.
- Banaei S. Novel role of microRNAs in renal ischemia reperfusion injury. Ren Fail. 2015;37(7):1073–1079.
- Song N, Zhang T, Xu X, et al. miR-21 protects against ischemia/reperfusion-induced acute kidney injury by preventing epithelial cell apoptosis and inhibiting dendritic cell maturation. Front Physiol. 2018;9:790.
- Wang K, Bei WJ, Liu YH, et al. miR-21 attenuates contrastinduced renal cell apoptosis by targeting PDCD4. Mol Med Rep. 2017;16(5):6757–6763.
- Xu X, Kriegel AJ, Liu Y, et al. Delayed ischemic preconditioning contributes to renal protection by upregulation of miR-21. Kidney Int. 2012;82(11):1167–1175.
- Wu S, Ju GQ, Du T, et al. Microvesicles derived from human umbilical cord Wharton’s jelly mesenchymal stem cells attenuate bladder tumor cell growth in vitro and in vivo. PloS One. 2013;8(4):e61366.
- Camussi G. Stem cell reviews and reports: microenvironment and extracellular microvesicles section. Stem Cell Rev. 2017;13(1):4.
- Nawaz M, Fatima F, Vallabhaneni KC, et al. Extracellular vesicles: evolving factors in stem cell biology. Stem Cells Int. 2016;2016:1073140.
- Du T, Zhu YJ. The regulation of inflammatory mediators in acute kidney injury via exogenous mesenchymal stem cells. Mediators Inflamm. 2014;2014:261697.
- Li Z, Deng X, Kang Z, et al. Elevation of miR-21, through targeting MKK3, may be involved in ischemia pretreatment protection from ischemia-reperfusion induced kidney injury. J Nephrol. 2016;29(1):27–36.
- Jiao X, Xu X, Fang Y, et al. miR-21 contributes to renal protection by targeting prolyl hydroxylase domain protein 2 in delayed ischaemic preconditioning. Nephrology (Carlton, Vic). 2017;22(5):366–373.
- Hu H, Jiang W, Xi X, et al. MicroRNA-21 attenuates renal ischemia reperfusion injury via targeting caspase signaling in mice. Am J Nephrol. 2014;40(3):215–223.