
ABSTRACT
Altered telomere maintenance mechanism (TMM) is linked to increased DNA damage at telomeres and telomere uncapping. We previously showed that HIV-1 latent cells have altered TMM and are susceptible to ligands that target G-quadruplexes (G4) at telomeres. Susceptibility of latent cells to telomere targeting could potentially be used to support approaches to eradicate HIV reservoirs. However, G4 ligands also target G-quadruplexes in promoters blocking gene transcription. Since HIV promoter sequence can form G-quadruplexes, we investigated whether G4 ligands interfere with HIV-1 promoter activity and virus reactivation from latency, and whether telomere targeting could be combined with latency reversing agents (LRAs) to promote elimination of HIV reservoirs. Our results indicate that Sp1 binding region in HIV-1 promoter can adopt G4 structures in duplex DNA, and that in vitro binding of Sp1 to G-quadruplex is blocked by G4 ligand, suggesting that agents targeting telomeres interfere with virus reactivation. However, our studies show that G4 agents do not affect HIV-1 promoter activity in cell culture, and do not interfere with latency reversal. Importantly, primary memory CD4 + T cells infected with latent HIV-1 are more susceptible to combined treatment with LRAs and G4 ligands, indicating that drugs targeting TMM may enhance killing of HIV reservoirs. Using a cell-based DNA repair assay, we also found that HIV-1 infected cells have reduced efficiency of DNA mismatch repair (MMR), and base excision repair (BER), suggesting that altered TMM in latently infected cells could be associated with accumulation of DNA damage at telomeres and changes in telomeric caps.
Introduction
HIV persistence in the form of latent viral reservoirs remains a critical limitation to HIV cure. One approach to eliminate this cell population is to reactivate virus expression with latency reversing agents (LRAs) in order to trigger apoptosis and to induce an antiviral immune response. However, clinical trials of LRAs have been disappointing, indicating that supporting approaches are needed [Citation1–Citation7]. We previously showed that cells infected with latent HIV have a deficiency in their DNA damage response (DDR) and altered telomere maintenance mechanisms (TMM) resulting in elongation of telomeres [Citation8–Citation10]. We also found that infected cells are susceptible to DDR targeting and to ligands targeting telomeres, such as BRACO19 and TMPyP4 that stabilize G-quadruplexes (G4) and promote telomere uncapping. Altered TMM is therefore a unique characteristic of cells infected with latent HIV that distinguishes them from healthy cells. Hence, targeting telomeres could potentially be used to support therapeutic approaches aiming at killing HIV reservoirs.
One concern with this approach is that G4 ligands targeting telomeres may also inhibit the activity of promoters that contain G-quadruplex structures [Citation11–Citation24]. This is especially important because the HIV promoter region is also enriched in G residues and has an ability to adopt G-quadruplexes in Sp1 and NF-κB binding elements that are critical for virus reactivation from latency [Citation25–Citation27]. This suggests that using strategies based on telomeres targeting may not result in increased killing of HIV reservoirs when combined with LRAs, since G4 ligands would interfere with virus reactivation from latency.
In the present study, we therefore investigated whether G4 ligands might affect HIV promoter activity and virus reactivation and whether telomere targeting could be combined with LRAs to increase the elimination of HIV reservoirs. We also further investigated whether HIV-1 infected cells have a deficiency in DDR, using our plasmid-based cellular assay. We found that although TMPyP4 affected in vitro binding of Sp1 to G-quadruplex, the G4 ligands did not interfere with the HIV-1 promoter activity in cells and virus reactivation from latency. Using a primary CD4 + T model of HIV latency, we further demonstrated that G4 ligands increased the levels of apoptosis and cell killing induced by LRAs, indicating that telomere targeting may represent a promising strategy to enhance the shock and kill approach to HIV elimination. Finally, we showed that cells infected with HIV have a reduced efficiency of DNA mismatch repair mediated by MMR, and repair of oxidative DNA damages mediated by base excision repair mechanism (BER). Deficiencies in BER and MMR are linked to defective telomeres and telomere elongation; therefore, this may offer a mechanistic explanation for altered TMM in cells with latent HIV infection.
Materials and methods
Reagents and cell culture
TMPyP4 was purchased from Calbiochem; BRACO19, Actinomycin D, TNFα were purchased from Sigma; Bryostatin 1 was purchased from Tocris Bioscience; SAHA was purchased from Selleck Chemical LLC. Recombinant Sp1 protein was purchased from Active Motif (Carlsbad, CA). Virus DHIV and HIV-1 env (X4-tropic) construct were obtained from Dr. Vicente Planelles (University of Utah). The CD4 + T lymphoid cell line Jurkat was obtained from the AIDS Research and Reference Reagent Program (National Institute of Allergy and Infectious Diseases [NIAID], National Institute of Health [NIH]). Latently infected CA5 T cells were obtained from Dr. Olaf Kutsch (University of Alabama at Birmingham). Jurkat and Jurkat-derived CA5 T cells were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum obtained from HyClone, 100 U of penicillin/mL, 100 μg of streptomycin/mL and 2 mM glutamine.
PEG-KCl gel assay
The dsDNA samples (500ng/20 μl) were resuspended in 10 mM Tris–HCl (pH 7.4) buffer containing 1 mM EDTA and the indicated concentrations of KCl (or LiCl) and PEG 200. The samples were heated at 95°C for 5 min and then cooled down to room temperature at a rate of 0.02°C per second. DNA samples were then loaded on 8% polyacrylamide gel containing 150 mM KCl, 40% (w/v) PEG 200 and subjected to electrophoresis at 4°C, 8 V/cm, in 1X TBE buffer containing 150 mM KCl. After electrophoresis the gel was stained with ethidium bromide. DNA products (DNA and smDNA forms) resolved in the gel were analyzed using the software ImageQuant (version 5.2).
Circular dichroism
CD spectra were obtained at 25°C over a wavelength range of 210–340 nm using an AVIV Circular Dichroism Spectrometer, Model 202. The DNA oligomer sample was at a concentration of 20 µM, in 10 mM Tris HCl, pH 7.5, 0.3 mM EDTA and 100 mM KCl. Before analysis, the sample was heated to 90°C for 10 min., and gently cooled at a rate of 1°C/5 min., and incubated at 4°C overnight. Spectra were recorded using a quartz cell of 1-mm optical path length, with data collected every nanometer at a bandwidth of 1 nm. Each spectrum was recorded three times and baseline-corrected for signal contributions from the buffer. The data were processed with AVIV Biomedical Inc software and reported as ellipticity (mdeg) versus wavelength (nm).
Pull-down of Sp1 and Western blotting
The Sp1 protein was selected with a biotinylated oligonucleotide (Integrated DNA Technologies, Inc.) and streptavidin-coated magnetic beads (Promega). The beads were first washed three times with 0.5 mL of 0.5× SSC buffer and three times with Buffer A (25 mM HEPES pH 7.5, 12.5 mM MgCl2, 20% v/v glycerol, 0.1% v/v Nonidet P-40, 1 mM dithiothreitol, 100 mM KCl) containing 3% BSA. Before binding of biotinylated DNA to the beads, DNA oligomer 20 was incubated overnight in Buffer A to form a G-quadruplex structure. In order to form dsDNA for protein selection, the pairs of oligomers 20/21 were incubated in buffer A in a ratio 1:3. 200 µL of biotinylated DNA samples (1 µM) were mixed with beads and incubated for 30 min at room temperature. Beads were then washed three times with 500 µL of Buffer A containing 3% BSA and blocked with the same buffer for 30 min. All subsequent procedures were performed at 4°C. Sp1 protein (80 ng from Active Motif) was added to 500 µL of Buffer A with 3% BSA. The mixture was added to the beads and incubated for 20 min., followed by washing six times with 200 µL of Buffer A. The beads were resuspended in 20 µL of Laemmli buffer and boiled for 2 min. After removing the beads, the samples were separated on a 4–12% gradient Tris–Glycine polyacrylamide gel (BioRad) and transferred to a PVDF membrane. Sp1 was identified by immunoblotting using a rabbit polyclonal antibody diluted 1:5000. A goat anti-rabbit secondary antibody linked with HRP was used for chemiluminescent detection. Sp1 polyclonal antibodies and anti-rabbit IgG, HRP-linked antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA). The effects of Actinomycin D and TMPyP4 on Sp1 binding to DNA were tested by first incubating the beads with DNA in Buffer A with Actinomycin D or TMPyP4 for 15 min., before Sp1 binding.
Generation of cultured TCM cells infected with latent HIV-1
Cultured TCM cells were generated and infected with DHIV pseudotyped with HIV-1 (X4-tropic) Env, as described [Citation8,Citation28,Citation29]. Briefly, naïve CD4 + T cells were isolated (negative selection) from peripheral blood mononuclear cells (PBMC) from healthy donors. The cells were activated with αCD3/αCD28 beads in the presence of αIL-4, αIL-12 and TGF-β1 for 3 days (Day-7 to −4), and then cultured in the presence of IL-2 to differentiate into a nonpolarized phenotype, resembling the central memory T cells (TCM) phenotype. Infection with DHIV was performed at day 0 (day 7 post-activation) and latency was established by progression of activated cells to a quiescent, memory-like state. Progression of cells into latency was monitored by flow cytometric detection of intracellular Gag p24. The percentage of cells infected with latent HIV was assessed by stimulation with αCD3/αCD28 beads and flow cytometric detection of intracellular Gag p24, 48–72 h later. Viable cell counts and viability were determined by trypan blue exclusion assay and the Vi-CELL Cell Viability Analyzer (Beckman Coulter). Statistical analysis was performed with GraphPad Prism (GraphPad Software, La Jolla, CA). A two-tailed paired-samples t test analysis was used to calculate the P value (significance P < 0.05).
Flow cytometry analysis
The phenotype of cultured primary central memory T cells was confirmed by staining with the following mAbs CD27-PE (BD Pharmingen) and CCR7-Alexa Fluor®647 (BD Pharmingen). Intracellular Gag p24 expression was detected with monoclonal antibody conjugated with RD1 (KC57-RD1, Coulter Clone) and analyzed as described [Citation28]. Cell apoptosis was analyzed by Annexin V-APC staining (BD Biosciences) followed by flow cytometric analysis of 10,000 cells, using an Accuri C6 cytometer (BD Biosciences). Data were analyzed using FlowJo software (Version 10.2) and statistical analysis was performed with GraphPad Prism (GraphPad Software, La Jolla, CA). A two-tailed paired-samples t test analysis was used to calculate the P value (significance P < 0.05).
DNA repair assay
All eGFP-Luc constructs (WT, MMR, BER-U, BER-GO/MMR) were produced by annealing of a synthetic oligonucleotide to a pre-created gap in CMVp-eGFP-Luc plasmid generated by Nb.BbvCI (New England Biolabs), as described previously [Citation30,Citation31]. Transfections of cells were performed with Lipofectamine LTX Plus according to the manufacturer’s protocol (Invitrogen). For transfection of Jurkat and CA5 T cells, the cells were seeded into 48-well plates (50,000 cells per well), whereas HEK 293 T cells were seeded into 12-well plates to achieve 60% confluence. For transfection of HEK 293 T cells, each eGFP-Luc construct (200 ng) was mixed with DHIV plasmid (200 ng), or empty pUC19 (200 ng). Transfections of Jurkat and CA5 cells were performed with 200 ng of each eGFP-Luc construct. Cells were collected 24 h after transfection and analyzed for eGFP expression or for luciferase activity. To determine the level of eGFP expression, 7,000 cells were counted and their eGFP intensity was measured using an Accuri C6 flow cytometer (BD Biosciences). Relative eGFP expression (RE) was calculated with the following formula RE = (M · IM)/(C · IC); where M and C are the percentages of green cells transfected with damaged plasmid and undamaged control plasmid, respectively. IM and IC are the median fluorescence intensities of eGFP positive cells for M and C, respectively. For luciferase activity, cells were sedimented at 400 g for 3 min and resuspended in 300 μl PBS. Cells were lysed by three-rounds of freeze-thawing, and debris was removed by centrifuging at 750 g for 3 min. Supernatants (75 μl) were transferred to a 96-well plate (Costar), and then firefly luciferase activity was measured using the Dual-glo Luciferase Reporter System (Promega) and a 96-well plate luminometer (Dynex). Statistical analysis was performed with GraphPad Prism (GraphPad Software, La Jolla, CA). A two-tailed paired-samples t test analysis was used to calculate the P value (significance P < 0.05).
Results
Sp1 binding motifs in HIV promoter can adopt G-quadruplex(es) in duplex DNA
We previously showed that latently HIV-1 infected cells are susceptible to G4 ligands that were described to inhibit telomerase activity and induce telomeres uncapping in cancer cells [Citation8,Citation32–Citation34]. However, many studies also showed that the G4 ligand TMPyP4 interferes with promoters that are enriched in G-residues due to tandem arrangements of binding motifs for transcription factors, such as Sp1 [Citation16,Citation20,Citation23,Citation35–Citation37]. The HIV promoter also contains a G-rich region that is composed of two binding sites for NF-κB and three binding sites for Sp1. This region is critical for virus reactivation from latency [Citation28].
It is currently unknown whether G4 structures are formed within the HIV promoter in latently infected cells, but we and others have shown that the single-stranded synthetic G-rich oligonucleotides adopt different forms of G-quadruplex structures in vitro [Citation25–Citation27]. To examine the ability of a synthetic double-stranded DNA (dsDNA) sequence corresponding to the HIV-1 promoter to fold into a G-quadruplex, we used a PEG-KCl gel migration assay [Citation38]. In this assay, a dsDNA containing a G4 structure can be distinguished as a slower migrating band during electrophoresis, in the presence of 40–60% PEG and 150 mM KCl simulating molecular crowding effect of a cellular environment. A 120-nt dsDNA fragment containing the two NF-κB and three Sp1 binding sites from the HIV promoter were subjected to heat denaturation/renaturation in 150 mM K+ solution containing different concentrations of PEG. For comparison, as a positive control, we used a 110-nt dsDNA sequence from the c-kit promoter region that has been shown to fold a G-quadruplex in a duplex DNA [Citation38]. The dsDNA samples were then resolved by electrophoresis in a native gel containing 150 mM KCl and 40% (w/v) PEG together with DNA samples that were not treated, and migrated in a gel as double-strand DNA duplexes (denoted here as “DNA”). ) shows that folding in the presence of 20% PEG did not induce formation of the slower migrating dsDNA form for the HIV and c-kit promoter regions. However, the sequences for both promoters adopted a slower migrating form (denoted here as “smDNA”) in the presence of PEG concentrations > 40%.
Figure 1. The Sp1 binding region in the HIV-1 promoter can adopt G-quadruplexes in a duplex DNA. (a) The PEG-KCl gel assay was used to analyze a synthetic double-stranded DNA (dsDNA) sequence corresponding to the NF-κB/Sp1 binding region in the HIV promoter (U3 region of 5ʹLTR) for its ability to adopt G-quadruplexes. DNA samples in 150 mM K+ and different concentrations of PEG 200 solution were subjected to heat denaturation/renaturation before electrophoresis. The U3 region in the 5ʹLTR (−3 to −123, NL4-3) adopted a slower migrating form (smDNA) in the presence of increasing concentrations of PEG, which facilitates the formation of G-quadruplexes. The promoter of the c-kit oncogene was previously described to form G-quadruplexes in a duplex DNA, and was used here as a positive control. The drawing at the right side of the gel image shows a schematic illustration of the structure associated with the corresponding DNA band. Below, adoption of smDNA form as a function of PEG concentration. (b) DNA samples were subjected to heat denaturation/renaturation (F) or were not heat-treated (C) before electrophoresis. The U3 region adopting smDNA species is compared to the R/U5 region not predicted to form any G-quadruplexes. The right side of the gel shows that the U3 region does not adopt the smDNA form in the presence of 150 mM Li+ indicating that folding properties of the U3 region are K+-dependent, which is consistent with requirements for formation of monomeric G-quadruplexes. (c)Mutational analyses showing that the smDNA form of the U3 region depends on the G runs present in three Sp1 binding motifs. The gray boxes indicate the G runs that were substituted for T, C or A residues in mutated DNA samples (M1-M9). The mutated DNA samples were heat-treated before electrophoresis and their gel migration characteristics were compared to wild type DNA that was heat-treated (WT) or not heat-treated (C) before electrophoresis. The gels were stained with ethidium bromide (EB). Below, graph shows the percentage of smDNA forms in a gel.
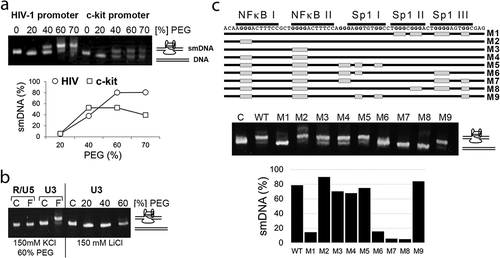
To confirm that this slower migrating dsDNA conformation was dependent on G-rich sequences, we first tested whether another region of the HIV genome not predicted to form G-quadruplexes would adopt a slow-migrating conformation in the PEG-KCl gel migration assay. For this analysis, we selected a 120-nt long sequence between +61 and +181 of the HIV-1 LTR (R/U5 region; numbering from the start of transcription). As shown in ), heat denaturation/renaturation in the presence of 150 mM KCl and 60% PEG did not cause the R/U5 region to adopt a smDNA form.
We next tested whether formation of the slower migrating conformation for the HIV promoter region was cation-dependent. To do this, we compared DNA folding in the presence of the physiologically relevant cations K+ or Na+, to that in presence of the non-physiologically relevant cation, Li+. Samples were then resolved electrophoretically in a PEG-KCl gel (150 mM KCl, 40% PEG). As shown in ), the 120-nt HIV promoter region did not adopt the smDNA form when folding was done in the presence of 150 mM Li+, indicating that folding properties of the HIV region are cation-dependent, which is consistent with the requirements for formation of monomeric G-quadruplexes.
Lastly, we evaluated whether elimination of G runs in the NF-κB and Sp1 binding sites affect formation of the slower migrating conformation. At least four G runs are needed for the sequence to form a G-quadruplex, therefore elimination of a single G run will affect formation of the structure. Nine runs of G residues are present in the region of the HIV-1 promoter that contains the NF-κB and Sp1 binding sites. To determine whether adoption of the smDNA form depends on these G runs, we generated mutated sequences in which each of these G runs was eliminated (), M1-M9). The DNA samples were then subjected to heat denaturation/renaturation in the presence of 150 mM KCl and 60% PEG, and resolved electrophoretically in the PEG-KCl gel. Elimination of G runs composed of two G residues in Sp1 III and Sp1 I (M9, )) did not affect adoption of the smDNA conformation (about 84% of smDNA), when compared to unmodified DNA (WT; 79% of the smDNA). Similarly, elimination of the G run in NF-κB II (M2) did not have any effect on formation of the smDNA (89%). Whereas elimination of the G run in NF-κB I (M3), or in both of the NF-κB binding sites (M4) only slightly affected formation of the smDNA conformation (70% and 67.5%, respectively). Also, additional elimination of the G runs in Sp1 III (M5) resulted in a small reduction in the level of the smDNA form (74.5%). However, elimination of the G runs in Sp1 II and Sp1 I (M1) abolished smDNA formation (14%), as did the elimination of the G runs in NF-κB I, NF-κB II, together with different sets of G runs within the three Sp1 binding sites (15% of smRNA form – M6, 5% – M7, and 4.5% – M8).
In summary, these results indicate that the Sp1 binding region in the HIV-1 promoter, which is critical for HIV expression, can adopt G-quadruplexes in dsDNA duplex, suggesting that these structures could also form in the HIV promoter in infected cells.
The G4 ligand TMPyP4 disrupts Sp1 binding to G-quadruplex structure formed by the HIV promoter sequence
We and others have shown that the transcription factor Sp1, which is critical for HIV reactivation from latency, can bind to its binding motif when it is folded into a G-quadruplex [Citation25,Citation39–Citation41]. We note, however, previous studies showing that G4 ligand TMPyP4 targeting telomeres can also downregulate activity of some of the oncogenes in cells by stabilizing the G-quadruplex structure [Citation12,Citation21–Citation24]. We therefore tested the effect of TMPyP4 on the Sp1 binding to G-quadruplex, and whether TMPyP4 can affect HIV-1 promoter activity in cells. The interaction between Sp1 protein and G-quadruplex in the presence of a G4 ligand was analyzed with the Sp1 II motif folded into a G-quadruplex, which we previously showed to be recognized by G4-specific antibodies and to form complex with Sp1 [Citation25]. The formation of the structure was confirmed by circular dichroism (CD) spectroscopy ()), which revealed a single positive absorption peak and a negative peak at 290 nm and 260 nm, respectively, consistent with the formation of an antiparallel G-quadruplex [Citation25,Citation42]. The effect of TMPyP4 on the Sp1 interaction with G-quadruplex was analyzed using an affinity selection approach, where pull-down of the protein is performed with a biotinylated Sp1 binding sequence immobilized on streptavidin-coated magnetic beads [Citation25,Citation39]. The binding of Sp1 to G-quadruplex was then analyzed by Western blotting. As a positive control of drug-mediated Sp1 protein displacement from the binding motif, we used Actinomycin D, which was shown to inhibit interactions of Sp1 with a dsDNA [Citation43]. Empty beads not coupled to any DNA were used as a negative control in the assay.
Figure 2. G4 ligand TMPyP4 disrupts binding of Sp1 protein to a G-quadruplex formed by the HIV promoter sequence. (a) CD spectra profile of the K+ – induced antiparallel G-quadruplex formed by a single-stranded DNA containing Sp1 binding site II of the HIV-1 promoter. (b) Western blot analysis of Sp1 protein selected by dsDNA, or ssDNA folded into a G-quadruplex structure (ss-GQ). A pull-down assay was performed in the presence of 5 μM, 15 μM and 30 μM of Actinomycin D (ActD). Actinomycin D has an affinity for the Sp1 binding site and displaces Sp1 from dsDNA. However, it is unable to inhibit the interaction between Sp1 and the G-quadruplex. Below, binding between Sp1 and ss-GQ is abolished by increasing concentrations of the G4 ligand TMPyP4 (1 μM and 15 μM), but not Actinomycin D (1 μM and 15 μM). C - control, pull-down assay performed using beads not coupled with any DNA.
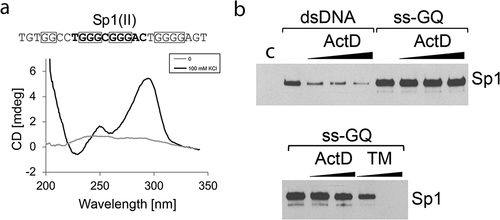
As shown in , the Sp1 was pulled down with the HIV-1 Sp1 binding site II dsDNA and with the oligomer folded into a G-quadruplex. Actinomycin D blocked the interaction of Sp1 with the HIV-1 Sp1 II site when present as native dsDNA, but had no effect on Sp1 binding to the same motif when it was folded into a G-quadruplex. In contrast, TMPyP4 blocked binding of Sp1 to its binding motif folded into a G-quadruplex, in a dose-dependent manner.
These data suggest that TMPyP4, and potentially other telomere targeting G4 ligands, may block Sp1 binding to the HIV promoter – thereby potentially interfering with viral reactivation from latency.
G4 ligands do not interfere with HIV promoter activity and do not block virus reactivation from latency
To test whether G4 ligands interfere with basal HIV promoter activity in cells, we constructed a plasmid reporter containing the HIV promoter (−455 to +334) placed upstream of the Renilla luciferase gene (). Since the test involves drugs that may affect cellular viability and transfection efficiency, we selected the psiCHECK-2 vector that also contains an independently transcribed firefly luciferase reporter gene, which can be used as a reference to normalize transfections. The level of HIV promoter-driven Renilla luciferase expression was at 60%, when compared to the expression level from original psiCHECK-2 vector. The effect of G4 ligands on HIV promoter activity was tested for TMPyP4 (15 μM) and BRACO19 (6 μM). The construct was transfected into HEK 293 T cells in the presence of G4 ligands, and luciferase activity was analyzed 24 h later. Both drugs affected viability of cells and the efficiency of transfection, but there was no reduction in the level of Renilla luciferase activity, when compared to firefly luciferase, indicating that both ligands did not interfere with basal HIV promoter activity in cells ().
Figure 3. G4 ligands do not interfere with HIV promoter activity and with virus reactivation from latency. (a) Schematic diagram of the expression cassette for Renilla luciferase (Ren Luc) under control of the HIV-1 promoter (HIVp-RLuc). The simian virus 40 early enhancer/promoter (SV40p) in psiCHECK-2 (SV40p-RLuc) was replaced with the HIV promoter (−455 to +334) derived from NL4-3. Firefly luciferase (FF Luc) expressed from herpes simplex virus thymidine kinase promoter (HSV-TKp) was used as a transfection control. Below, dual-luciferase assay (n 3) of HEK 293 T lysates after transfection of HIVp-RLuc. Left panel: Renilla Luciferase expression levels from the HIV promoter are about 60% of those from the SV40 promoter in the psiCHECK-2 vector. Right panel: HIV promoter activity is not affected by TMPyP4 (TM; 15 μM) and BRACO19 (BR; 6 μM). RLU, relative light units; NT, not treated. (b) Schematic diagram of the NL4-3-based green fluorescent protein (GFP) reporter virus, which was used to infect Jurkat T cells and to generate latently infected CA5 T cells [Citation44]. (c) Latency reversal in latently infected CA5 T cells is not affected by BRACO19 (35 μM) and TMPyP4 (20 μM). Flow cytometry plots show no change in the level of GFP expressed in CA5 T cells reactivated with TNFα (10 ng/ml), or latency reversing agents (LRAs), SAHA (100 nM) and Bryostatin (BRY; 20 nM), in the presence of G4 ligands. Numbers in gates indicate the GFP positive population. The expression of GFP was determined for the same number of cells in each sample.
![Figure 3. G4 ligands do not interfere with HIV promoter activity and with virus reactivation from latency. (a) Schematic diagram of the expression cassette for Renilla luciferase (Ren Luc) under control of the HIV-1 promoter (HIVp-RLuc). The simian virus 40 early enhancer/promoter (SV40p) in psiCHECK-2 (SV40p-RLuc) was replaced with the HIV promoter (−455 to +334) derived from NL4-3. Firefly luciferase (FF Luc) expressed from herpes simplex virus thymidine kinase promoter (HSV-TKp) was used as a transfection control. Below, dual-luciferase assay (n 3) of HEK 293 T lysates after transfection of HIVp-RLuc. Left panel: Renilla Luciferase expression levels from the HIV promoter are about 60% of those from the SV40 promoter in the psiCHECK-2 vector. Right panel: HIV promoter activity is not affected by TMPyP4 (TM; 15 μM) and BRACO19 (BR; 6 μM). RLU, relative light units; NT, not treated. (b) Schematic diagram of the NL4-3-based green fluorescent protein (GFP) reporter virus, which was used to infect Jurkat T cells and to generate latently infected CA5 T cells [Citation44]. (c) Latency reversal in latently infected CA5 T cells is not affected by BRACO19 (35 μM) and TMPyP4 (20 μM). Flow cytometry plots show no change in the level of GFP expressed in CA5 T cells reactivated with TNFα (10 ng/ml), or latency reversing agents (LRAs), SAHA (100 nM) and Bryostatin (BRY; 20 nM), in the presence of G4 ligands. Numbers in gates indicate the GFP positive population. The expression of GFP was determined for the same number of cells in each sample.](/cms/asset/5f2d4d00-feae-43ea-b108-a878b39f8bda/kccy_a_1796268_f0003_b.gif)
Next, we tested whether G4 ligands interfere with virus reactivation from latency using Jurkat-derived, latently infected CA5 T cells, which harbor a single integrated copy of a replication competent HIV-1 provirus that contains a GFP reporter gene () [Citation44]. Tumor necrosis factor-alpha (TNFα) induced latency reversal in these cells was unaffected by G4 ligands even at higher concentrations (TMPyP4, 20 μM; BRACO19, 35 μM). The effect of G4 ligands on virus reactivation in CA5 T cells was also analyzed for cells treated with two LRAs, the histone deacetylase inhibitor (HDACi) SAHA, also known as Vorinostat [Citation1,Citation7,Citation45–Citation47], and protein kinase C activator Bryostatin [Citation48–Citation50]. ) shows that after 24 h SAHA (100 nM) and Bryostatin (20 nM) induced virus expression in CA5 T cells in about 90% and 35% of the CA5 T cell population, respectively. As observed for TNFα-mediated latency reversal, virus reactivation with both LRAs was not affected by TMPyP4, and BRACO19.
In summary, these results indicate that G4 ligands TMPyP4 and BRACO19 do not interfere with HIV promoter activity, or virus reactivation from latency.
Latently infected cells are more susceptible to combined treatment with LRAs and G4 ligands
We next tested whether G4 ligands might enhance LRA-mediated cell killing in a primary CD4+ TCM cell latency model. Briefly, central memory T cells (TCM) were established from human primary CD4 + T lymphocytes, as described [Citation28,Citation29], and then infected with single-cycle HIV-1NL4-3; latency was established by day 10–12 post-infection (). The level of established latency in the population varies from about 15% to 50%, and can be assessed by antigenic stimulation (CD3/CD28) that allows virus reactivation [Citation28]. SAHA is not effective in latency reversal with this HIV-1 latency model, but Bryostatin can reactivate virus in about 50% of the population, when compared to anti-CD3/CD28 stimulation [Citation51]. We generated cultured CD4 TCM cells prepared from five blood donors and infected them with single cycle HIV-1NL4-3 (DHIV). At day 11 post-infection, cultures were exposed to BRACO19 (20 μM) or TMPyP4 (15 μM) in the presence of SAHA (110 nM) or Bryostatin (10 nM), and analyzed for apoptosis and viable cell count 24 h later.
Figure 4. G4 ligands enhance LRA-mediated killing of HIV latently infected primary CD4 TCM cells. (a) Procedure used to generate primary CD4 TCM cells latently infected with HIV-1. dpi, days post-infection. (b) The level of established latency in primary CD4 TCM cells was assessed by the intracellular staining for Gag p24 after αCD3/αCD28 activation. Latency reversal mediated by SAHA (110 nM) and Bryostatin (BRY; 10 nM) is shown for comparison. (c) On day 11 post-infection, cultures of infected and uninfected primary CD4 TCM cells were treated with BRACO19 (20 μM) or TMPyP4 (15 μM) in the presence of SAHA (110 nM) or Bryostatin (BRY, 10 nM), and analyzed for the rate of apoptosis and viable cell count 24 h later. Cells were stained with APC-conjugated Annexin V. The left panel shows representative flow cytometry histograms for uninfected and HIV-1 infected (+ DHIV) CD4 TCM cultures from the same blood donor treated with different drugs. The numbers in the histograms indicate the percentage of cells that stained positively with Annexin V (AV+). The right panel shows the mean percentage of apoptotic (Annexin V+) cells (n = 5 donors) after treatment with the following drugs: NT – not treated cells; TM – TMPyP4; BR – BRACO19. (d) Graphs show viable cell counts determined for uninfected and HIV-1 infected CD4 TCM cultures treated with LRAs together with G4 binding ligands. Viable cell counts and viability were analyzed by trypan blue exclusion assay and Vi-CELL Cell Viability Analyzer. Significance (P < 0.05) was determined by 2-tailed, paired-samples t test. *P < 0.05.
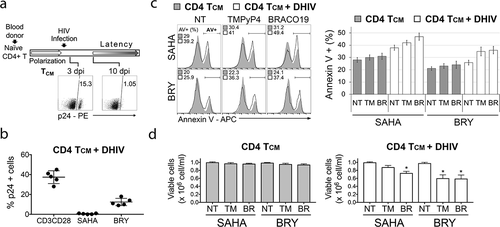
The results summarized in show that combining LRAs with G4 ligands resulted in greater reductions in cell populations in cultures infected with latent HIV. The level of established latency in cultures was about 38% (±SD 7), as determined by anti-CD3/CD28 stimulation (48 h). Bryostatin reactivated 13% of cells from latency, and SAHA had no effect.
When latently infected cells were exposed to both LRAs and G4 ligands, cell viability decreased and the percentage of apoptotic cells increased. For example, the combination of Bryostatin and BRAC019 reduced cell viability by 41%, compared to Bryostatin alone, while the combination of Bryostatin and TMPyP4 reduced cell viability by 40%, compared to Bryostatin alone; the combination of Bryostatin and G4 ligands also increased the percentage of apoptotic cells by 9–10% relative to Bryostatin alone. Similar results were obtained with SAHA. Combining SAHA with G4 ligands reduced cell viability by 27% and 12% for BRACO19 and TMPyP4, respectively, when compared to SAHA only. The effects were accompanied by an increase in the percentage of apoptotic cells by 4–9%. In contrast, treatment of uninfected cells with the combination of LRAs and G4 ligands resulted in only a minimal increase in cell death (by 2.5–3.5% for SAHA, and by 4.5–5.5% for Bryostatin).
These results show that the killing of latently infected cells by LRAs is enhanced in the presence of G4 ligands, even though the extent of virus reactivation is unaltered, indicating that drugs targeting telomeres could enhance killing of HIV reservoirs in a shock and kill therapeutic approach.
Efficiency of DNA repair is reduced in HIV-infected cells
Increased susceptibility to G4 ligands and telomere elongation indicates that HIV latently infected cells have altered TMM. Changes in telomeric caps and telomere elongation were previously shown to be associated with increased DNA damage at telomeres resulting from inefficient DNA damage repair mediated by BER and MMR mechanisms [Citation52–Citation57]. To determine efficiency of BER and MMR in infected cells, we used our plasmid system that allows us to measure in cells the efficiency of repair of particular damage. The strategy relies on using plasmids bearing DNA damages in a reporter gene expressing eGFP-Luc fusion protein, and monitoring DNA repair in transfected cells. Briefly, the reporter plasmid contains a damaged residue (denoted by a star in ) in the transcribed strand within a short cassette attached to the N terminus of the eGFP-Luc gene. A nicking enzyme is used to create a gap and replace the original ssDNA fragment with a synthetic DNA oligonucleotide containing the desired modification.
Figure 5. Plasmid constructs used to analyze DNA repair in a cell-based system. (a) Reporter system design and method for site-specific incorporation of DNA damage. The eGFP-Luc fusion reporter gene contains a nicking cassette (nick cst) at the beginning of the ORF that is used to incorporate DNA modifications in the transcribed strand (T). (b) Sequences of altered regions in plasmids generated to analyze mismatch repair mechanism (MMR), base excision repair (BER) of 8-oxoG, and BER of uracil. BER-U, MMR, and BER-GO/MMR refer to the eGFP-Luc plasmids containing: uracil residues creating a stop codon, a mismatch creating a stop codon, and 8-oxoG (GO) plus a mismatch creating a stop codon, respectively. WT refers to the eGFP-Luc plasmid created with an unaltered synthetic oligonucleotide.
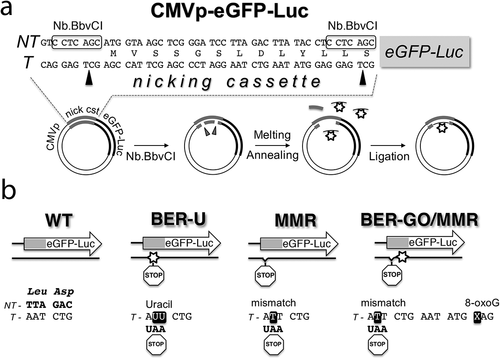
The damages and modifications in the eGFP-Luc reporter gene were designed to inactivate eGFP-Luc expression, unless the repairs were performed ()). To analyze MMR, we placed a mismatch generating a stop codon in the MMR eGFP-Luc construct, so deficiency of mismatch repair is detected by reduced level of reporter gene expression, when compared to not infected cells. The same approach was used to analyze uracil removal through BER. Efficiency of 8-oxoG repair was measured using also a mismatch that created a stop codon placed on the 3ʹ side of the lesion in the BER-GO/MMR eGFP-Luc construct. For this construct, the repair of the inactivating stop codon is processed by MMR and through a long-patch BER (LP-BER) mechanism that utilizes strand displacement synthesis initiated by repair of 8-oxoG. The levels of eGFP and luciferase activity expressed from the damaged plasmids were analyzed with reference to an eGFP-Luc plasmid created with an unaltered synthetic oligonucleotide (WT eGFP-Luc construct).
We first measured the repair of these damaged eGFP-Luc constructs in HEK 293 T cells co-transfected with plasmid expressing HIV (+DHIV), or empty plasmid (-DHIV). By analysis of eGFP expression and luciferase activity, we determined about 7–8% reductions in efficiency of uracil removal for HEK 293 T cells co-transfected with HIV expressing plasmid, when compared to cells co-transfected with empty plasmid (). However, repair of a mismatch (MMR) was reduced for about 20–25%. Whereas, for the BER-GO/MMR construct, the reductions in eGFP level and luciferase activity were about 15%.
Figure 6. Efficiency of DNA repair is reduced in cells infected with HIV. (a,b) HEK 293 T cells were co-transfected with eGFP-Luc reporter plasmids bearing modifications inactivating eGFP-Luc expression, and a HIV-1 vector plasmid (+DHIV), or an empty vector (-DHIV). (a) The left graph shows relative eGFP expression (measured using flow cytometry) by damaged eGFP-Luc constructs (plasmids BER-U, MMR, and BER-GO/MMR), calculated in reference to eGFP expression from the WT eGFP-Luc plasmid (100%) (n = 3 experiments). The right graph shows relative eGFP expression by damaged eGFP-Luc constructs, in HEK 293 T cells co-transfected with a HIV-1 vector plasmid (+DHIV), when compared to cells transfected with an empty plasmid (-DHIV; set to 100%). The lower panel shows representative flow cytometry contour plots. The numbers in the gates indicate the percentage of eGFP positive cells within the population analyzed. The median fluorescence intensities for the eGFP+ population are indicated by a cross and were used to calculate relative eGFP expression. All plots were gated on live cells. (b) The left graph shows relative luciferase expression by damaged eGFP-Luc constructs, calculated in reference to luciferase expression from the WT eGFP-Luc plasmid (100%) (n = 4 experiments). The right graph shows relative luciferase expression by damaged eGFP-Luc constructs, in HEK 293 T cells co-transfected with a HIV-1 vector plasmid (+DHIV), when compared to cells transfected with an empty plasmid (-DHIV; set to 100%). (c)The left graph shows relative luciferase expression by damaged eGFP-Luc constructs in Jurkat and CA5 cell lysates, calculated in reference to luciferase expression from the WT eGFP-Luc plasmid (100%) (n = 3 experiments). The right graph shows relative luciferase expression by damaged eGFP-Luc constructs, in CA5 cells, when compared to Jurkat cells (set to 100%). Significance (P < 0.05) was determined by 2-tailed, paired-sample t test. *P < 0.05; **P < 0.01.
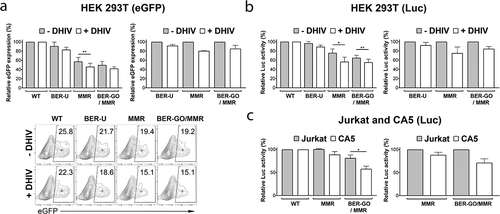
We also analyzed the efficiency of DNA repair in latently infected CA5 T cells, using the same approach. In this case, we transfected Jurkat and CA5 T cells with reporter plasmids MMR and BER-GO/MMR, and compared their repair efficiency to undamaged eGFP-Luc (WT). HIV-1 provirus in latent CA5 T cells contains an eGFP reporter, therefore we analyzed DNA repair of the eGFP-Luc constructs by measuring only luciferase activity. As shown in ), in CA5 cells there were about 12% and 29% reductions in efficiency of DNA repair by MMR and BER-GO/MMR, respectively, when compared to repairs in Jurkat cells.
In summary, these results show that cells infected with HIV have deficiencies in repair of mismatches mediated by MMR, and damages repaired by BER mechanisms, suggesting that cells infected with latent HIV experience accumulation of unrepaired DNA damages at telomeres that affect TMM and telomeric caps.
Discussion
Alterations in TMM are generally associated with cancer cells, and thus are considered as an attractive target for development of anticancer drugs. Our studies indicate that cells infected with latent HIV also have altered TMM. We have previously shown that HIV-infected cells have elongated telomeres and show increased susceptibility to agents that are linked to targeting TMM, such as nucleoside analogs and G4 ligands [Citation8,Citation10]. This unique characteristic of latently infected cells provides a potential platform for developing drugs targeting HIV reservoirs.
G4 ligands are one of the most studied agents targeting TMM as they can induce genomic instability and apoptosis by stabilizing G-quadruplexes at telomeres, inducing uncapping and inhibiting telomerase. This is attractive from the standpoint of targeting latently infected cells. However, G4 ligands may also inhibit the expression of genes carrying in their promoters a sequence capable of forming G-quadruplex [Citation11–Citation24,Citation58,Citation59], such as the HIV-1 LTR. Indeed, we have previously demonstrated that a single-stranded DNA containing the Sp1 binding motifs from the HIV promoter can adopt G4 structures recognized by G4-specific antibodies, and that the Sp1 protein binds efficiently to G-quadruplex [Citation25]. Here, we show that the Sp1 binding region also adopts G4 structure(s) in a duplex DNA and that the Sp1-G4 complex can be disrupted by the G4 ligand, suggesting the possibility that G4 ligands interfere with virus reactivation from latency. We therefore examined the transcriptional activity of the HIV promoter (−455/+344) in the presence of G4 ligands, following transient transfection of an LTR-driven luciferase reporter gene into HEK 293 T cells. Neither BRACO19 (6 μM) nor TMPyP4 (15 μM) reduced HIV promoter-driven luciferase expression, suggesting that G4 ligands do not reduce the transcriptional activity of the HIV-1 LTR in HEK 293 T cells. These findings conflict with data reported by Perrone et al., who showed that 6 μM BRACO19 caused a 30% reduction in HIV-1 LTR-driven expression of a GFP reporter, also in HEK 293 T cells [Citation26]. However, these prior studies used only a partial fragment of the HIV LTR (−381/+83) [Citation26].
It is currently unknown whether in cells the HIV promoter is regulated by G-quadruplexes, and what is the function of Sp1 in regulating HIV expression when it binds to G-quadruplex. However, here we demonstrate that G4 ligands targeting telomeres do not interfere with virus reactivation from latency. Moreover, combining G4 ligands with LRAs resulted in enhanced killing of latently infected cells. This suggests that G4 ligands, or other agents designed to target telomeric G-quadruplexes, should not interfere with latency reversal, and that elimination of HIV reservoirs could be more effective when LRAs are combined with a strategy based on targeting TMM.
Finally, we also examined the mechanisms that were found to be related to altered TMM. Previous studies showed that accumulation of oxidized DNA bases and uracil at telomeres due to BER deficiency and oxidative stress is associated with uncapping and telomere elongation [Citation52–Citation57,Citation60]. Deficiency in MMR was also found to accelerate telomere elongation through ALT (alternative lengthening of telomeres), a mechanism that relies on homologous recombination events [Citation61–Citation63]. Here, we show that infected cells are less efficient at repairing mismatches and damages mediated by BER, such as oxidative damage to G residues and removal of uracil resulting from deamination of C. Since telomeres are rich in G and C residues, we hypothesize that altered TMM in infected cells could be related to increased DNA damage at telomeres that triggers changes in telomeric caps and results in more “open” conformation and better accessibility of agents that promote uncapping, such as G4 ligands.
Deficiency in repairs mediated by BER and MMR in HIV-infected cells could be related to virus-mediated targeting of DDR factors that are involved in maintaining telomeres. Previous studies found that Tat protein downregulates DNA-PKs important for maintaining proper telomeric caps [Citation64–Citation66]. Whereas Vpr protein targets UNG for degradation, a DNA glycosylase responsible for removal of uracil [Citation67–Citation69]. UNG prevents accumulation of uracil in telomeres, which is linked to recombination-based telomere lengthening [Citation55]. Vpr also targets other DNA repair factors important for TMM, such as HLTF, MUS81, and Tet2 [Citation67,Citation70–Citation74]. It is currently unknown whether HIV interferes with any factors of MMR system, which is not only responsible for repairing DNA replication errors, but also class-switch recombination and somatic hypermutation of V(D)J and S regions of Ig genes. Studies indicate that the role of MMR in TMM is prevention of homologous recombination in telomeres; therefore, MMR deficiency can be associated with induction of ALT-like telomere elongation [Citation61,Citation62].
It is currently unknown how the type and the level of DNA damage at telomeres influence activation of telomerase-mediated and the ALT-mediated TMM, but studies suggest that DNA damages in telomeres have a direct effect on DNA replication, formation of G-quadruplexes, and association of shelterin proteins that form a telomeric cap. For example, in assays in vitro 8-oxoG and Tg (thymine glycol, oxidative damage to T) interfere with formation of the G4 structure, and increase binding of telomerase to the telomeric sequence [Citation52,Citation75]. 8-oxoG is also an efficient chain terminator in telomere elongation by telomerase [Citation52,Citation53], whereas unremoved Tg is a severe block to continued synthesis by eukaryotic replicative DNA polymerases [Citation76]. Cells with defective OGG1, which mediates 8-oxoG repair, show telomere shortening or telomere elongation [Citation56,Citation77,Citation78]. Whereas, cells with defective NTH1, which mediates repairs of Tg, 5-hC and 5-hU, show increased telomere shortening and aberrations, including telomere sister chromatid exchange via homologous recombination, known as T-SCE, which is also a hallmark of ALT [Citation79]. Recombination-based telomere lengthening is also induced in cells with increased accumulation of uracil in telomeres due to defective UNG [Citation55].
In conclusion, it is important to determine what factors and mechanisms are associated with changes in TMM, since telomere elongation is related to greater longevity and greater clonal expansion of HIV reservoirs. Currently, there are no G4 ligands that have therapeutic application in anticancer treatments, however approaches that are based on TMM targeting may provide an effective strategy to purge hidden HIV reservoirs.
Supplemental Material
Download MS Word (23.1 KB)Acknowledgments
We are grateful to Dr. Olaf Kutsch (University of Alabama at Birmingham) for providing the latently infected cell line CA5 used in these studies. We would also like to thank Dr. Vicente Planelles for providing the DHIV and env (X4-tropic) constructs.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Archin NM, Kirchherr JL, Sung JA, et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J Clin Invest. 2017;127:3126–3135.
- Gutierrez C, Serrano-Villar S, Madrid-Elena N, et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS. 2016;30:1385–1392.
- Elliott JH, McMahon JH, Chang CC, et al. Short-term administration of disulfiram for reversal of latent HIV infection: a phase 2 dose-escalation study. Lancet HIV. 2015;2:e520–529.
- Sogaard OS, Graversen ME, Leth S, et al. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog. 2015;11:e1005142.
- Elliott JH, Wightman F, Solomon A, et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014;10:e1004473.
- Rasmussen TA, Tolstrup M, Brinkmann CR, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV. 2014;1:e13–21.
- Archin NM, Liberty AL, Kashuba AD, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485.
- Piekna-Przybylska D, Maggirwar SB. CD4+ memory T cells infected with latent HIV-1 are susceptible to drugs targeting telomeres. Cell Cycle. 2018;17:2187–2203.
- Piekna-Przybylska D, Nagumotu K, Reid DM, et al. HIV-1 infection renders brain vascular pericytes susceptible to the extracellular glutamate. J Neurovirol. 2019;25:114–126.
- Piekna-Przybylska D, Sharma G, Maggirwar SB, et al. Deficiency in DNA damage response, a new characteristic of cells infected with latent HIV-1. Cell Cycle. 2017;16:968–978.
- Hu MH, Wu TY, Huang Q, et al. New substituted quinoxalines inhibit triple-negative breast cancer by specifically downregulating the c-MYC transcription. Nucleic Acids Res. 2019;47:10529–10542.
- Lavrado J, Brito H, Borralho PM, et al. KRAS oncogene repression in colon cancer cell lines by G-quadruplex binding indolo[3,2-c]quinolines. Sci Rep. 2015;5:9696.
- Bidzinska J, Cimino-Reale G, Zaffaroni N, et al. G-quadruplex structures in the human genome as novel therapeutic targets. Molecules. 2013;18:12368–12395.
- Balasubramanian S, Hurley LH, Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov. 2011;10:261–275.
- Brown RV, Danford FL, Gokhale V, et al. Demonstration that drug-targeted down-regulation of MYC in non-Hodgkins lymphoma is directly mediated through the promoter G-quadruplex. J Biol Chem. 2011;286:41018–41027.
- Seenisamy J, Rezler EM, Powell TJ, et al. The dynamic character of the G-quadruplex element in the c-MYC promoter and modification by TMPyP4. J Am Chem Soc. 2004;126:8702–8709.
- Gonzalez V, Hurley LH. The C-terminus of nucleolin promotes the formation of the c-MYC G-quadruplex and inhibits c-MYC promoter activity. Biochemistry. 2010;49:9706–9714.
- Gonzalez V, Guo K, Hurley L, et al. Identification and characterization of nucleolin as a c-myc G-quadruplex-binding protein. J Biol Chem. 2009;284:23622–23635.
- Thakur RK, Kumar P, Halder K, et al. Metastases suppressor NM23-H2 interaction with G-quadruplex DNA within c-MYC promoter nuclease hypersensitive element induces c-MYC expression. Nucleic Acids Res. 2009;37:172–183.
- Sun D, Liu WJ, Guo K, et al. The proximal promoter region of the human vascular endothelial growth factor gene has a G-quadruplex structure that can be targeted by G-quadruplex-interactive agents. Mol Cancer Ther. 2008;7:880–889.
- Mikami-Terao Y, Akiyama M, Yuza Y, et al. Antitumor activity of G-quadruplex-interactive agent TMPyP4 in K562 leukemic cells. Cancer Lett. 2008;261:226–234.
- Cogoi S, Xodo LE. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006;34:2536–2549.
- Siddiqui-Jain A, Grand CL, Bearss DJ, et al. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci U S A. 2002;99:11593–11598.
- Grand CL, Han H, Munoz RM, et al. The cationic porphyrin TMPyP4 down-regulates c-MYC and human telomerase reverse transcriptase expression and inhibits tumor growth in vivo. Mol Cancer Ther. 2002;1:565–573.
- Piekna-Przybylska D, Sullivan MA, Sharma G, et al. U3 region in the HIV-1 genome adopts a G-quadruplex structure in its RNA and DNA sequence. Biochemistry. 2014;53:2581–2593.
- Perrone R, Nadai M, Frasson I, et al. A dynamic G-quadruplex region regulates the HIV-1 long terminal repeat promoter. J Med Chem. 2013;56:6521–6530.
- Amrane S, Kerkour A, Bedrat A, et al. Topology of a DNA G-quadruplex structure formed in the HIV-1 promoter: a potential target for anti-HIV drug development. J Am Chem Soc. 2014;136:5249–5252.
- Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood. 2009;113:58–65.
- Bosque A, Planelles V. Studies of HIV-1 latency in an ex vivo model that uses primary central memory T cells. Methods. 2011;53:54–61.
- Piekna-Przybylska D, Bambara RA, Balakrishnan L. Acetylation regulates DNA repair mechanisms in human cells. Cell Cycle. 2016;15:1506–1517.
- Piekna-Przybylska D. Reporter Assays for BER Pathway. Methods Mol Biol. 2019;1999:145–160.
- Zhou G, Liu X, Li Y, et al. Telomere targeting with a novel G-quadruplex-interactive ligand BRACO-19 induces T-loop disassembly and telomerase displacement in human glioblastoma cells. Oncotarget. 2016;7:14925–14939.
- Temime-Smaali N, Guittat L, Sidibe A, et al. The G-quadruplex ligand telomestatin impairs binding of topoisomerase IIIalpha to G-quadruplex-forming oligonucleotides and uncaps telomeres in ALT cells. PLoS One. 2009;4:e6919.
- Gomez D, O’Donohue MF, Wenner T, et al. The G-quadruplex ligand telomestatin inhibits POT1 binding to telomeric sequences in vitro and induces GFP-POT1 dissociation from telomeres in human cells. Cancer Res. 2006;66:6908–6912.
- Yan J, Zhao X, Liu B, et al. An intramolecular G-quadruplex structure formed in the human MET promoter region and its biological relevance. Mol Carcinog. 2016;55:897–909.
- Zidanloo SG, Hosseinzadeh Colagar A, Ayatollahi H, et al. Downregulation of the WT1 gene expression via TMPyP4 stabilization of promoter G-quadruplexes in leukemia cells. Tumour Biol. 2016;37:9967–9977.
- Li Y, Syed J, Suzuki Y, et al. Effect of ATRX and G-quadruplex formation by the VNTR sequence on alpha-globin gene expression. Chembiochem. 2016;17:928–935.
- Zheng KW, Chen Z, Hao YH, et al. Molecular crowding creates an essential environment for the formation of stable G-quadruplexes in long double-stranded DNA. Nucleic Acids Res. 2010;38:327–338.
- Raiber EA, Kranaster R, Lam E, et al. A non-canonical DNA structure is a binding motif for the transcription factor SP1 in vitro. Nucleic Acids Res. 2012;40:1499–1508.
- Kong JN, Zhang C, Zhu YC, et al. Identification and characterization of G-quadruplex formation within the EP0 promoter of pseudorabies virus. Sci Rep. 2018;8:14029.
- Tsukakoshi K, Saito S, Yoshida W, et al. CpG methylation changes g-quadruplex structures derived from gene promoters and interaction with VEGF and SP1. Molecules. 2018;23. DOI:https://doi.org/10.3390/molecules23040944
- Vorlickova M, Kejnovska I, Sagi J, et al. Circular dichroism and guanine quadruplexes. Methods. 2012;57:64–75.
- Czyz M, Gniazdowski M. Actinomycin D specifically inhibits the interaction between transcription factor Sp1 and its binding site. Acta Biochim Pol. 1998;45:67–73.
- Shishido T, Wolschendorf F, Duverger A, et al. Selected drugs with reported secondary cell-differentiating capacity prime latent HIV-1 infection for reactivation. J Virol. 2012;86:9055–9069.
- Bouchat S, Gatot JS, Kabeya K, et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4(+) T cells from HIV-1-infected HAART-treated patients. AIDS. 2012;26:1473–1482.
- Reuse S, Calao M, Kabeya K, et al. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: implications for treatment of latent infection. PLoS One. 2009;4:e6093.
- Archin NM, Espeseth A, Parker D, et al. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retroviruses. 2009;25:207–212.
- Grau-Exposito J, Luque-Ballesteros L, Navarro J, et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PLoS Pathog. 2019;15:e1007991.
- Laird GM, Bullen CK, Rosenbloom DI, et al. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J Clin Invest. 2015;125:1901–1912.
- Darcis G, Kula A, Bouchat S, et al. An In-depth comparison of latency-reversing agent combinations in various in vitro and ex vivo HIV-1 latency models identified bryostatin-1+JQ1 and ingenol-B+JQ1 to potently reactivate viral gene expression. PLoS Pathog. 2015;11:e1005063.
- Spina CA, Anderson J, Archin NM, et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013;9:e1003834.
- Lee HT, Bose A, Lee CY, et al. Molecular mechanisms by which oxidative DNA damage promotes telomerase activity. Nucleic Acids Res. 2017;45:11752–11765.
- Fouquerel E, Lormand J, Bose A, et al. Oxidative guanine base damage regulates human telomerase activity. Nat Struct Mol Biol. 2016;23:1092–1100.
- Sarkar J, Liu Y. The origin of oxidized guanine resolves the puzzle of oxidation-induced telomere-length alterations. Nat Struct Mol Biol. 2016;23:1070–1071.
- Vallabhaneni H, Zhou F, Maul RW, et al. Defective repair of uracil causes telomere defects in mouse hematopoietic cells. J Biol Chem. 2015;290:5502–5511.
- Lu J, Liu Y. Deletion of Ogg1 DNA glycosylase results in telomere base damage and length alteration in yeast. Embo J. 2010;29:398–409.
- Opresko PL, Fan J, Danzy S, et al. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 2005;33:1230–1239.
- Eddy J, Maizels N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006;34:3887–3896.
- Marsico G, Chambers VS, Sahakyan AB, et al. Whole genome experimental maps of DNA G-quadruplexes in multiple species. Nucleic Acids Res. 2019;47:3862–3874.
- Coluzzi E, Buonsante R, Leone S, et al. Transient ALT activation protects human primary cells from chromosome instability induced by low chronic oxidative stress. Sci Rep. 2017;7:43309.
- Bechter OE, Zou Y, Walker W, et al. Telomeric recombination in mismatch repair deficient human colon cancer cells after telomerase inhibition. Cancer Res. 2004;64:3444–3451.
- Rizki A, Lundblad V. Defects in mismatch repair promote telomerase-independent proliferation. Nature. 2001;411:713–716.
- Bellacosa A. Functional interactions and signaling properties of mammalian DNA mismatch repair proteins. Cell Death Differ. 2001;8:1076–1092.
- Sun Y, Huang YC, Xu QZ, et al. HIV-1 Tat depresses DNA-PK(CS) expression and DNA repair, and sensitizes cells to ionizing radiation. Int J Radiat Oncol Biol Phys. 2006;65:842–850.
- Zhang SM, Zhang H, Yang TY, et al. Interaction between HIV-1 Tat and DNA-PKcs modulates HIV transcription and class switch recombination. Int J Biol Sci. 2014;10:1138–1149.
- Sui J, Zhang S, Chen BPC. DNA-dependent protein kinase in telomere maintenance and protection. Cell Mol Biol Lett. 2020;25:2.
- Hrecka K, Hao C, Shun MC, et al. HIV-1 and HIV-2 exhibit divergent interactions with HLTF and UNG2 DNA repair proteins. Proc Natl Acad Sci U S A. 2016;113:E3921–3930.
- Schrofelbauer B, Yu Q, Zeitlin SG, et al. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J Virol. 2005;79:10978–10987.
- Bouhamdan M, Benichou S, Rey F, et al. Human immunodeficiency virus type 1 Vpr protein binds to the uracil DNA glycosylase DNA repair enzyme. J Virol. 1996;70:697–704.
- Lv L, Wang Q, Xu Y, et al. Vpr targets TET2 for degradation by CRL4(VprBP) E3 ligase to sustain IL-6 expression and enhance HIV-1 replication. Mol Cell. 2018;70:961–970 e965.
- Lahouassa H, Blondot ML, Chauveau L, et al. HIV-1 Vpr degrades the HLTF DNA translocase in T cells and macrophages. Proc Natl Acad Sci U S A. 2016;113:5311–5316.
- Sandhu S, Wu X, Nabi Z, et al. Loss of HLTF function promotes intestinal carcinogenesis. Mol Cancer. 2012;11:18.
- Lu F, Liu Y, Jiang L, et al. Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev. 2014;28:2103–2119.
- Zeng S, Xiang T, Pandita TK, et al. Telomere recombination requires the MUS81 endonuclease. Nat Cell Biol. 2009;11:616–623.
- Vorlickova M, Tomasko M, Sagi AJ, et al. 8-oxoguanine in a quadruplex of the human telomere DNA sequence. Febs J. 2012;279:29–39.
- McNulty JM, Jerkovic B, Bolton PH, et al. Replication inhibition and miscoding properties of DNA templates containing a site-specific cis-thymine glycol or urea residue. Chem Res Toxicol. 1998;11:666–673.
- Wang Z, Rhee DB, Lu J, et al. Characterization of oxidative guanine damage and repair in mammalian telomeres. PLoS Genet. 2010;6:e1000951.
- Fouquerel E, Barnes RP, Uttam S, et al. Targeted and persistent 8-oxoguanine base damage at telomeres promotes telomere loss and crisis. Mol Cell. 2019;75:117–130 e116.
- Vallabhaneni H, O’Callaghan N, Sidorova J, et al. Defective repair of oxidative base lesions by the DNA glycosylase Nth1 associates with multiple telomere defects. PLoS Genet. 2013;9:e1003639.