
ABSTRACT
Necrotizing enterocolitis (NEC) is a major cause of mortality and morbidity in newborns, characterized by inflammatory intestinal necrosis. Sirtuin-1 (SIRT1), a NAD-dependent deacetylase, is involved in multiple biological functions. It has been reported that SIRT1 was downregulated in NEC tissues. However, the precise role of SIRT1 in NEC progress remains unknown. In this study, we found that SIRT1 was decreased in serum samples of NEC patients, associated with an inflammation response. an in vitro model was established by using LPS-induced NEC-like cell in this study. The results indicate that overexpression of SIRT1 inhibited the cell apoptosis induced by LPS. Besides, overexpression of SIRT1 suppressed the high expression of proinflammatory factors (IL-6, IL-8, and TNF-α), the decrease of transepithelial electrical resistance (TEER), and the decline expression of tight junction proteins (ZO-1, ZO-2, and Claudin-4) induced by LPS in Caco-2 cells. What is more, serum HIF-1α was increased in NEC patients. SIRT1 overexpression suppressed the expression and activity of HIF-1a, while knockdown of SIRT1 made the opposite effect. In summary, this study indicates that overexpression of SIRT1 alleviates the inflammation response and intestinal epithelial barrier dysfunction through regulating the expression and inactivation of HIF-1a.
KEYWORDS:
1. Introduction
Necrotizing Enterocolitis (NEC) is a common and severe intestinal disease in preterm infants [Citation1]. It is characterized by inflammation, hemorrhagic, and intestinal tissue necrosis [Citation2]. Statistically, the incidence of NEC was about 3% in newborns, while it may reach to 13% in premature infants <2500 g, with an average mortality of 20–30% [Citation3]. The pathogenesis of NEC is complicated, including hypoxia, prematurity, formula-feeding, and bacterial colonization of the gut [Citation4].
Lipopolysaccharide (LPS), a component of the outer wall of Gram-negative bacteria, that reported could increase paracellular permeability in the lung through activation of proinflammatory factors, such as IL-6, TNF-α, and NO [Citation5]. Besides, circulating LPS and proinflammatory cytokines were increased in NEC patients [Citation6]. What is more, LPS leads to intestinal epithelial barrier dysfunction through regulating the tight junction between epithelial cells [Citation7].
Sirtuin-1 (SIRT1), an NAD+-dependent deacetylase that catalyzes the deacetylation of acetyl-lysine residues of proteins such as p53 and FOXO3a [Citation8]. It regulates a member of cellular responses, such as cell senescence, apoptosis, tumor progression, inflammation, endocrine metabolism, and glucose homeostasis [Citation9–Citation13]. It has been reported that SIRT1 shuttles between the nucleus and cytoplasm, and could be tested in the serum [Citation14]. Increasing evidence has suggested that the level of SIRT1 in serum was associated with aging, neurological, and many other diseases [Citation15,Citation16]. It was consistently decreased with aging and exhibited low level in Alzheimer’s disease, frailty, chronic obstructive pulmonary disease, and obesity sufferers [Citation16–Citation21]. In addition, SIRT1 was increased in patients with asthma [Citation15]. These studies suggested that serum SIRT1 may be a potential biomarker for diseases. What is more, SIRT1 has been reported to downregulated in Necrotizing Enterocolitis (NEC) patients [Citation22]. However, its function in NEC has not been explored.
HIF-1 (Hypoxia Inducible Factor-1), a master regulator of oxygen homeostasis, is a heterodimer consisting of HIF-1α and HIF-1β (ARNT) subunits [Citation23]. In normal conditions, HIF-1α is typically and rapidly degraded by the ubiquitin/proteasomal pathway [Citation24]. Under hypoxic conditions, HIF-1α degradation was inhibited and lead to its accumulation, and then it translocated into the nucleus and formed a dimer with HIF-1β, activates the downstream targets [Citation25]. HIF-1α was upregulated in vivo model of NEC [Citation1]. Besides, HIF-1α deficiency ameliorated hypoxia-reoxygenation injury-induced increases in intestinal permeability [Citation1,Citation26]. In the present study, the role of SIRT1 in LPS‑induced NEC cell model was investigated through transfection with overexpression vectors and shRNA. It is interesting to note that the expression of HIF-1α was influenced by SIRT1 in LPS‑stimulated Caco-2 cells. The results of this study indicate that SIRT1 participated in the progress of NEC through regulating the activity and expression of HIF-1α.
2. Materials and methods
2.1. Blood specimens
Thirty-four patients between days 14 and 28 of ages with NEC were selected from the Department of the Affiliated Children Hospital of Xi’an Jiaotong University (Xi’an, China) between 2016–2018 years. NEC patients are characteristic with bloating, hematochezia, and Abdominal X-ray plain film appeared pneumatosis cystoides intestinalis, and they were diagnosed by professionals. All of them were premature infants cultured in incubators and feed with baby milk. In the early manifestations of disease, they exhibited nonspecific feeding intolerance, gastric retention, and apnea. At the same, 34 healthy volunteers between days 14 and 28 of ages were employed in this study form the same Hospital. Their blood samples were collected and stored at −20 °C after centrifugal settling. The study was approved by the ethics committee of the Affiliated Children Hospital of Xi’an Jiaotong University, and the guardians of all subjects provided informed consent for participation. The research has been carried out in accordance with the World Medical Association Declaration of Helsinki, and that all subjects provided written informed consent.
2.2. Cell culture and treatment
Human epithelial colorectal adenocarcinoma Caco-2 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; HyClone, Salt Lake City, UT) supplemented with 10% fetal bovine serum (FBS; HyClone). And maintained in a humidified incubator with 5% CO2 in 37°C. LPS treatment: cells were treated for 12 h with lipopolysaccharide (LPS, 1.0 ug/mL) [Citation27,Citation28].
2.3. Cell transfection
Overexpression plasmid of SIRT1 (ov-SIRT1), SIRT1 shRNA (sh-SIRT1), empty plasmid and scramble shRNA were chemically constructed and purchased from GenePharma (Shanghai, China). Lipofectamine 3000 kit was adopted to transfect ov-SIRT1(50 nM), sh-SIRT1 (40 nM), empty plasmid (50 nM), and scramble shRNA (40 nM) into cells according to the manufacturer’s instructions (Invitrogen, CA, USA).
2.4. Quantitative real-time PCR analysis
Total RNA was isolated by TRIzol reagent according to the manufacturer’s protocol (Invitrogen). Complementary DNA was reverse-transcribed from RNA by M-MLV Reverse Transcriptase. The expression level of SITR1 mRNA was detected by Quantitative RT-PCR using an SYBR ® Premix Ex Taq™ Kit in Thermal Cycler Dice® Real-time System TP800.
2.5. Cell viability assay
Cell viability was investigated using a Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies) according to the manufacturers’ instructions. In brief, Caco-2 cells were plated into 96-well plates with a density of 2 × 104/mL for overnight, and transfected with ov-SIRT1 or sh-SIRT1, after 48 h of culturation, LPS (1.0 ug/mL) was added into cell culture. After culturing for 12 h, 10 μL CCK-8 reagents were added into the culture medium and cultured for 3 h in 37°C with 5% CO2. The cell viability was tested by a microplate reader at 450 nm.
2.6. Enzyme-linked immunosorbent assays (ELISA)
The content of SIRT1 in serum samples was detected through a human SIRT1 ELISA kit (ab171573, Abcam, Cambridge, UK). The content of HIF-1α in serum and culture medium was detected by a HIF-1α ELISA kit (Cusabio Biotech Co., LTD, Wuhan, China). The levels of proinflammatory factors in the culture medium of Caco-2 cells and in serum were tested with ELISA kits (Wuhan USCN Life Science Inc, China) according to the manufacturers’ instructions.
2.7. Cell apoptosis assay
Cell apoptosis was examined using the Annexin V/propidium iodide (PI) detection kit (Beyotime, Shanghai, China) and analyzed by flow cytometry. Caco-2 cells were plated into 6-well plates and treated by LPS (1.0 ug/mL,12 h) following transfected with ov-SIRT1 or sh-SIRT1 (48 h). Then, cells were collected and incubated in 500 μL binding buffer with 5 μL Annexin V-FITC for 25 min in dark at room temperature, 5 μL PI was added before flow cytometry detection.
2.8. Western blot analysis
Western blot analysis was performed as previously described [Citation5]. Cell lysates were separated in 10% SDS-PAGE and transferred onto nitrocellulose membranes (Millipore, Boston, MA, USA). After blocked with 5% skim milk, the blots were incubated with primary antibodies against ZO-1 (ab216880, Abcam), TJP2/ZO2 (ab53156, Abcam), Claudin 4 (ab53156, Abcam) and SIRT1 (#2496, CST) for overnight at 4 °C. After washed by TBST, the bands were incubated with corresponding HRP-conjugated secondary antibody for 1 h at room temperature. At last, proteins were detected using the enhanced chemiluminescence detection kit (Millipore) and quantified using Quantity One software (Bio‑Rad, Hercules, CA, USA).
2.9. Dual-luciferase reporter assay
The activity of HIF-1α was detected through a Dual-luciferase reporter assay as described in the previous report [Citation29]. Overexpression plasmid of SIRT1 (ov-SIRT1) and HIF-1α luciferase reporter p2.1 plasmid were co-transfected into HEK293 T cells using Lipofectamine 3000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Dual-luciferase reporter assays were performed with the Dual-Luciferase Assay kit from Promega (Madison, WI) according to the manufacturers’ instructions.
2.10. Transepithelial electrical resistance (TEER)
Caco-2 cells were plated on Transwell inserts (membrane area 0.33 cm2, pore size 0.4 μm) and achieved a polarized and differentiated state within 21 days after seeding. The tight junction permeability of the Caco-2 monolayer was evaluated by measuring TEER using the Millicell-ERS system (Millipore, Manassas, USA). TEER was recorded with before the addition of LPS with or without vector transfection (time zero) and then at various time intervals (3 h, 6 h, 9 h, 12 h, and 24 h), and expressed as the ratio of TEERt/TEER0.
2.11. Statistical analysis
The data are presented as the means ± standard deviation. Statistical analyses were performed with ANOVA with Bonferroni’s posttest and a Student’s t-test using prism 5.0 and SPSS software version 19.0 (SPSS Inc., Chicago, IL, USA). P < 0.05 was considered to indicate statistically significant.
3. Results
3.1. SIRT1 was downregulated in NEC patients
The content of SIRT1 in serum was measured using a SIRT1 ELISA kit, the results showed that the levels of serum SIRT1 in NEC patients were significantly decreased compared with healthy control peoples ()). In addition, the expression of proinflammatory factors IL-6, IL-8, and TNF-α was markedly increased in NEC patients ()).
Figure 1. Serum SIRT1 was downregulated in NEC patients, and overexpression of SIRT1 inhibited LPS-induced cell apoptosis. (a) Serum SIRT1 in NEC patients (n = 34) were examined by an ELISA kit. (b) The contents of IL-6, IL-8 and TNF-α in NEC patients were examined through ELISA kits. *P < 0.05, **P < 0.01. (c) The expression of SIRT1 in Caco-2 cells was tested by Western blotting after stimulated by LPS. **P < 0.01, compared with 0 h treatment group; #P < 0.05, compared with 6 h treatment group. (d) and (e) The transfection efficiency of overexpression vector of SIRT1 (ov-SIRT1) in Caco-2 cells was tested. (f) Cell viability were measured after transfection with ov-SIRT1. (g) and (h) Cell apoptosis were measured after transfection with ov-SIRT1. *P < 0.05, compared with the empty vector transfection group; #P < 0.05, compared with the control group, &P < 0.05, compared with the LPS treatment group.
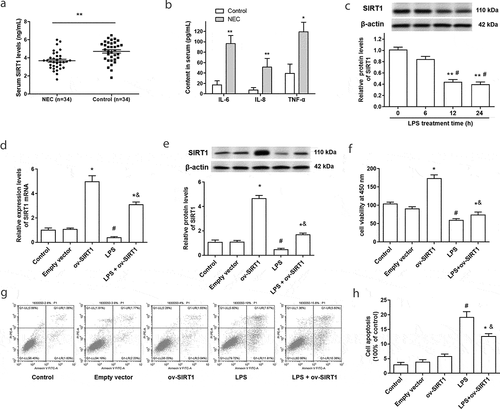
3.2. Overexpression of SIRT1 inhibited cell apoptosis induced by LPS
To investigate the role of SIRT1 in NEC, an in vitro model in which Caco-2 cell monolayers were treated with LPS (1.0 ug/mL) was adopted. As shown in ), the expression of SIRT1 was downregulated in Caco-2 cells after being stimulated by LPS for 12 and 24 h. Overexpression of SIRT1 in Caco-2 cells was achieved through transfection with SIRT1 overexpression vectors ( and e)). At the same time, we found SIRT1 overexpression promoted the cell viability, and suppressed the decline of cell viability and the increase of apoptosis induced by LPS treatment (-h)).
3.3. Overexpression of SIRT1 suppressed LPS-induced inflammatory response and tight junction dysfunction in Caco-2 cells
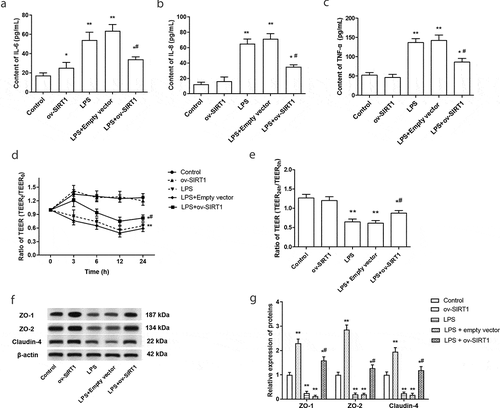
It is known that inflammation and increased permeability of intestinal epithelial barrier were associated with the progress of NEC. Hence, the effect of overexpression of SIRT1 on the intestinal epithelial cell barrier and inflammatory response were explored. We found that overexpression of SIRT1 markedly inhibited the increase of IL-6, IL-8, and TNF-α in Caco-2 cells that induced by LPS treatment (-c)). In addition, the tight-junction permeability of the Caco-2 monolayer was evaluated by measuring TEER. The results suggested that LPS treatment induced a dramatic decrease in TEER of polarized Caco-2 monolayer, overexpression of SIRT1 inhibited the decrease of TEER induced by LPS ( and e)). Besides, the expression of tight junction proteins ZO-1, ZO-2, and Claudin-4 was increased in LPS-stimulated Caco-2 cells, while this increase was significantly inhibited by SIRT1 overexpression ( and g)).
Figure 2. The effect of overexpression of SIRT1 on intestinal epithelial barrier dysfunction and inflammatory response. (a-c) The expression levels of IL-6 (a), IL-8 (b) and TNF-α (c) in Caco-2 cells were examined after stimulated by LPS or overexpression of SIRT1. (d) TEER of different times was determined. (e) TEER was determined after stimulated by LPS or overexpression of SIRT1 for 24 h. (f) and (g) The expression of tight junction proteins was measured through Western blotting. *P < 0.05, **P < 0.01, compared with the control group; #P < 0.05, compared with the LPS treatment group and LPS + empty vector transfection treatment group.
3.4. Overexpression of SIRT1 inhibited the activation of HIF-1α in LPS-treated Caco-2 cell
Further, the underlying mechanisms of SIRT1 relieve inflammation and intestinal epithelial barrier disorder were studied. We found the content of serum HIF-1α was significantly upregulated in NEC patients ()). Meanwhile, LPS treatment notably enhanced the expression of HIF-1α, while SIRT1 overexpression inhibited this increase in LPS-treated Caco-2 cell ()). What is more, Dual-luciferase reporter assay showed that overexpression of SIRT1 suppressed the transcriptional activity of HIF-1 ()). Hence, it is predicted that the protective effect of SIRT1 on NEC may attribute to the downregulation of HIF-1 activity and expression.
Figure 3. The effect of SIRT1 overexpression or downregulation on the activation of HIF-1α in LPS-treated Caco-2 cells. (a) The expression of serum HIF-1α in NEC patients (n = 34) were examined by an ELISA kit. **P < 0.01, compared with the control group. (b) The content of HIF-1α in Caco-2 cells was tested after stimulated by LPS or overexpression of SIRT1. **P < 0.01, compared with the empty vector transfection group; #P < 0.05, compared with the LPS treatment group. (c) The activity of HIF-1α was tested after transfection with ov-SIRT1 through Dual-luciferase reporter assay. **P < 0.01, compared with the empty vector transfection group. (d) The transfection efficiency of SIRT1 shRNA (sh-SIRT1) was tested. (e-g). Cell viability and apoptosis were measured after transfected with sh-SIRT1. (h) The content of HIF-1α in Caco-2 cells was tested after knockdown of SIRT1. *P < 0.05, compared with the scramble shRNA transfection group (shRNA-NC); #P < 0.05, compared with the LPS treatment group. (i) The activity of HIF-1α was tested after transfection with sh-SIRT1 by Dual-luciferase reporter assay. *P < 0.05, compared with scramble shRNA transfection group.
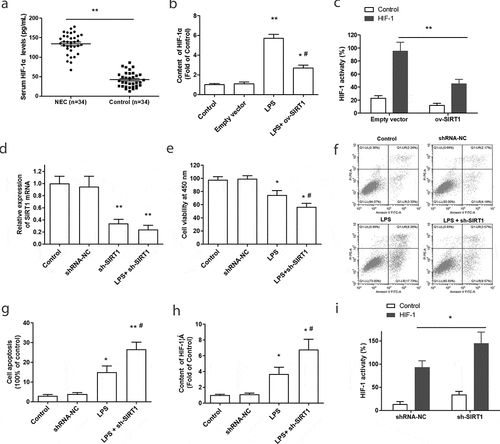
3.5. Silence of SIRT1 promoted the activation of HIF-1α in LPS-treated Caco-2 cells
To further verify this mechanism, SIRT1 shRNA (sh-SIRT1) was synthesized and transfected into Caco-2 cells. The transfection efficiency of sh-SIRT1 in Caco-2 cell was detected through RT-PCR, and the result showed that transfection with sh-SIRT1 significantly inhibited the expression of SIRT1, compared with the control group and scrambled shRNA (shRNA-NC) transfection group ()). Next, the effect of knockdown of SIRT1 on the cell viability and cell apoptosis was examined. As shown in -g), transfection with sh-SIRT1 further aggravated the decline of cell viability and the increase of cell apoptosis that induced by LPS. More importantly, silence of SIRT1 markedly promoted the increasing of HIF-1α expression in LPS-stimulated Caco-2 cells ()). Downregulation of SIRT1 significantly promoted the activity of HIF-1 ()). These results further suggested that SIRT1 participated in the progress of NEC through the regulation of HIF-1α activity and expression.
3.6. HIF-1 inhibitor suppressed the tight junction proteins dysfunction and inflammatory response in LPS-stimulated Caco-2 cells
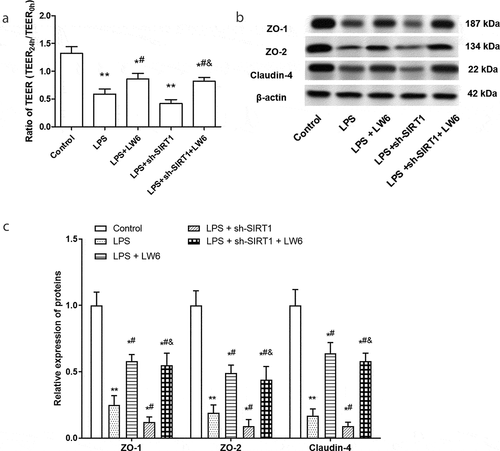
What is more, LW6, a specific HIF-1 inhibitor, which promotes the proteasomal degradation of HIF-1a, was adopted in this study [Citation30]. As shown in ), the content of HIF-1a was significantly decreased after treated by LW6 in LPS-stimulated Caco-2 cells. Further, the effect of HIF-1a inhibition on the inflammation and tight junction proteins was examined. The results showed that LW6 treatment suppressed the increase of proinflammatory cytokine IL-6, IL-8, and TNF-α, and reversed the promoting effect of sh-SIRT1 on the expression of IL-6, IL-8, and TNF-α in LPS-stimulated Caco-2 cells (-d)). At the same time, LW6 treatment significantly increased the ratio of TEER and the expression of ZO-1, ZO-2, and Claudin-4 in LPS-stimulated Caco-2 cells (-c)). Besides, silence of SIRT1 exacerbated the tight junction protein dysfunction in Caco-2 cells that induced by LPS, while LW6 treatment effectively reversed this effect (-c)). These results suggest that LW6 suppressed the inflammatory response and tight junction dysfunction that induced by LPS, and reversed the effect of SIRT1 silence.
Figure 4. Inhibition of HIF-1 relieved the inflammation in LPS-stimulated Caco-2 cells. (a) The inhibitory effect of LW6 on HIF-1α was detected through an ELISA kit. (b-e). The effect of LW6 on the levels of IL-6 (b), IL-8 (c), and TNF-α (d) in Caco-2 cells were examined after stimulated by LPS and downregulation of SIRT1. *P < 0.05, **P < 0.01, compared with the control group; #P < 0.05, compared with the LPS treatment group; & P < 0.05, compared with the LPS + sh-SIRT1 treatment group.
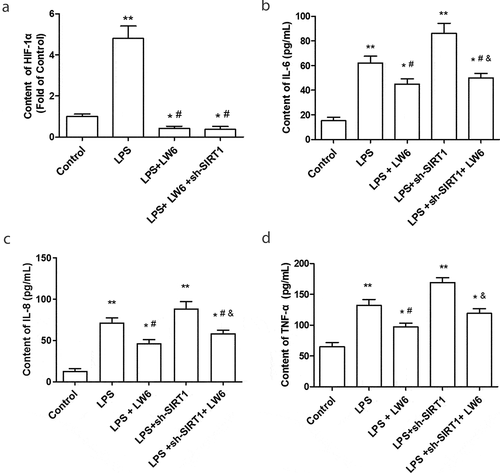
Figure 5. Inhibition of HIF-1 relieved the tight junction proteins dysfunction in LPS-stimulated Caco-2 cells. (a) The effect of LW6 on TEER in Caco-2 cells were examined after stimulated by LPS and downregulation of SIRT1. (b) and (c) The effect of LW6 on the expression of tight junction proteins. *P < 0.05, **P < 0.01, compared with the control group; #P < 0.05, compared with the LPS treatment group; & P < 0.05, compared with the LPS + sh-SIRT1 treatment group.
4. Discussion
The current study focused on the role of SIRT1 in NEC. Our study found that SIRT1 was downregulated in the peripheral blood samples of NEC patients, consistent with the report that SIRT1 mRNA was decreased in NEC tissue samples [Citation22]. It is known that NEC is characterized by the presence of a number of proinflammatory cytokines and a disorder of intestinal barrier [Citation2,Citation31]. Tight junctions are composed of large protein complexes that are physiologically responsible for maintaining the intestine impermeable to different substances and organisms [Citation32]. ZO-1 (TJP1) and ZO-2 (TJP2) are important scaffolding proteins of tight junction [Citation33,Citation34]. As peripheral membrane proteins, they bind to the cytoplasmic C termini of junctional transmembrane proteins and link them to the actin cytoskeleton [Citation33,Citation34]. Claudin-4, a member of Claudins family, was downregulated in NEC and is a component of tight junction strands [Citation22,Citation32]. In this study, we found that overexpression of SIRT1 improved the intestinal epithelial barrier dysfunction via upregulating the expression of tight junction proteins ZO-1, ZO-2, and claudin-4. A previous study has been reported that activation of SIRT1 could attenuate LPS-induced lung hyperpermeability and increased tight junction protein expression, including occludin, claudin-5, ZO-1, and ZO-2 [Citation35]. Another study indicated that SIRT1 activator resveratrol could improve the disorder of blood-brain barrier permeability through increasing the expression of tight junction proteins ZO‑1, Occludin, and Claudin‑5 in brain injury [Citation36]. Hence, the current findings are consistent with previous work indicating that SIRT1 overexpression improved the intestinal epithelial barrier dysfunction via regulating the expression of tight junction proteins.
With further respect to NEC pathogenesis, we note that HIF-1α was markedly upregulated in NEC. A previous report revealed that HIF-1 activation exacerbates the intestinal ischemia-reperfusion injury-induced gut-and lung-derived inflammatory response, and HIF-1α deficiency ameliorated intestinal permeability, bacterial translocation, and cell apoptosis in lung injury [Citation37]. Furthermore, it has been reported that SIRT1 inactivated HIF-1a by blocking p300 recruitment and consequently repressed HIF-1 target genes [Citation38,Citation39]. On the contrary, another study reported that SIRT1 overexpression enhances hypoxic stabilization of HIF-1a protein, knockdown of SIRT1 decreases HIF-1 transcriptional activity, and inhibits HIF-1a protein accumulation [Citation29]. In our study, SIRT1 overexpression inhibited the increase in expression of HIF-1α that induced by LPS treatment, while downregulating SIRT1 made the opposite effect. In addition, we found inhibition of HIF-1a suppressed the inflammatory response and tight junction protein disorder that induced by LPS.
In summary, our study identified the protective role of SIRT1 in NEC. Overexpression of SIRT1 could alleviate the inflammation response and intestinal epithelial barrier dysfunction through regulating the expression and the inactivation of HIF-1.
Author contributions
Wang Q and Bai M designed the study. Bai M, Lu Z.X, An L, Gao Q, Xie W.K, Miao F, Chen X. F, and Pan Y.K finished the cell experiment section. Bai M wrote this manuscript and Wang Q oversaw language edit. All authors read and approved the final manuscript.
Disclosure statement
The authors declare that they have no conflict of interest.
References
- Baregamian N, Rychahou PG, Hawkins HK, et al. Phosphatidylinositol 3-kinase pathway regulates hypoxia-inducible factor-1 to protect from intestinal injury during necrotizing enterocolitis. Surgery. 2007;142(2):295–302.
- Mϋller M, Paul T, Seeliger S. Necrotizing enterocolitis in premature infants and newborns. J Neonatal Perinatal Med. 2016;9:233–242.
- Niño DF, Sodhi CP, Hackam DJ. Necrotizing enterocolitis: new insights into pathogenesis and mechanisms. Nat Rev Gastroenterol Hepatol. 2016;13:590.
- Niño DF, Sodhi CP, Hackam DJ. Necrotizing enterocolitis: new insights into pathogenesis and mechanisms. Nat Clin Pract Gastroenterol Hepatol. 2016;13:590.
- Bein A, Zilbershtein A, Golosovsky M, et al. LPS induces hyper‐permeability of intestinal epithelial cells. J Cell Physiol. 2017;232:381–390.
- Duffy LC, Zielezny MA, Carrion V, et al. Concordance of bacterial cultures with endotoxin and interleukin-6 in necrotizing enterocolitis. Dig Dis Sci. 1997;42:359–365.
- Nighot M, Al-Sadi R, Guo S, et al. Lipopolysaccharide-induced increase in intestinal epithelial tight permeability is mediated by toll-like receptor 4/myeloid differentiation primary response 88 (MyD88) activation of myosin light chain kinase expression. Am J Pathol. 2017;187:2698–2710.
- Eaton EN. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159.
- Tomomi I, Ryuji H, Kensuke S, et al. Sirtuin 1 activator SRT1720 suppresses inflammation in an ovalbumin-induced mouse model of asthma. Respirology. 2013;18:332–339.
- Picard F, Kurtev M, Chung N, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776.
- Kim D, Nguyen MD, Dobbin MM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. Embo J. 2014;26:3169–3179.
- Rodgers JT, Lerin C, Haas W, et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118.
- Alcendor RR, Gao S, Zhai P, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512.
- Masaya T, Jun S, Tetsuji M, et al. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem. 2007;282:6823–6832.
- Wang Y, Li D, Ma G, et al. Increases in peripheral SIRT1: a new biological characteristic of asthma. Respirology. 2015;20:1066–1072.
- Rahul K, Prasun C, Sharma PK, et al. Sirtuin1: a promising serum protein marker for early detection of Alzheimer’s disease. PLoS ONE. 2013;8:e61560.
- Julien C, Tremblay C, Emond V, et al. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:48–58.
- Schug TT, Li X. Sirtuin 1 in lipid metabolism and obesity. Ann Med. 2011;43:198–211.
- Yanagisawa S, Papaioannou A, Papaporfyriou A, et al. Decreased serum sirtuin-1 in chronic obstructive pulmonary disease. chest. 2017;152:343–352.
- Zhong Y, Chen AF, Zhao J, et al. Serum levels of cathepsin D, sirtuin1, and endothelial nitric oxide synthase are correlatively reduced in elderly healthy people. Aging Clin Exp Res. 2016;28:641–645.
- Rahul K, Navinath M, Ashish Datt U, et al. Identification of serum sirtuins as novel noninvasive protein markers for frailty. Aging Cell. 2015;13:975–980.
- Bein A, Eventov-Friedman S, Arbell D, et al. Intestinal tight junctions are severely altered in NEC preterm neonates. Pediatr Neonatol. 2018;59:464–473.
- Li G, Lu W-H, Ai R, et al. The relationship between serum hypoxia-inducible factor 1α and coronary artery calcification in asymptomatic type 2 diabetic patients. Cardiovasc Diabetol. 2014;13:52.
- Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-to the von Hippel-Lindau Ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472.
- Koyasu S, Kobayashi M, Goto Y, et al. Regulatory mechanisms of hypoxia‐inducible factor 1 activity: two decades of knowledge. Cancer Sci. 2018;109:560–571.
- Feinman R, Deitch EA, Watkins AC, et al. HIF-1 mediates pathogenic inflammatory responses to intestinal ischemia-reperfusion injury. Am J Physiol. 2010;299:833–843.
- Ling X, Linglong P, Weixia D, et al. Protective effects of bifidobacterium on intestinal barrier function in LPS-induced enterocyte barrier injury of Caco-2 monolayers and in a rat NEC model. PLoS ONE. 2016;11(8):e0161635.
- Jian C, Ren Z, Jian W, et al. Protective effects of baicalin on LPS-induced injury in intestinal epithelial cells and intercellular tight junctions. Can J Physiol Pharmacol. 2015;93:233–237.
- Alexander L, Antje L, Vincent R, et al. Inhibition of SIRT1 impairs the accumulation and transcriptional activity of HIF-1α protein under hypoxic conditions. PLoS ONE. 2012;7:e33433.
- Lee K, Kang JE, Park SK, et al. LW6, a novel HIF-1 inhibitor, promotes proteasomal degradation of HIF-1α via upregulation of VHL in a colon cancer cell line. Biochem Pharmacol. 2010;80:982–989.
- Ford H, Watkins S, Reblock K, et al. The role of inflammatory cytokines and nitric oxide in the pathogenesis of necrotizing enterocolitis. J Pediatr Surg. 1997;32:275–282.
- Pearce SC, Aljawadi A, Kishida K, et al. Marked differences in tight junction composition and macromolecular permeability among different intestinal cell types. BMC Biol. 2018;16:19.
- Itoh M, Nagafuchi A, Moroi S, et al. Involvement of ZO-1 in Cadherin-based cell adhesion through its direct binding to α catenin and actin filaments. J Cell Biol. 1997;138(1):181–192.
- Itoh M, Morita K, Tsukita S. Characterization of ZO-2 as a MAGUK family member associated with tight as well as adherens junctions with a binding affinity to occludin and alpha catenin. J Biol Chem. 1999;274:5981–5986.
- Fu C, Hao S, Xu X, et al. Activation of SIRT1 ameliorates LPS-induced lung injury in mice via decreasing endothelial tight junction permeability. Acta Pharmacol Sin. 2019;40:630–641.
- Qian C, Jin J, Chen J, et al. SIRT1 activation by resveratrol reduces brain edema and neuronal apoptosis in an experimental rat subarachnoid hemorrhage model. Mol Med Rep. 2017;16:9627–9635.
- Feinman R, Deitch EA, Watkins AC, et al. HIF-1 mediates pathogenic inflammatory responses to intestinal ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2010;299:G833–G843.
- Lim JH, Lee YM, Chun YS, et al. Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1α. Mol Cell. 2010;38:864–878.
- Ryu DR, Yu MR, Kong KH, et al. Sirt1-hypoxia-inducible factor-1α interaction is a key mediator of tubulointerstitial damage in the aged kidney. Aging Cell. 2019;18:e12904.