
ABSTRACT
The 3-hydroxy-3-methylglutaryl reductase degradation (HRD1) is an E3 ubiquitin ligase that can preserve heart structure and function, but its role in endothelial dysfunction and atherosclerosis (AS) is unclear. The aim of this study was to explore the role and biological function of HRD1 in AS. HRD1 expression was significantly decreased in atherosclerotic intima and ox-LDL led to a decrease of HRD1 level in endothelial cells (ECs). Forced expression of HRD1 inhibited the endothelial apoptosis induced by ox-LDL. The transcription factor KLF2 specifically bound to the HRD1 promoter and positively regulated HRD1 expression. KLF2 up-regulation could reverse the decrease of HRD1 level in ECs treated with ox-LDL. Further analysis showed that HRD1 interacted with LOX-1 and promoted ubiquitination and degradation of LOX-1 by the proteasome. Deletion of LOX-1 attenuated the ECs apoptosis induced by HRD1 downregulation. Pravastatin, which protected EC from damage via a KLF2-dependent mechanism, could dose-dependently enhanced HRD1 expression in EC exposed to ox-LDL. Interestingly, interference of HRD1 abolished the cytoprotective effect of pravastatin. Collectively, our data indicate that decreased HRD1 expression leads to apoptosis of ECs and restoration of HRD1 expression could represent a novel strategy for human AS therapy.
Introduction
Atherosclerosis (AS) is a hyperlipidemia-induced chronic inflammatory vascular disease and is now one of the leading causes of morbidity and mortality worldwide [Citation1]. Many types of cells, including endothelial cell, vascular smooth muscle cells, leukocytes, etc., participate in the development of atherosclerotic lesions [Citation2]. Endothelial cells (ECs) dysfunction, e.g., apoptosis, is the initial and crucial step in the pathogenesis of AS [Citation3]. Therefore, research on the mechanism of EC dysfunction has opened up a useful new avenue for the prevention of AS [Citation4].
Oxidized low-density lipoproteins (oxLDL) are thought to be involved in inflammatory and vascular pathologies, such as the development and progression of retinitis pigmentosa and AS [Citation5,Citation6]. The binding and uptake of ox-LDL is mediated by several receptors including SRA, SRBI, CD36, and lectin-like oxidized LDL receptor (LOX-1). Among these, LOX-1 is the major receptor of ox-LDL in ECs [Citation7,Citation8]. LOX-1 expression and activity can be enhanced by ox-LDL, which then contributes to vascular endothelial dysfunction [Citation9–Citation11]. Overexpression of LOX-1 in ECs resulted in vascular lipid deposition of ApoE−/- mice [Citation12], whereas knockout of LOX-1 resisted to the development and progression of AS and improved endothelial function in vivo [Citation13,Citation14]. Thus, inhibition of LOX-1 expression is viewed as an attractive therapeutic target for the treatment of human atherosclerotic diseases [Citation15]. However, the molecular mechanism regulating LOX-1 expression in ECs is not well defined.
The zinc finger transcription factor Krüppel-like factor 2 (KLF2), which is specifically expressed in ECs of vasculature, plays a pivotal role in regulating endothelial function, hemostasis, inflammatory response and angiogenesis [Citation16]. Suppression of KLF2 expression may be involved in the endothelial activation triggered by many stimuli, including cytokines, lipopolysaccharide, thrombin, and ox-LDL [Citation17]. Recent research revealed that the absence of in KLF2 increased atherosclerotic lesion formation in ApoE−/- Mice [Citation18], whereas overexpression of KLF2 in ECs exerted an anti-thrombotic effect by regulating multiple factors in the thrombotic pathway [Citation19]. Therefore, KLF2 has been regarded as part of the endothelial “atheroprotective phenotype” [Citation20].
The 3-hydroxy-3-methylglutaryl reductase degradation (HRD1) protein is an ER-associated degradation (ERAD)-associated E3 ubiquitin ligase, which is involved in the clearance of of misfolded proteins from the ER and controlling the availability of specific proteins, such as p53, Nrf2, and IGF-1 R [Citation21–Citation24]. Recently, Doroudgar et al have reported the protective effect of HRD1 on heart structure and function [Citation25]. We found that HRD1 was significantly downregulated in human atherosclerotic intimas. We also found ox-LDL could decrease HRD1 expression in human umbilical vein endothelial cells. We hypothesized that HRD1 might play a role in endothelial dysfunction and atherosclerosis.
Materials and methods
Human carotid lesion samples
Human atherosclerotic lesion specimens were obtained from 30 patients undergoing carotid endarterectomy at the Second Affiliated Hospital of Nanjing Medical University, Nanjing, China, between 2014 and 2018. Histologically normal internal mammary arteries (n = 24) from patients undergoing aortocoronary bypass surgery were used as controls. Exclusion criteria were: severely impaired renal function, thromboembolism, advanced liver disease, acute infection, and administration of anti-platelet, anticoagulant and anti-inflammatory drugs therapy in the previous two weeks. There were no significant difference in age, gender, body mass index (BMI), hypertension, obesity, hyperlipoproteinemia, diabetes, heart rate (HR), and ejection fraction (EF) between atherosclerotic patients and controls. One-half of each sample was immediately stored in RNAlater (Life Technologies), and the other half was fixed in paraformaldehyde. The human arterial vessel study was approved by the ethical committees at Nanjing Medical University, Nanjing, China. All the patients provided written consent for the collection and use of samples in this study.
Immunofluorescence staining
Coronary arterioles were fixed in 4% paraformaldehyde and sectioned at 5 μm onto glass slides. Samples were blocked in 5% (w/v) BSA for 1 h, then, incubated at 4°C overnight with the following primary antibodies against HRD1 (1:50, Abgent, Cat. no. AP2184A), CD31 (1: 50, Cell Signaling Technology, Cat. no. 3528) or LOX-1 (1:50, abcam, Cat. no. ab81709). After washing, specimens were incubated with secondary fluorescent antibodies for 1 h at room temperature (RT) against the light. Nuclei were stained with DAPI and images were acquired with a laser scanning microscope (Olympus).
Cell cultures
Human umbilical vein endothelial cells (HUVECs) were isolated using collagenase, and cultured in 25 mmol/l HEPES-buffered M199, containing 10% FBS, 2 mmol/l glutamine, 0.75 g/l human epidermal growth factor, and 75 mg/l hydrocortisone, as previously described [Citation26].
Small interfering RNA transfection and adenoviruse infection
For small interfering RNA (siRNA) transfection, HUVECs was transfected with 20 nmol/l si-HRD1 or si-KLF2 (Ribobio, Guangzhou, China) into HUVECs using Lipofectamine 2000. The recombinant adenoviral vector Ad-HRD1 or Ad-KLF2 were generated as previously described [Citation24]. HUVECs were infected with Ad-HRD1 or Ad-KLF2 in diluted medium at a multiplicity of infection (MOI) of 50.
Co-Immunoprecipitation (Co-IP) assay and Western blot analysis
HUVECs (5 × 107) were lysed in 1 ml lysis buffer and Co-IP was performed by standard procedures, as previously described [Citation24]. After centrifugation, 10% supernatants were used as input, the remaining were immunoprecipitated with protein A/G agarose beads, IgG (negative control) or antibody for target proteins at 4°C overnight. The pellets were washed 3 times in wash buffer and resuspended in 2× SDS, and then subjected to western blot. Western blot was performed as previously reported [Citation27]. Antibody against LOX-1 (Cat. no. 3659 R-100) was from Biovision (Biovision Inc, Milpitas, CA, USA). Antibodies against myc-Tag (Cat. no. 2276), HA-Tag (Cat. no. 3724) and cleaved caspase-3 (Cat. no. 9661) were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies against HRD1 (Cat. no. ABS1623) and KLF2 (Cat. no. SAB2108355) were obtained from Sigma Aldrich (St. Louis, MO, USA). Antibody against β-Actin (Cat. no. sc-47,778) was acquired from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The intensity of protein blots was analyzed by Image J software.
Cell viability assay
Cell viability was measured using WST-1 assays. Briefly, HUVECs (4 × 104cells/well) were seeded in 48-well dishes. Then, each well was supplemented with 20 ml WST-1 (Roche, Nonenwald, Germany) and incubated for 3 h. The absorbance of samples was detected with a spectrophotometer reader.
Cell apoptosis assay
Apoptotic cells were evaluated by flow cytometry analysis using an Annexin V-FITC/PI staining kit (Invitrogen, USA). After washes with cold PBS, the cells were stained with Annexin V-FITC/PI at RT in darkness for 15 min. Apoptotic cells were then evaluated by gating PI and Annexin V-positive cells on a FACSCalibur instrument (BD Bio-sciences).
Real-time PCR assay
The mRNA level of target genes was measured by SYBR green real-time PCR kit (TakaRa, RR420 A) using a LightCycler480 II Sequence Detection System (Roche, Basel, Switzerland). Primers used to identify HRD1 were: forward, 5ʹ-AAC CCC TGG GAC AAC AAG G-3ʹ and reverse, 5ʹ-GCG AGA CAT GAT GGC ATC TG-3ʹ. β-Actin was used as an internal control: forward, 5ʹ-GCA AGT GCT TCT AGG CGG AC-3ʹ and reverse, 5ʹ-AAG AAA GGG TGT AAA ACG CAG C-3ʹ.
ChIP-qPCR assay
The ChIP assay was performed using a ChIP assay kit following the manufacturer’s protocol. HUVECs (2 × 107) were harvested and fixed in 1% formaldehyde for 10 min at RT. The cells were then collected in lysis buffer and the chromatin was sonicated. The chromatin was incubated overnight at 4°C with anti-KLF2 antibody or with IgG (negative control). ChIP-qPCR assay was used to examine whether ox-LDL could decrease the binding activity of KLF2 on the promoter of HRD1. The fold difference between the KLF2 antibody-immunoprecipitated samples and those immunoprecipitated with IgG was calculated using 2−ΔΔCt.
Statistical analysis
Statistical analyzes were performed using statistical analysis software SPSS 19.0. All the experiments above were performed independently at least 3 times. Data were expressed as the mean ± SD. Analysis of variance (ANOVA) was used to determine the statistical differences among the groups followed by Tukey’s post-hoc test as appropriate. P values of less than 0.05 and 0.01 are provided in the figures. A P value < 0.05 or p < 0.01 was considered statistically significant.
Results
HRD1 expression is decreased in human atherosclerotic intima and ECs treated with ox-LDL
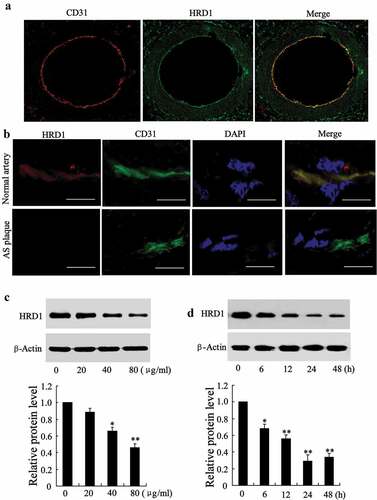
HRD1 staining, along with immunofluorescent staining of the endothelial marker CD31 (red) ()), indicated that HRD1 was primarily localized to the endothelial layer of coronary arterioles. To examine whether HRD1 expression was altered atherosclerotic intima, HRD1 expression was investigated in normal and atherosclerotic segments of morphologically mapped aortic walls. Reduced endothelial expression of HRD1 in the atherosclerotic vessel was identified by dual immunostaining of HRD1 and the endothelial marker CD31 ()). Next, we treated HUVECs with ox-LDL (0, 20, 40 and 80 μg/ml) for 24 h. The ox-LDL treatment inhibited HRD1 expression in a dose-dependent manner (40 μg/ml ox-LDL, p < 0.05; 80 μg/ml ox-LDL, p < 0.01) ()) and time-dependent manner (6 h, p < 0.05; 12 h, 24 h and 48 h, p < 0.01) ()).
Figure 1. HRD1 expression is decreased in human atherosclerotic intima and ECs treated with ox-LDL. (a) Immunofluorescence staining for HRD1 (green) and CD31 (red) in normal arterial walls. Original magnification, ×100. (b) Immunostaining of HRD1 (red) and the endothelial marker CD31 (green) in normal arterial intima and atherosclerotic arterial intima. Bar = 20 μm. (c) Human umbilical vein endothelial cells (HUVECs) were treated with ox-LDL (0, 20, 40 and 80 μg/ml) for 24 h, followed by measurement of the protein levels of HRD1 and LOX-1. (d) HUVECs were treated with ox-LDL (80 μg/ml) for 0, 6, 12, 24 or 48 h, followed by measurements of the protein levels of HRD1 and LOX-1. Values are means ± SD and are representative of three individual experiments. *P < 0.05 and **P < 0.01, compared to 0 μg/ml or 0 h.
Ox-LDL-induced ECs apoptosis can be attenuated by HRD1 overexpression
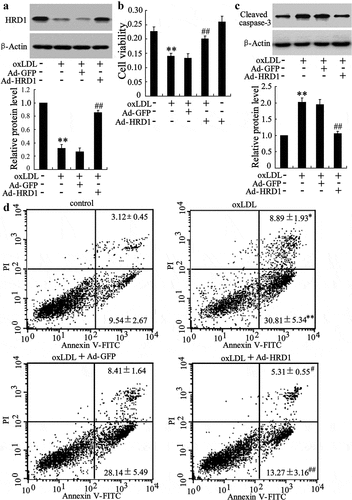
The finding that HRD1 expression was significantly decreased by ox-LDL stimulated us to investigate the effect of HRD1 on ECs viability and apoptosis. We found that HRD1 protein levels were significantly elevated in HUVECs after infected with Ad-HRD1 (p < 0.01) ()). The treatment of ox-LDL resulted in a clear inhibition of growth of HUVECs, which could be abolished by HRD1 overexpression (p < 0.01) ()). Besides, HRD1 upregulation could abolish the increase of the cleaved fragment of caspase-3 in HUVECs exposed to ox-LDL ()). When HUVECs were exposed to oxLDL, the percentage of apoptotic cells increased significantly compared with control (early apoptosis: oxLDL, 30.81 ± 5.34% vs. control, 9.54 ± 2.67%, p < 0.01; late apoptosis: oxLDL, 8.89 ± 1.93% vs. control, 3.12 ± 0.45%, p < 0.05), in agreement with a previous report [Citation24]. Overexpression of HRD1 prevented ox-LDL-induced ECs apoptosis (early apoptosis: oxLDL+Ad-HRD1, 13.27 ± 3.16% vs. oxLDL+Ad-GFP, 28.14 ± 5.49%, p < 0.01; late apoptosis: oxLDL+Ad-HRD1, 5.31 ± 0.55% vs. oxLDL+Ad-GFP, 8.41 ± 1.64%, p < 0.05) ()).
Figure 2. Ox-LDL-induced apoptosis of endothelial cells can be attenuated by HRD1 overexpression. HUVECs were transfected with Ad-GFP or Ad-HRD1 for 24 h, followed by exposure to ox-LDL (80 μg/ml) for 48 h. HRD1 expression (a), cell viability (b), cleaved caspase-3 levels (c) and HUVEC apoptosis (d) were then measured. Values are means ± SD and are representative of three individual experiments. *P < 0.05 and **P < 0.01, compared to control. #P < 0.05 and ##P < 0.01, compared to ox-LDL + Ad-GFP.
HRD1 expression is regulated by KLF2 in ECs treated with ox-LDL
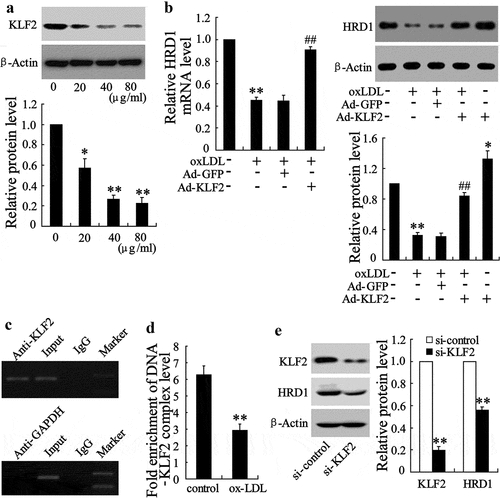
The Genomatix databases predicted that KLF2 could bind to the HRD1 gene promoter. We explored the possible role of KLF2 in inhibition of HRD1 expression in ECs treated with ox-LDL. KLF2 expression was dramatically decreased in HUVECs exposed to ox-LDL (20 μg/ml ox-LDL, p < 0.05; 40 or 80 μg/ml ox-LDL, p < 0.01) ()). Importantly, overexpression of KLF2 could reverse ox-LDL-induced decrease of HRD1 expression (p < 0.01) ()). A specific association of KLF2 with the HRD1 promoter was confirmed by ChIP assays ()). In addition, ox-LDL treatment decreased the activity of KLF2 binding to the HRD1 promoter (p < 0.01) ()), and downregulation of KLF2 clearly reduced HRD1 expression (p < 0.01) ()). These results indicated that inhibition of KLF2 is responsible for the downregulation of HRD1 expression in ECs treated with ox-LDL.
Figure 3. HRD1 expression is regulated by KLF2 in endothelial cells treated with ox-LDL. (a) Human umbilical vein endothelial cells (HUVECs) were treated with ox-LDL (0, 20, 40 and 80 μg/ml) for 24 h, followed by measurement of the KLF2 expression. (b) HUVECs were transfected with Ad-GFP or Ad-KLF2 for 24 h followed by exposure to ox-LDL (80 μg/ml) for 24 h and measurement of HRD1 mRNA and protein levels. (c) KLF2 bound to the HRD1 promoter in HUVECs in a ChIP analysis. (d) ChIP-qPCR analysis was performed to measure the capacity of KLF2 binding to the HRD1 promoter in HUVECs treated with ox-LDL. (e) HUVECs were transfected with KLF2 siRNA (si-KLF2) for 24 h, followed by measurement of KLF2 and HRD1 expression. Values are means ± SD and are representative of three individual experiments. *P < 0.05 and **P < 0.01, compared to control. ##P < 0.01, compared to ox-LDL + Ad-GFP.
HRD1 promotes LOX-1 ubiquitination for degradation
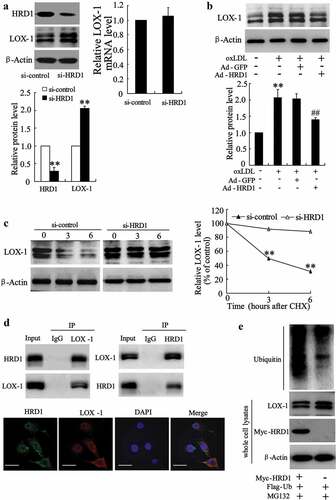
Significant increases in LOX-1 expression and activity have been reported in ECs treated with ox-LDL [Citation8]. We observed a negative correlation between the LOX-1 and HRD1 expression levels in ECs treated with ox-LDL (data not shown), indicating a potential relationship between LOX-1 and HRD1. Immunoblot results revealed accumulation of LOX-1 in HRD1-knock down ECs but no significant change was seen in its mRNA levels (p < 0.01) ()). Overexpression of HRD1 clearly attenuated the increase of LOX-1 protein level induced by ox-LDL (p < 0.01) ()). Moreover, HRD1 knockdown resulted in inhibition of LOX-1 degradation in HUVECs treated with cycloheximide (CHX) (p < 0.01) ()). The results of co-IP and Immunofluorescence staining demonstrated that HRD1 could interact with LOX-1 ()). In addition, HRD1 overexpression resulted in an increase in LOX-1 ubiquitination but a decrease of LOX-1 protein level in HUVECs ()). These results suggest that HRD1 serves as an E3 ligase that promotes LOX-1 ubiquitination for degradation by the proteasome. However, we also found that upregulation of expression of a dominant-negative HRD1 mutant (C291 S) had no effect on LOX-1 expression in HUVECs treated with ox-LDL (data not shown). These results indicated that HRD1-mediated LOX-1 degradation was dependent on HRD1 enzyme activity.
Figure 4. HRD1 promotes LOX-1 ubiquitination for degradation. (a) The expression of HRD1 and LOX-1 in si-HRD1 transfected human umbilical vein endothelial cells (HUVECs) was measured by immunoblotting. (b) HUVECs were transfected with Ad-GFP or Ad-HRD1 for 24 h, followed by exposure to ox-LDL (80 μg/ml) for 24 h and measurement of LOX-1 expression. (c) HUVECs were transfected with si-HRD1 or si-control for 48 h, followed by exposure to cycloheximide (CHX 50 mg/ml) for 0, 3, or 6 h and measurement of the LOX-1 protein levels in whole cell lysates by immunoblotting. The intensity of the LOX-1 protein bands was analyzed by densitometry, after normalization to the corresponding β-Actin level. (d) HUVECs were pretreated with MG132 (10 μM) for 6 h, followed by determination of endogenous protein-protein interactions between HRD1 and LOX-1 by immunoprecipitation (IP) with HRD1 or LOX-1 antibodies and subsequent immunoblotting. IgG was used as a negative control for IP. HUVECs were stained with Hrd1 (green) and LOX-1 (red). HRD1 and LOX-1 merged appear as orange/yellow. Bar = 10 μm. (e) Ubiquitination of LOX-1 was induced by HRD1. Flag-ubiquitin was coexpressed in HUVECs with myc-HRD1 or vector control with treatment with MG132 (10 μmol/l) for 6 h. Ubiquitinated LOX-1 protein was immunoprecipitated using Flag-Tag antibody and further detected with Anti-LOX-1 antibody. The endogenous levels of LOX-1 and myc-HRD1 in the whole cell lysates were examined by anti-LOX-1 and anti-myc antibodies. Values are means ± SD and are representative of three individual experiments. **P < 0.01, compared to si-control. ##P < 0.01, compared to ox-LDL + Ad-GFP.
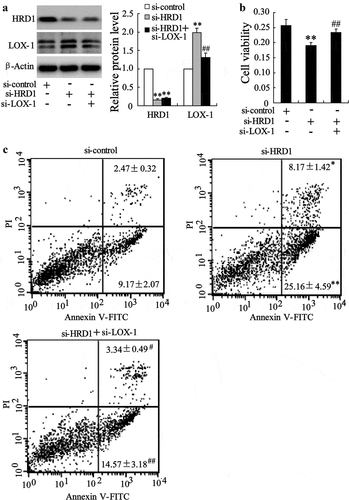
LOX-1 is required for the EC apoptosis induced by HRD1 downregulation
We also measured the effect of HRD1 on EC apoptosis. Transfection of si-HRD1 or si-LOX-1 could effectively downregulate the expression of HRD1 and LOX-1, respectively (p < 0.01) ()). Downregulation of HRD1 inhibited the viability of HUVECs, which could be reversed by deletion of LOX-1 (p < 0.01) ()). In addition, knockdown of HRD1 significantly increased the percentage of apoptotic cells (early apoptosis: si-HRD1, 25.16 ± 4.59% vs. si-control, 9.17 ± 2.07%, p < 0.01; late apoptosis: si-HRD1, 8.17 ± 1.42% vs. si-control, 2.47 ± 0.32%, p < 0.05). Overexpression of HRD1 prevented ox-LDL-induced ECs apoptosis. Knockdown of LOX-1 abolished the increase of HUVEC apoptosis induced by HRD1 downregulation (early apoptosis: si-HRD1+ si-LOX-1, 14.57 ± 3.18% vs. si-HRD1, p < 0.01; late apoptosis: si-HRD1+ si-LOX-1, 3.34 ± 0.49% vs. si-HRD1, p < 0.05) ()).
Figure 5. LOX-1 is required for endothelial cell apoptosis induced by HRD1 downregulation. Human umbilical vein endothelial cells (HUVECs) were transfected with si-HRD1 or si-HRD1 plus si-LOX-1 for 48 h, followed by measurement of HRD1 and LOX-1 expression (a), cell viability (b), and HUVEC apoptosis (c). Values are means ± SD and are representative of three individual experiments. *P < 0.05 and **P < 0.01, compared to si-control. #P < 0.05 and ##P < 0.01, compared to si-HRD1.
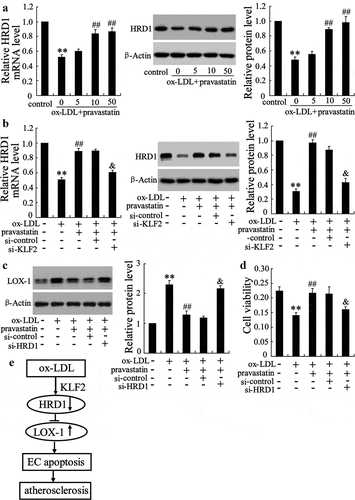
Pravastatin stimulated HRD1 expression via a KLF2-dependent mechanism
We performed a further exploration of the regulatory effect of KLF2 on HRD1 expression using various concentrations of pravastatin (a HMG-CoA reductase inhibitors), which could stimulate KLF2 expression to prevent ECs dysfunction. As shown in ), ox-LDL treatment significantly decreased the expression of HRD1, and this effect was reversed by pravastatin in a dose-dependent manner (p < 0.01). Remarkably, interference with KLF2 abrogated the effect of pravastatin on HRD1 expression (p < 0.01) ()). Furthermore, specific inhibition of HRD1 significantly attenuated pravastatin-mediated LOX-1 downregulation (p < 0.01) ()) and cytoprotection against ox-LDL-induced EC damage (p < 0.01) ()). Together, these results identified HRD1 as a novel prognostic or progression marker for AS, as depicted in ). Downregulation of KLF2 was responsible for the decrease in HRD1 levels observed in ox-LDL-treated ECs, which directly upregulated LOX-1 expression. Following the elevation of LOX-1 expression, EC apoptosis was induced, which resulted in AS development.
Figure 6. Pravastatin stimulated HRD1 expression via a KLF2-dependent mechanism. (a) After pretreatment with pravastatin (0, 5, 10 or 50 mmol/L) for 2 h, human umbilical vein endothelial cells (HUVECs) were treated with ox-LDL for additional 24 h, followed by measurement of HRD1 mRNA and protein levels. (b) HUVECs were transfected with si-KLF2 for 24 h and then treated with ox-LDL and/or pravastatin, followed by measurement of HRD1 mRNA and protein levels. (c) HUVECs were transfected with si-HRD1 for 24 h, followed by treatment with ox-LDL and/or pravastatin and measurement of LOX-1 protein levels. (d) HUVECs were transfected with si-HRD1 for 24 h, followed by treatment with ox-LDL and/or pravastatin and measurement of cell viability. (e) Diagram depicting the role of HRD1 in EC apoptosis and AS development. Values are means ± SD and are representative of three individual experiments. **P < 0.01, compared to control. ##P < 0.01, compared to ox-LDL. &P < 0.01, compared to ox-LDL+pravastatin+ si-control.
Discussion
The present study demonstrated a protective role for HRD1, an E3 ubiquitin ligase, against endothelial dysfunction and AS. These effects of HRD1 were mediated by promoting the ubiquitination of LOX-1 for degradation. HRD1 expression levels were downregulated in ECs treated with ox-LDL due to the inhibition of KLF2.
EC dysfunction is the initial and crucial step in the pathogenesis of AS [Citation28]. We observed a significantly lower HRD1 expression in the atherosclerotic intima than in normal intima and an inhibition of HRD1 expression in ECs treated with ox-LDL. Thus, we hypothesized that HRD1 was involved in EC dysfunction and AS. We used loss-of-function and gain-of-function strategies to demonstrate that HRD1 acts as an anti-atherosclerosis regulator. Upregulation of HRD1 effectively alleviates EC apoptosis induced by ox-LDL, whereas HRD1 deletion increased EC apoptosis. These findings indicated that HRD1 expression was essential for preventing EC injury and death and implicated HRD1 as a novel future prognostic or progression marker for AS.
The increase in LOX-1 expression and activity contributes to vascular endothelial dysfunction [Citation29]. Downregulation of LOX-1 expression is viewed as an attractive therapeutic target for the treatment of human atherosclerotic diseases [Citation15]. In the present study, we demonstrated that HRD1 inhibited LOX-1 expression at the post-transcriptional level. Further study showed that HRD1could interact with LOX-1 by acting as a ubiquitin E3 ligase that targets LOX-1 for degradation via the ubiquitin proteasome pathway. The inversed correlation between LOX-1 and HRD1 in the atherosclerotic intima provided clinical evidence of HRD1 regulation of LOX-1 expression.
The present study demonstrated that HRD1 is a transcriptional target of KLF2, which has been regarded as the part of the endothelial “atheroprotective phenotype” [Citation30]. Our results showed that KLF2 could directly bind to the HRD1 promoter and that downregulation of KLF2 remarkably decreased HRD1 expression. Notably, ox-LDL treatment of ECs decreased KLF2 and HRD1 expression, but the inhibition of HRD1 was reversed by overexpression of KLF2. Based on these finding, we hypothesized that KLF2 played a direct role in the downregulation of HRD1 expression in ox-LDL-treated ECs.
Previous studies have indicated that statins (HMG-CoA reductase inhibitors) prevent EC dysfunction via a KLF2-dependent mechanism [Citation31,Citation32]. In addition, KLF2 has been shown to protect ECs against ox-LDL-induced damage [Citation33]. The present study provides for the first demonstration of a KLF2-dependent mechanism for pravastatin-mediated regulation of HRD1 expression. Furthermore, inhibition of HRD1 expression reversed the protective effect of pravastatin on ECs exposed to ox-LDL. These findings may be sufficient to indicate that the KLF2-HRD1 regulatory axis contributes significantly to the protective effect of statins.
In conclusion, our findings indicated that HRD1 expression may be a novel prognostic or progression marker for AS. Downregulation of KLF2 was responsible for the decrease in HRD1 levels observed in ox-LDL-treated ECs. Overexpression of HRD1 prevented ox-LDL-induced apoptosis by degradation of LOX-1. Based on our findings, we propose that restoration of HRD1 expression could represent a novel strategy for human AS therapy.
Author contribution statement
Q.L., W.X., and Z.J. designed and conducted the research, contributed to the writing of the manuscript. H.L., M.L. and X.L. conducted experiments, contributed to the acquisition and interpretation of data. D.S. contributed to conception and design of the study and critical analysis of the results and critically reviewed the drafts of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The human arterial vessel study was approved by the ethical committees at Nanjing Medical University, Nanjing, China. All the patients provided written consent for the collection and use of samples in this study.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Mäkinen PI, Ylä-Herttuala S. Therapeutic gene targeting approaches for the treatment of dyslipidemias and atherosclerosis. Curr Opin Lipidol. 2013;24:116–122.
- Hopkins PN. Molecular biology of atherosclerosis. Physiol Rev. 2013;93:1317–1542.
- Meyers MR, Gokce N. Endothelial dysfunction in obesity: etiological role in atherosclerosis. Curr Opin Endocrinol Diabetes Obes. 2007;14:365–369.
- Cahill PA, Redmond EM. Vascular endothelium - Gatekeeper of vessel health. Atherosclerosis. 2016;248:97–109.
- Valente AJ, Irimpen AM, Siebenlist U, et al. OxLDL induces endothelial dysfunction and death via TRAF3IP2: inhibition by HDL3 and AMPK activators. Free Radic Biol Med. 2014;70:117–128.
- Donato L, Bramanti P, Scimone C, et al. miRNA expression profile of retinal pigment epithelial cells under oxidative stress conditions. FEBS Open Bio. 2018;8:219–233.
- Al-Banna N, Lehmann C. Oxidized LDL and LOX-1 in experimental sepsis. Mediators Inflamm. 2013;2013:761789.
- Mitra S, Goyal T, Mehta JL. Oxidized LDL, LOX-1 and atherosclerosis. Cardiovasc Drugs Ther. 2011;25:419–429.
- Hermonat PL, Zhu H, Cao M, et al. LOX-1 transcription. Cardiovasc Drugs Ther. 2011;25:393–400.
- Rinaldi C, Bramanti P, Famà A, et al. GLYOXALASE I A111E, PARAOXONASE 1 Q192R AND L55M POLYMORPHISMS IN ITALIAN PATIENTS WITH SPORADIC CEREBRAL CAVERNOUS MALFORMATIONS: A PILOT STUDY. Scimone C, Donato L, Antognelli C, Alafaci C, Tomasello F, D’Angelo R, Sidoti A. J Biol Regul Homeost Agents. 2015;29:493–500.
- Donato L, Scimone C, Nicocia G, et al. GLO1 gene polymorphisms and their association with retinitis pigmentosa: a case-control study in a Sicilian population. Mol Biol Rep. 2018;45:1349–1355.
- Akhmedov A, Rozenberg I, Paneni F, et al. Endothelial overexpression of LOX-1 increases plaque formation and promotes atherosclerosis in vivo. Eur Heart J. 2014;35:2839–2848.
- Hu C, Dandapat A, Sun L, et al. LOX-1 deletion decreases collagen accumulation in atherosclerotic plaque in low-density lipoprotein receptor knockout mice fed a high-cholesterol diet. Cardiovasc Res. 2008;79:287–293.
- Mehta JL, Sanada N, Hu CP, et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesteroldiet. Circ Res. 2007;100:1634–1642.
- Chistiakov DA, Orekhov AN, Bobryshev YV. LOX-1-mediated effects on vascular cells in atherosclerosis. Cell Physiol Biochem. 2016;38:1851–1859.
- Novodvorsky P, Chico TJ. The role of the transcription factor KLF2 in vascular development and disease. Prog Mol Biol Transl Sci. 2014;124:155–188.
- Atkins GB, Jain MK. Role of Krüppel-like transcription factors in endothelial biology. Circ Res. 2007;100:1686–1695.
- Yang L, Zhou X, Guo R, et al. Role of Krüppel-like factor 2 and protease-activated receptor-1 in vulnerable plaques ofApoE(-/-) mice and intervention with statin. Can J Cardiol. 2013;29:997–1005.
- Lin Z, Kumar A, SenBanerjee S, et al. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res. 2005;96:e48–57.
- Parmar KM, Nambudiri V, Dai G, et al. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J Biol Chem. 2005;280:26714–26719.
- Yagishita N, Yamasaki S, Nishioka K, et al. Synoviolin, protein folding and the maintenance of joint homeostasis. Nat Clin Pract Rheumatol. 2008;4:91–97.
- Yamasaki S, Yagishita N, Sasaki T, et al. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase ‘Synoviolin’. Embo J. 2007;26:113–122.
- Wu T, Zhao F, Gao B, et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28:708–722.
- Xu YM, Wang HJ, Chen F, et al. HRD1 suppresses the growth and metastasis of breast cancer cells by promoting IGF-1R degradation. Oncotarget. 2015;6:42854–42867.
- Doroudgar S, Völkers M, Thuerauf DJ, et al. Hrd1 and ER-associated protein degradation, ERAD, are critical elements of the adaptive ER stress response in cardiac myocytes. Circ Res. 2015;117:536–546.
- Tang Y, Zhang YC, Chen Y, et al. The role of miR-19b in the inhibition of endothelial cell apoptosis and its relationship with coronary artery disease. Sci Rep. 2015;5:15132.
- Jia L, Xing J, Ding Y, et al. Hyperuricemia causes pancreatic β-cell death and dysfunction through NF-κB signaling pathway. PLoS One. 2013;8:e78284.
- Maier JA. Endothelial cells and magnesium: implications in atherosclerosis. Clin Sci (Lond). 2012;122:397–407.
- Lubrano V, Balzan S. Roles of LOX-1 in microvascular dysfunction. Microvasc Res. 2016;105:132–140.
- Doddaballapur A, Michalik KM, Manavski Y, et al. Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler Thromb Vasc Biol. 2015;35:137–145.
- Ali F, Hamdulay SS, Kinderlerer AR, et al. Statin-mediated cytoprotection of human vascular endothelial cells: a role for Kruppel-like factor 2-dependent induction of heme oxygenase-1. J Thromb Haemost. 2007;5:2537–2546.
- Panigrahi S, Freeman ML, Funderburg NT, et al. SIV/SHIV infection triggers vascular inflammation, diminished expression of krüppel-like factor 2 and endothelial dysfunction. J Infect Dis. 2016;213:1419–1427.
- Li M, Wang X, Fu W, et al. CD4+CD25+Foxp3+ regulatory T cells protect endothelial function impaired by oxidized low density lipoprotein via the KLF-2 transcription factor. Cell Physiol Biochem. 2011;28:639–648.