
ABSTRACT
Kinases form the major part of the druggable genome and their selective inhibition in human cancers has had reasonable clinical success. In contrast to tumorigenesis, the role of kinases in mediating immune responses is poorly understood. However, synergistic therapeutic regimens combining targeted therapy and immune therapy have been found to increase the median survival of tumor patients. In this context, we uncovered that RAF and MEK1/2 kinases, which are the integral parts of the classical MAPK cascade, have unique roles in driving DC differentiation and activation. RAF kinases are stabilized in their protein levels during DC differentiation and are obligatory for normal functioning of DCs. But, the targeting of MEK1/2 kinases with specific inhibitors did not phenocopy the effects observed with RAF inhibitors suggesting that RAF and MEK1/2 kinases may have specific and unique roles in driving immune responses, which deserves further studies to successfully administer these inhibitors in clinics.
Introduction
Aberrant protein kinase activities are directly or indirectly linked to more than 400 human diseases including cancer [Citation1]. Protein kinases display a major group of the druggable genome and targeting kinases has proven to be a successful therapy for many cancers [Citation2]. Imatinib was the first tyrosine kinase inhibitor approved by the US Food and Drug Administration (FDA) for the treatment of Philadelphia chromosome-positive chronic myeloid leukemia, thus opening an avenue of targeted therapeutics in clinical oncology [Citation3]. Due to the success of ATP competitive and noncompetitive inhibitors against oncogenic kinases in genetically defined human cancers, lots of studies were carried out to uncover the role of these kinases in tumorigenesis. Moreover, the list of potential kinase targets, of their inhibitors and the corresponding clinical trials is expanding rapidly [Citation4]. Novel inhibitors that target the kinases outside the kinase domains like allosteric inhibitors have garnered significant interest as they reduce the side effects. To date, the FDA has approved 48 kinase inhibitors, 36 of which are primarily employed to treat malignancies, and seven are used to treat immune disorders like rheumatoid arthritis, Crohn’s disease and ulcerative colitis [Citation5].
Thus, kinase inhibitors developed for cancer are also used for the treatment of disorders involving immune cell hyperactivation and in some cases for the selective reactivation of immune cell function [Citation4]. The most notable inhibitors being employed for the treatment of patients with immunologic diseases are inhibitors targeting Janus kinase (JAK) 2 and JAK3 [Citation4]. An interesting example of how a kinase inhibitor that has been already FDA-approved for the treatment of non-small cell bronchial carcinomas can be used to modulate immune responses is provided by gefitinib. In preclinical studies, it limited the growth of Mycobacterium tuberculosis by promoting targeting of the bacteria to the host cell lysosomes [Citation4,Citation6]. Yet, though oncogenic kinases do obviously have fundamental importance in immune cell signaling, their functions in this context are often only insufficiently investigated. The need to further clarify the role of kinases in the immune response is underlined by the fact, that synergetic treatment regimens have been a norm to treat cancer patients in the clinics [Citation7]. It is therefore pertinent to understand the biological function of the “druggable targets” in the immune cells to manifest successful combinatorial treatments.
MAPK pathways – signaling modules contributing to tumorigenesis and immune responses
Mitogen-Activated Protein Kinases (MAPKs) are key enzymes that regulate fundamental cellular processes like proliferation, migration and differentiation. There are 14 mammalian MAPKs that define seven distinct pathways and they are broadly classified into atypical and typical MAPKs [Citation8]. The classical MAPK includes the highly conserved RAF-MEK1/2-ERK1/2 cascade, the members of which are found to be deregulated in approximately one-third of all human cancers [Citation9,Citation10]. The RAF-MEK1/2-ERK1/2 pathway is activated in response to growth factors via the protooncogene RAS, which is mutated in nearly 30% of all human cancers making it to one of the most frequently activated oncogenes [Citation11]. RAFs are serine/threonine kinases and members of the classical MAPK cascade. Among the three RAF isoforms (ARAF, BRAF and CRAF), BRAF is the most frequently mutated isoform in human cancers [Citation12]. Therefore, extensive studies were carried out to elucidate the role of the ERK1/2 pathway in tumorigenesis and to develop targeting strategies. These efforts have led to the FDA-approved BRAF inhibitors, vemurafenib [Citation13] and dabrafenib [Citation14], as well as the MEK inhibitors, trametinib and cobimetinib [Citation15]. While studies pertaining to inhibition of the RAF-MEK1/2-ERK1/2 pathway in order to impede tumor growth are pursued, it is prudent to investigate the role of these cascades in modulating immune responses to mitigate the unexpected side effects and to maximize the anti-tumor immune responses. In this mini-review, we aim to summarize the role of MAPKs in the immune system and then discuss the role of the cascade components in controlling immune responses.
Role of MAPKs in dendritic cells
The anti-tumor immune response is a complex interplay of a variety of signals and interactions so that an efficient immune response relies on a series of events and on the functionality of different immune cells. An effective anti-tumor immune response, which results in the elimination of cancer cells, requires, among other things, functional dendritic cells (DCs). Tumor-associated DCs capture and transport cancer-associated antigens to the draining lymph nodes in order to induce priming and activation of T cells [Citation16]. Activated T cells subsequently traffic to the tumor, specifically recognize cancer cells and mediate their killing [Citation16]. But due to genetic instability and constant cell division, cancer cells can exert immune-suppressive effects, which result in a reduced immunogenicity and subsequently in the evasion of immune surveillance. The mechanistic basis and strategies of how cancer cells can suppress the anti-tumor response are already extensively reviewed elsewhere [Citation17,Citation18] and are only briefly summarized here. The immune-suppressive effects induced by the tumors are multilayered and include the loss of target antigen expression [Citation19], the production of immune-suppressive mediators like the secretion of the cytokine transforming growth factor (TGF)-β [Citation20], the recruitment of immune-suppressive cells like regulatory T cells to the tumor microenvironment [Citation21] and recruitment of inflammatory cells, which might promote for tumor growth. In this context, myeloid-derived suppressor cells (MDSCs) [Citation22] and alternatively activated M2 macrophages play a pivotal role [Citation17,Citation23].
As already mentioned, the activation of DCs is required to initiate a profound T cell-mediated anti-tumor immune response as DCs are the central regulators of the adaptive immune system [Citation24,Citation25]. The activation of DCs is mediated through pattern recognition receptors (PRRs), which are able to sense pathogen-associated molecular patterns (PAMPs) and antigens associated with immunogenic or tolerogenic cell death [Citation16,Citation26]. The best-studied family of PRRs is represented by Toll-like receptors (TLRs). The three major signaling pathways activated by TLR signaling are MAPKs, nuclear factor-κB (NF-κB) and interferon regulatory factors (IRFs) [Citation26]. Activation of MAPK and NF-κκB pathways is also common with other classes of PRRs, including RIG-I-like receptors (RLRs), NOD-like receptors (NLRs) and C-type lectin receptors (CLRs) [Citation27]. Consequently, these signaling modules are crucial to dictate the functional consequences of DC activation [Citation28]. Studies have focused especially on the three MAPKs: p38, JNK and ERK1/2, and their roles in translating the PRR signals into the correct physiological responses. Concerning DC maturation, it has been suggested that p38 MAPK is essential for the maturation of DCs into fully functional DCs, while JNK positively influences the surface antigen expression and cytokine secretion without having major effects on the allostimulatory functions of DCs [Citation29]. Previous studies have shown that DC precursors with increased p38 MAPK activation and production of autocrine cytokines such as IL-6, as found in myeloma patients, differentiate into phenotypically and functionally defective moDCs [Citation30]. The induction of p38 activation in DC precursors by tumor-derived suppressive factors and the resulting impaired differentiation and function of DCs has been demonstrated in both murine tumor models and in cancer patients [Citation30–Citation33]. In these cases, treatment with p38 MAPK inhibitors has been beneficial to restore DC function [Citation30–Citation33]. Furthermore, inhibition of p38 MAPK activity during DC differentiation enhances their stimulatory function and these DCs even compromise the inhibitory effects of Tregs on effector T cells [Citation31]. The pronounced stimulatory capacity of DCs after inhibiting p38 MAPK is partially mediated through the upregulation of OX40L expression and OX40 L/OX40 interactions between DCs and T cells further inhibit the conversion of effector T cells to Tregs [Citation31]. The beneficial effects of p38 MAPK inhibition during DC differentiation have also been shown to enhance anti-tumor immune responses after immunization with inhibitor-treated bone-marrow-derived DCs (BMDCs) [Citation31].
In the case of ERK1/2, it has been shown that ERK1/2 activation during DC maturation induces the production of inflammatory cytokines like TNFα and IL-1β [Citation27,Citation34,Citation35], whereas it negatively regulates IL-12 production [Citation29]. Regarding DC differentiation, it has been demonstrated that DCs from ERK1−/- mice develop normally with viability [Citation36]. However, in the DC precursors from myeloma patients, the enhanced p38 MAPK activation is accompanied by a reduced ERK1/2 activation, and inhibition of p38 MAPK has restored the activity of ERK1/2 signaling [Citation30]. In our own study, we observed oscillating ERK1/2 activation during the differentiation from human monocytes to monocyte-derived DCs (moDCs) [Citation37]. Thus, it is likely that ERK1/2 signaling contributes to the differentiation of functional DCs.
MAPK signaling not only plays a role in the signal transduction of PRRs in innate immune cells but also induced by T cell receptor (TCR) stimulation [Citation38,Citation39]. In particular, the ERK pathway represents one of the important MAPK pathways downstream of TCR signaling and is involved in the regulation of the priming and proliferation of naïve T cells [Citation40,Citation41]. But, while ERK1/2 signaling is crucial for T cell priming, a previous study has shown that the inhibition of the pathway by MEK1/2 inhibitors promotes the survival of tumor-infiltrating CD8+ T cells [Citation42]. Thus, ERK1/2 activation does not only play an important role in tumorigenesis but it is also part of signal transductions in cells of the immune system. Therefore, it has to be carefully evaluated, in which relevant steps within an anti-tumor immune response cascade are affected upon the inhibition of the pathway.
RAF-MEK-ERK activation upon TLR signaling
While it is believed that the signal transduction from the TCR to ERK1/2 is mediated through the small GTPase RAS and the RAF-MEK1/2-ERK1/2 cascade [Citation43], the activation of the ERK1/2 pathway upon PRR signaling in other immune cells still raises some questions. It has been initially shown in macrophages, that ERK1/2 but not p38 MAPK and JNK activation by TLRs is mediated by the MAP3K tumor progression locus 2 (Tpl-2) [Citation34]. It has been demonstrated that, under Tpl-2 deficient conditions, lipopolysaccharide (LPS)-induced ERK1/2 activation is impaired in both bone-marrow-derived DCs (BMDCs) and macrophages [Citation35]. Further, specific knockout studies showed that Tpl-2−/- BMDMs as well as Tpl-2−/- BMDCs produce elevated levels of IL-12p40 and IL-12p70 protein, which matched the effect of pharmacological inhibitors against MEK1/2 leading to ERK1/2-inhibition [Citation35]. Since previous studies examining the influence of MAPK signaling pathway on DCs mainly applied MEK inhibitors to block the pathway, we were curious to evaluate the direct role of RAF kinases in DCs. Hence, in the following sections, we are now focusing more on the intrinsic regulation and activation of RAF kinases in moDCs.
RAF kinases are stabilized during DC differentiation
RAF kinases are serine/threonine kinases and this family comprises three isoforms, ARAF, BRAF and CRAF. They are direct RAS effector proteins and thus playing the role of the proximal MAP3K to activate the classical MAPK cascade. We found that all three RAF kinases were stabilized on the protein levels during moDC differentiation and that they were active in moDCs [Citation37]. The underlying mechanism that regulates the proteostasis of RAF proteins and leads to the upregulation of RAF proteins in moDCs remains to be investigated. Early studies have already demonstrated that RAF kinases can be degraded by the proteasome [Citation44,Citation45]. In line with this, E3 ligases labeling RAF kinases with ubiquitin are described [Citation46–Citation48]. Additionally, studies from the lab demonstrated that X-linked inhibitor of apoptosis protein (XIAP) and cellular inhibitor of apoptosis protein (cIAP) promote CRAF degradation by promoting the binding of the E3 ligase CHIP to the Hsp90-CRAF complex [Citation49]. So far we were not able to detect significant changes in the poly-ubiquitination of the RAF proteins during moDC differentiation. One reason for this could be technical limitations to enrich ubiquitinated species of endogenous proteins from moDCs. It would be interesting to further elucidate whether the stability of RAF kinases is regulated by the ubiquitin-proteasome machinery or whether other degradation mechanisms like the lysosomal pathway play a role. Since all three RAF kinases are upregulated during moDC differentiation, a common regulatory mechanism is possible. Additionally, it is tempting to propose that activation of RAF kinases may play a direct role in regulating the stability of these kinases in DCs as in tumor cells [Citation50].
RAF activation during DC maturation
Concerning RAF activation, we uncovered that upon LPS stimulation the activation kinetics of RAF kinases differ with that of MEK1/2 kinases [Citation37]. Intriguingly, pan-RAF inhibition reduced ERK1/2 activation under non-stimulating conditions, but not under LPS-stimulating conditions [Citation37]. In contrast, both pan-RAF- and MEK1/2-inhibition prevented ERK1/2 activation in stimulated CD4+ T cells [Citation37]. Our results are in line with the published observations that in DCs, MAPK activation upon TLR signaling is promoted through other kinases possibly through the aforementioned Tpl-2 kinase and suggest that RAF proteins are here probably not required for MAPK signaling (). But this raises the question of how the activation of RAF kinases is regulated in DCs. Since RAF is an effector protein for activated RAS, it is important to directly examine the activation of RAS and interaction with CRAF upon LPS stimulation in these cell types. So far it is not precisely understood whether TLR4 signaling results in RAS activation [Citation51] and if there is any isoform specificity as RAS presents three isoforms: KRAS, HRAS and NRAS. Another regulatory step in RAF activation is the dimerization of RAF kinases. In our study, we observed a complex formation especially between ARAF and BRAF at steady state in moDCs and an induced interaction between ARAF and CRAF under LPS-stimulating conditions [Citation37]. But in the case of ARAF, we only detected a marginal activity in a kinase assay [Citation37]. RAF dimerization is not necessarily linked to catalytic activity [Citation52], which provides new functional possibilities for ARAF. As described by Rebocho et al., ARAF can function as a scaffold to stabilize BRAF-CRAF heterodimers [Citation53] and the study by Mooz et al. suggested that RAF heterodimers can compete for each other thus regulating the downstream signaling [Citation54]. Consequently, further studies are needed to not only clarify how RAF kinases are activated but also how the different heterodimers differ in their function in the context of DC activation.
Figure 1. Differences in RAF-MEK1/2-ERK1/2 signaling in moDCs and T cells.
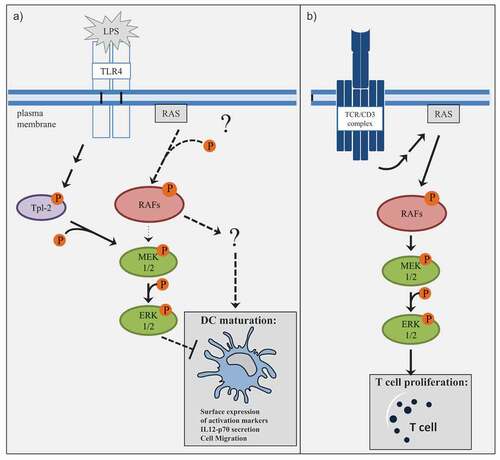
MEK-independent functions of RAF kinases
Consistent with the different activation kinetics of RAF and MEK1/2 kinases, we detected that pan-RAF inhibition and MEK1/2 inhibition exhibited opposing effects on the LPS-induced surface expression of activation markers and IL-12p70 secretion of moDCs [Citation37]. Consistent with published data, inhibition of MEK1/2 has compromised moDC maturation. In contrast, pan-RAF inhibition had a negative effect on DC maturation, which in turn resulted in a reduced ability to activate CD4+ T cells [Citation37]. These observations obviously strengthen the argument that RAF kinases probably have MEK1/2-independent functions in stimulated moDCs (). While BRAF is the original RAF precursor and is considered the primary MEK1/2 kinase, MEK1/2-independent functions are described for ARAF and CRAF. For instance, it has been reported that CRAF activates adenylyl cyclases by direct phosphorylation [Citation55,Citation56], which could serve as a negative feedback event, since the generation of cAMP leads to the activation of the negative regulator PKA [Citation57]. Another study conducted in primary cardiac myocytes has shown that an induced form of activated CRAF regulates the expression of a hypertrophic marker differently compared to activated MEK1/2 [Citation58]. But also kinase-independent functions have been described for RAF proteins: Ehrenreiter et al. described that the physical interaction of CRAF with the Rho effector Rok-α is required to regulate its localization and activation [Citation59]. Loss of CRAF results in hyperactivity and incorrect localization of Rok-α to the plasma membrane, which leads to defects in adhesion and motility [Citation59]. Although MEK1/2 is the only commonly accepted RAF substrate it is tempting to speculate that RAF proteins probably recruit or regulate other substrates to fulfill their functions in moDCs (). As most of these observations were made in other cell types, further studies are clearly warranted in order to identify new potential RAF substrates and other interaction partners which may contribute to the RAF-driven DC activation phenotype.
The differential effects of BRAF- and pan-RAF-inhibitors on DC function
As mentioned above there are currently two FDA-approved RAF inhibitors, vemurafenib [Citation13] and dabrafenib [Citation14], which are ATP-competitive inhibitors targeting BRAF V600E mutant prevalent in many cancers. Several studies were carried out addressing the question of how the BRAF inhibitors affect isolated immune cell populations. It has been demonstrated that vemurafenib and dabrafenib do not have detrimental effects on human moDC function [Citation60,Citation61]. In human primary DCs, vemurafenib but not dabrafenib impaired the R848-induced maturation and cytokine secretion, although the inhibitory effect was absent when vemurafenib was applied to total PBMCs [Citation62]. In murine BMDCs, Hajek et al. identified a common off-target effect of vemurafenib and dabrafenib leading to inflammasome activation and IL-1β production [Citation63]. In human moDCs, both inhibitors moderately activated the caspase-1-inflammasome, but IL-1β levels were not elevated. In the case of human primary DCs, most samples exhibited increased IL-1β levels after treatment with dabrafenib, but not with vemurafenib [Citation63]. Their finding that only a fraction of primary DCs respond to dabrafenib is in agreement with a clinical observation that the side effects of BRAF inhibitors markedly differ between patients treated with these inhibitors. While patients treated with vemurafenib suffer from pronounced rash, dabrafenib treatment often causes fever attacks. This led the authors to hypothesize that there might exist a genetic polymorphism that controls the immunomodulatory effects of BRAF inhibitors [Citation63].
Effects of pan-RAF inhibitors on DCs
Despite the remarkable clinical success of BRAF inhibitors in the treatment of melanomas [Citation3,Citation64–Citation67], patients rapidly develop drug resistance and secondary malignancies. Extensive studies revealed that this is caused by paradoxical activation of ERK1/2, which occurs in the absence of BRAF-activating mutations [Citation68]. Consequently, next-generation inhibitors are being developed to prevent the paradoxical ERK activation. Pan-RAF inhibitors are third-generation inhibitors, which have the benefit to bind monomeric and dimeric RAF proteins with equal efficiency [Citation69]. Currently, several pan-RAF inhibitors like LY3009120 [Citation70], TAK632 [Citation71] and TAK580 [Citation72] have been developed and assessed preclinically [Citation73].
In our study already discussed above, we employed the inhibitor LY3009120 in order to investigate how pan-RAF inhibition affects DCs [Citation37]. The observations that vemurafenib and dabrafenib do not compromise DC function and T cell proliferation [Citation60,Citation61] are in contrast to our study, which showed that pan-RAF inhibition by LY3009120 impairs the LPS-induced surface expression of activation markers and IL-12p70 secretion of moDCs as well as the ability of moDCs to activate CD4+ T cells () [Citation37]. In an in vivo experiment, where we treated C57BL/6 J mice with tumor-inhibiting concentrations of LY3009120, we additionally observed a reduction of LPS-mediated activation of DCs in the spleen [Citation37]. Therefore, our study raises the question whether pan-RAF inhibitors might have other adverse effects in the clinics like the BRAF inhibitors, vemurafenib or dabrafenib. Interestingly, consistent with our study Müller et al. assigned a special role to pan-RAF inhibition since only pan-RAF inhibition in multiple myeloma cells prevents their survival [Citation74]. Intriguingly, they also observed different effects after pan-RAF or MEK1/2 inhibition suggesting RAF-dependent and MEK-independent functions, which is exemplified by RAF-dependent regulation of PI3 K signaling [Citation74]. More studies are needed to elucidate whether MEK-independent functions of RAF kinases are also evident in other cell types after the application of pan-RAF inhibitors.
Effects of BRAF- and pan-RAF-inhibitors on T cells
In addition to the studies investigating the effects of RAF inhibitors on DCs, it has been examined how these inhibitors further influence T cells. Studies revealed that in contrast to the pharmacological inhibition of MEK1/2, which results in an impaired T cell proliferation, BRAF inhibition did not affect T cell responses [Citation61,Citation75]. This was explained by a paradoxical activation of MAPK signaling in T cells upon RAF inhibitor treatment [Citation76]. Consequently, it seems as if BRAF inhibitors do not have detrimental effects on DCs and T cells. In contrast, the direct treatment of CD4+ T cells with the pan-RAF inhibitor LY3009120 inhibited CD4+ T cell proliferation in the same way as the MEK1/2 inhibitor trametinib [Citation37]. This suggests that pan-RAF inhibitors exhibit different effects as BRAF inhibitors in a cell-type-dependent manner. On the other hand, these observations allude to a model where MAPK signaling acts linear in activated T cells but not in DCs.
Although the negative impact of MEK1/2 inhibitors on T cell priming is known, no difference in intratumoral CD8+ T cells is observed in patients treated with BRAF inhibitors alone in comparison to patients treated with a combination of BRAF and MEK1/2 inhibitors [Citation77]. The assumption that MEK1/2 inhibitors do not affect T cell infiltration and expansion was strengthened by the aforementioned study of Ebert et al. revealing that MEK1/2 inhibition had a beneficial effect on the survival of tumor-infiltrating CD8+ T cells [Citation42]. These studies again warrant careful evaluation of the role of the various MAPK constituents in controlling immune responses.
Conclusion and perspectives
For an effective cancer treatment, it is not only important to know how the key immune cells are affected by the administered inhibitors but it is pertinent to examine the overall response contributing to the anti-tumor immune response. Studies have previously demonstrated a link between MAPK pathway activation in cancer and the suppression of the anti-tumor immunity. For instance, BRAF inhibition in melanoma cell lines decreases immune-suppressive factors like IL-10, VEGF and IL-6 and enhances the expression of melanocyte differentiation antigens (MDA) to improve recognition by MDA-specific T cells [Citation78]. In biopsies of patients, a decrease of intratumoral IL-6 and IL-8 as well as an increased CD8+ T cell infiltration was confirmed [Citation77]. But exhaustion markers like TIM-3, PD-1 and PD-L1 are elevated and probably contribute to the observed phenomenon that the CD8+ T cell infiltrate returns again to pre-treatment levels [Citation77,Citation79].
The altered tumor microenvironment after BRAF inhibitor treatment provides the scientific rationale to combine BRAF inhibitors with immune checkpoint inhibitors. As only recently reviewed by Pelster et al., combination of BRAF and MEK inhibitors with immunotherapeutics becomes a promising approach for cancer treatment since preclinical work has demonstrated a potential therapeutic benefit in mouse models [Citation80]. Initial clinical trials with melanoma patients receiving BRAF inhibitors alone or in combination with MEK inhibitors as targeted therapeutics and immune checkpoint inhibitors targeting CTLA-4, PD-1 or PD-L1, further suggest an increase in response, but also reveal additional toxicities [Citation80]. More clinical trials are currently carried out to deepen the knowledge about the duration of the responses, the resulting toxicities and how to handle the treatment regimes [Citation80]. Additionally, other combinatorial strategies are tested in the clinics, in which especially vemurafenib is combined with other forms of immunotherapies. For instance, clinical studies have been going on to evaluate a combination of vemurafenib with cytokines like IL-2 and IFNα2 in melanoma patients [Citation80,Citation81]. But in the case of the combination of vemurafenib with IL-2 alone, a lack of synergism was reported [Citation80,Citation82,Citation83]. Another approach is currently being pursued by two clinical studies in metastatic melanoma patients, in which vemurafenib is combined with an adoptive transfer of tumor-infiltrating lymphocytes (TIL). To improve the efficiency of treatment, patients are additionally given lymphodepleting chemotherapy before the TIL infusion and a high dose of IL-2 after the infusion to support the activation and expansion of T cells in vivo [Citation80,Citation81]. First, results of one of the clinical studies suggest that this treatment is a well-tolerated therapy with the potential for a durable response [Citation80,Citation84].
Besides the establishment of combinatorial treatments with targeted inhibitors and immunotherapeutics an additional approach to improve cancer treatment is the development of next-generation inhibitors like the aforementioned pan-RAF inhibitors to solve the problem of paradoxical ERK1/2 activation, which is induced by BRAF inhibitors in the absence of BRAF mutations [Citation68]. An initial phase I clinical study with LY3009120 in patients with advanced or metastatic cancer has been terminated due to a lack of sufficient clinical efficacy [Citation81]. However, there are also other pan-RAF inhibitors such as TAK580 and BGB283, which are currently tested in the clinics. TAK580 is clinically evaluated as a single treatment for metastatic melanoma and low-grade glioma, but also in combination with other inhibitors such as MLN0128, Alisertib, Paclitaxel, Cetuximab and Irinotecan [Citation81]. In the case of BGB283, an inhibitor for RAF and EGFR, clinical studies have been conducted to assess the safety and pharmacokinetics in patients with solid tumors, while another clinical study is recruiting patients in order to investigate the combination of BGB283 with the MEK inhibitor PD-0325901 [Citation81]. Only recently it has been reported that BGB283 has an acceptable risk-benefit profile and antitumor activity has been shown for this inhibitor in patients with BRAF V600E mutated solid tumors [Citation85].
As we outlined in our review, we observed fundamental differences between the pan-RAF inhibitor LY3009120 and the clinically approved BRAF inhibitors at the level of individual immune cell types. Thus, supposedly small changes in the targeting strategy, as it is the case with pan-RAF inhibitors or RAF dimer breaker, can eventually result in new unknown side effects. Therefore, it is advisable that new types of inhibitors need to be reevaluated not only for their effectiveness in blocking the MAPK pathway and tumor growth but also for their effects on the immune system.
Based on the discussed studies on RAF kinases, our review further highlights that there are still many questions to be answered about druggable kinases and their functions in the immune system. The following questions still need to be addressed: How are RAF isoforms stabilized in DCs during differentiation? How are RAF kinases activated? What is the role of RAS in this process? As inhibitors directly targeting specific mutations of RAS (especially the KRASG12 C mutants) have entered clinical trials, their effects on immune cells need further characterization. What are the substrates of RAF kinases in DCs? Do patients treated with pan-RAF inhibitors show any adverse effects? How do RAF kinases contribute toward the migration and polarity of DCs? Answering these questions will not only enhance the current understanding of how these molecules mediate immune responses but also help us to adroitly administer RAF inhibitors in the clinic.
Acknowledgments
The authors would like to thank CRC1292-DFG for support. KR is a Heisenberg Professor of the DFG (RA1739/7-1) and a GFK fellow. Christiane Schönfeld is acknowledged for critical reading of the manuscript.
Disclosure statement
The authors declare that they have no conflict of interest.
Additional information
Funding
References
- Melnikova I, Golden J. Targeting protein kinases. Nat Rev Drug Discov. 2004;3(12):993–994.
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1(9):727–730.
- Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov Today. 2016;21(1):5–10.
- Pandey R, Kapur R. Kinase inhibitors in clinical practice: an expanding world. J Allergy Clin Immunol. 2018;141(2):522–524.
- Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol Res. 2019;144:19–50.
- Sogi KM, Lien KA, Johnson JR, et al. The tyrosine kinase inhibitor gefitinib restricts mycobacterium tuberculosis growth through increased lysosomal biogenesis and modulation of cytokine signaling. ACS Infect Dis. 2017;3(8):564–574.
- Gotwals P, Cameron S, Cipolletta D, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer. 2017;17(5):286–301.
- Coulombe P, Meloche S. Atypical mitogen-activated protein kinases: structure, regulation and functions. Biochim Biophys Acta. 2007;1773(8):1376–1387.
- Yang SH, Sharrocks AD, Whitmarsh AJ. Transcriptional regulation by the MAP kinase signaling cascades. Gene. 2003;320:3–21.
- Krishna M, Narang H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci. 2008;65(22):3525–3544.
- Fernandez-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer. 2011;2(3):344–358.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954.
- Kim G, McKee AE, Ning YM, et al. FDA approval summary: vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin Cancer Res. 2014;20(19):4994–5000.
- Ballantyne AD, Garnock-Jones KP. Dabrafenib: first global approval. Drugs. 2013;73(12):1367–1376.
- Cheng Y, Tian H. Current development status of MEK inhibitors. Molecules. 2017;22:10.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10.
- Vinay DS, Ryan EP, Pawelec G, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35(Suppl):S185–S198.
- Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550.
- Maeurer MJ, Gollin SM, Martin D, et al. Tumor escape from immune recognition: lethal recurrent melanoma in a patient associated with downregulation of the peptide transporter protein TAP-1 and loss of expression of the immunodominant MART-1/Melan-A antigen. J Clin Invest. 1996;98(7):1633–1641.
- Pasche B. Role of transforming growth factor beta in cancer. J Cell Physiol. 2001;186(2):153–168.
- Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949.
- Yang L, DeBusk LM, Fukuda K, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6(4):409–421.
- Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22(2):231–237.
- Gardner A, Ruffell B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016;37(12):855–865.
- Salmon H, Idoyaga J, Rahman A, et al. Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. 2016;44(4):924–938.
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801.
- Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13(9):679–692.
- Dalod M, Chelbi R, Malissen B, et al. Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming. Embo J. 2014;33(10):1104–1116.
- Nakahara T, Moroi Y, Uchi H, et al. Differential role of MAPK signaling in human dendritic cell maturation and Th1/Th2 engagement. J Dermatol Sci. 2006;42(1):1–11.
- Wang S, Hong S, Yang J, et al. Optimizing immunotherapy in multiple myeloma: restoring the function of patients’ monocyte-derived dendritic cells by inhibiting p38 or activating MEK/ERK MAPK and neutralizing interleukin-6 in progenitor cells. Blood. 2006;108(13):4071–4077.
- Lu Y, Zhang M, Wang S, et al. p38 MAPK-inhibited dendritic cells induce superior antitumour immune responses and overcome regulatory T-cell-mediated immunosuppression. Nat Commun. 2014;5:4229.
- Wang S, Yang J, Qian J, et al. Tumor evasion of the immune system: inhibiting p38 MAPK signaling restores the function of dendritic cells in multiple myeloma. Blood. 2006;107(6):2432–2439.
- Oosterhoff D, Lougheed S, van de Ven R, et al. Tumor-mediated inhibition of human dendritic cell differentiation and function is consistently counteracted by combined p38 MAPK and STAT3 inhibition. Oncoimmunology. 2012;1(5):649–658.
- Dumitru CD, Ceci JD, Tsatsanis C, et al. TNF-alpha induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell. 2000;103(7):1071–1083.
- Kaiser F, Cook D, Papoutsopoulou S, et al. TPL-2 negatively regulates interferon-beta production in macrophages and myeloid dendritic cells. J Exp Med. 2009;206(9):1863–1871.
- Dillon S, Agrawal A, Van Dyke T, et al. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J Immunol. 2004;172(8):4733–4743.
- Riegel K, Schloder J, Sobczak M, et al. RAF kinases are stabilized and required for dendritic cell differentiation and function. Cell death and differentiation. 2019.
- Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619.
- Cantrell D. T cell antigen receptor signal transduction pathways. Annu Rev Immunol. 1996;14:259–274.
- DeSilva DR, Jones EA, Favata MF, et al. Inhibition of mitogen-activated protein kinase kinase blocks T cell proliferation but does not induce or prevent anergy. J Immunol. 1998;160(9):4175–4181.
- D’Souza WN, Chang CF, Fischer AM, et al. The Erk2 MAPK regulates CD8 T cell proliferation and survival. J Immunol. 2008;181(11):7617–7629.
- Ebert PJR, Cheung J, Yang Y, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity. 2016;44(3):609–621.
- Kortum RL, Rouquette-Jazdanian AK, Samelson LE. Ras and extracellular signal-regulated kinase signaling in thymocytes and T cells. Trends Immunol. 2013;34(6):259–268.
- Schulte TW, An WG, Neckers LM. Geldanamycin-induced destabilization of Raf-1 involves the proteasome. Biochem Biophys Res Commun. 1997;239(3):655–659.
- Grbovic OM, Basso AD, Sawai A, et al. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc Natl Acad Sci U S A. 2006;103(1):57–62.
- Schneider T, Martinez-Martinez A, Cubillos-Rojas M, et al. The E3 ubiquitin ligase HERC1 controls the ERK signaling pathway targeting C-RAF for degradation. Oncotarget. 2018;9(59):31531–31548.
- Li Z, Zhou L, Prodromou C, et al. HECTD3 mediates an HSP90-dependent degradation pathway for protein kinase clients. Cell Rep. 2017;19(12):2515–2528.
- Hong SW, Jin DH, Shin JS, et al. Ring finger protein 149 is an E3 ubiquitin ligase active on wild-type v-Raf murine sarcoma viral oncogene homolog B1 (BRAF). J Biol Chem. 2012;287(28):24017–24025.
- Dogan T, Harms GS, Hekman M, et al. X-linked and cellular IAPs modulate the stability of C-RAF kinase and cell motility. Nat Cell Biol. 2008;10(12):1447–1455.
- Noble C, Mercer K, Hussain J, et al. CRAF autophosphorylation of serine 621 is required to prevent its proteasome-mediated degradation. Mol Cell. 2008;31(6):862–872.
- Bauerfeld C, Samavati L. Role of MEK1 in TLR4 mediated signaling. J Cell Signal. 2017;2:1.
- Shaw AS, Kornev AP, Hu J, et al. Kinases and pseudokinases: lessons from RAF. Mol Cell Biol. 2014;34(9):1538–1546.
- Rebocho AP, Marais R. ARAF acts as a scaffold to stabilize BRAF:CRAF heterodimers. Oncogene. 2013;32(26):3207–3212.
- Mooz J, Oberoi-Khanuja TK, Harms GS, et al. Dimerization of the kinase ARAF promotes MAPK pathway activation and cell migration. Sci Signal. 2014;7(337):ra73.
- Beazely MA, Alan JK, Watts VJ. Protein kinase C and epidermal growth factor stimulation of Raf1 potentiates adenylyl cyclase type 6 activation in intact cells. Mol Pharmacol. 2005;67(1):250–259.
- Ding Q, Gros R, Gray ID, Taussig R, Ferguson SS, Feldman RD. Raf kinase activation of adenylyl cyclases: isoform-selective regulation. Mol Pharmacol. 2004;66(4):921–928.
- Matallanas D, Birtwistle M, Romano D, et al. Raf family kinases: old dogs have learned new tricks. Genes Cancer. 2011;2(3):232–260.
- Jette C, Thorburn A, Raf-induced A. MEK-independent signaling pathway regulates atrial natriuretic factor gene expression in cardiac muscle cells. FEBS Lett. 2000;467(1):1–6.
- Ehrenreiter K, Piazzolla D, Velamoor V, et al. Raf-1 regulates Rho signaling and cell migration. J Cell Biol. 2005;168(6):955–964.
- Ott PA, Henry T, Baranda SJ, et al. Inhibition of both BRAF and MEK in BRAF(V600E) mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol Immunother. 2013;62(4):811–822.
- Vella LJ, Pasam A, Dimopoulos N, et al. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunol Res. 2014;2(4):351–360.
- Tel J, Koornstra R, de Haas N, et al. Preclinical exploration of combining plasmacytoid and myeloid dendritic cell vaccination with BRAF inhibition. J Transl Med. 2016;14:88.
- Hajek E, Krebs F, Bent R, et al. BRAF inhibitors stimulate inflammasome activation and interleukin 1 beta production in dendritic cells. Oncotarget. 2018;9(47):28294–28308.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516.
- Lee JT, Li L, Brafford PA, et al. PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment Cell Melanoma Res. 2010;23(6):820–827.
- Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–714.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365.
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–430.
- Durrant DE, Morrison DK. Targeting the Raf kinases in human cancer: the Raf dimer dilemma. Br J Cancer. 2018;118(1):3–8.
- Vakana E, Pratt S, Blosser W, et al. LY3009120, a panRAF inhibitor, has significant anti-tumor activity in BRAF and KRAS mutant preclinical models of colorectal cancer. Oncotarget. 2017;8(6):9251–9266.
- Okaniwa M, Hirose M, Arita T, et al. Discovery of a selective kinase inhibitor (TAK-632) targeting pan-RAF inhibition: design, synthesis, and biological evaluation of C-7-substituted 1,3-benzothiazole derivatives. J Med Chem. 2013;56(16):6478–6494.
- Sun Y, Alberta JA, Pilarz C, et al. A brain-penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low-grade astrocytomas. Neuro Oncol. 2017;19(6):774–785.
- Pan JH, Zhou H, Zhu SB, et al. Development of small-molecule therapeutics and strategies for targeting RAF kinase in BRAF-mutant colorectal cancer. Cancer Manag Res. 2018;10:2289–2301.
- Muller E, Bauer S, Stuhmer T, et al. Pan-Raf co-operates with PI3K-dependent signalling and critically contributes to myeloma cell survival independently of mutated RAS. Leukemia. 2017;31(4):922–933.
- Boni A, Cogdill AP, Dang P, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70(13):5213–5219.
- Callahan MK, Masters G, Pratilas CA, et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol Res. 2014;2(1):70–79.
- Frederick DT, Piris A, Cogdill AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19(5):1225–1231.
- Reddy SM, Reuben A, Wargo JA. Influences of BRAF inhibitors on the immune microenvironment and the rationale for combined molecular and immune targeted therapy. Curr Oncol Rep. 2016;18(7):42.
- Cooper ZA, Juneja VR, Sage PT, et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol Res. 2014;2(7):643–654.
- Pelster MS, Amaria RN. Combined targeted therapy and immunotherapy in melanoma: a review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther Adv Med Oncol. 2019;11:1758835919830826.
- ClinicalTrials.gov. Accesse 17 April 2020. https://clinicaltrials.gov/ct2/home.
- Clark JI, Singh J, Ernstoff MS, et al. A multi-center phase II study of high dose interleukin-2 sequenced with vemurafenib in patients with BRAF-V600 mutation positive metastatic melanoma. J Immunother Cancer. 2018;6(1):76.
- Mooradian MJ, Reuben A, Prieto PA, et al. A phase II study of combined therapy with a BRAF inhibitor (vemurafenib) and interleukin-2 (aldesleukin) in patients with metastatic melanoma. Oncoimmunology. 2018;7(5):e1423172.
- Deniger DC, Kwong ML, Pasetto A, et al. A pilot trial of the combination of vemurafenib with adoptive cell therapy in patients with metastatic melanoma. Clin Cancer Res. 2017;23(2):351–362.
- Desai J, Gan H, Barrow C, et al. Phase I, open-label, dose-escalation/dose-expansion study of lifirafenib (BGB-283), an RAF family kinase inhibitor, in patients with solid tumors. J Clin Oncol. 2020;38(19):2140–2150.;JCO1902654.