
ABSTRACT
During evolution, cells have developed a plethora of mechanisms to optimize survival in a changing and unpredictable environment. In this regard, they have evolved networks that include environmental sensors, signaling transduction molecules and response mechanisms. Hog1 (yeast) and p38 (mammals) stress-activated protein kinases (SAPKs) are activated upon stress and they drive a full collection of cell adaptive responses aimed to maximize survival. SAPKs are extensively used to learn about the mechanisms through which cells adapt to changing environments. In addition to regulating gene expression and metabolism, SAPKs control cell cycle progression. In this review, we will discuss the latest findings related to the SAPK-driven regulation of mitosis upon osmostress in yeast.
Introduction
The budding yeast Saccharomyces cerevisiae has been used as a model to study environmental signal transduction pathways. Yeasts have the HOG (High Osmolarity Glycerol) pathway to sense, transduce and respond to an external increase in osmolarity. The cornerstone of the HOG pathway is the stress-activated protein kinase (SAPK) Hog1 (p38 in mammalian cells), which belongs to the mitogen-activated protein kinase (MAPK) family. Upon osmostress, the HOG pathway is rapidly activated to orchestrate a full set of actions to protect cells and ensure their fitness and survival. This response involves various aspects of cell biology, ranging from gene transcription regulation and metabolism control to cell cycle progression [Citation1,Citation2].
Beyond cyclin-dependent kinases (CDKs) and cyclins, the main proteins involved in cell cycle progression, cells have evolved to develop regulatory mechanisms aimed at ensuring their faithful duplication and, consequently, perpetuation. Safe and accurate cell duplication has many threads, both internal and external, and adaptation to a changing environment is a remarkable one. Osmostress in yeast cells has been used as a model to study the mechanisms used by cells to protect their progeny in a changing environment [Citation3,Citation4]. Among these mechanisms, the transient arrest of cell cycle progression has attracted the attention of several research groups, which have unveiled several mechanisms that govern G1, S and G2 phases. Recently, the molecular mechanism responsible for regulating cell cycle progression in mitosis has been reported. In this review, we will focus on the SAPK-dependent molecular mechanisms that regulate transient arrest of the cell cycle in response to stress, with a particular emphasis on mitosis, using osmostress as a prototypical case study.
1. The SAPK stress signaling pathway
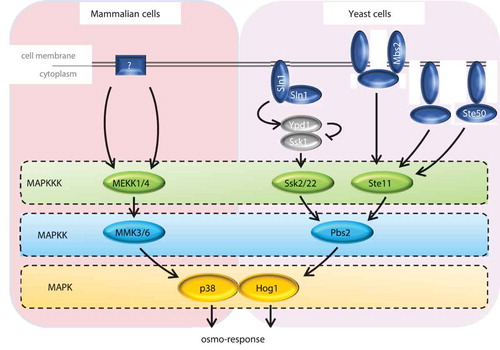
The HOG pathway of the yeast S. cerevisiae is the paradigm of a SAPK signaling pathway. One of the five MAPK cascades present in this organism, the HOG pathway is the main signaling system responsible for cellular adaptation to osmostress [Citation5]. Upon osmostress, this pathway is rapidly and transiently activated by two upstream branches: Sln1 and Sho1 [Citation6,Citation7]. The MAPK core module has three MAPKKKs (Ssk2, Ssk22 and Ste11) [Citation8–Citation11], which activate the MAPKK component (Pbs2) by phosphorylation. Subsequently, Pbs2 phosphorylates and activates the MAPK Hog1 () (reviewed in [Citation2]. Upon activation, Hog1 rapidly translocates into the nucleus [Citation12], where it exerts some of its main functions, such as reprogramming gene expression (reviewed in [Citation1,Citation13] and delaying cell cycle progression (focus of this review). p38 is the mammalian SAPK ortholog of Hog1. p38 responds to an increase in extracellular osmolarity and is essential for adaptation to osmostress [Citation14]. However, it is activated and responds to other stimuli such as cytokines, DNA damage, oxidative and heat stress [Citation15,Citation16]. The core structure of the p38 pathway is similar to that of HOG in yeast [Citation17], although the activation mechanism is not totally understood. In vivo replacement of components of the HOG pathway in S. cerevisiae by their mammalian counterparts demonstrated that there is a strong functional preservation of these MAPK pathways from yeast to mammals [Citation14,Citation18]. It is also worth mentioning the pleiotropic function of p38, which is also pivotal in regulating differentiation, proliferation, apoptosis, cell morphology and immune response [Citation15,Citation19,Citation20].
Figure 1. Schematic representation of the HOG and p38 SAPK pathways. In mammalian cells (left panel), the sensors are unclear. In budding yeast (right panel), two independent osmosensing mechanisms, the Sln1 and Sho1 branches, converge in the MAPK module. Activation of the sensors leads to the phosphorylation of the MAPK (p38 and Hog1) by specific MAPKKK and MAPKK, triggering the osmo-adaptive response by phosphorylation of multiple substrates.
2. Regulation of G1, S and G2 phases by SAPKs upon stress
Upon activation, Hog1 rapidly and transiently migrates into the nucleus, where it phosphorylates substrates to regulate cell cycle progression. This serves to prevent cells having to deal with cell cycle progression in stress conditions where successful accomplishment cannot be assured and thus viability is compromised. All the mechanisms presented below share the same strategy: arresting the cell cycle to provide cells with time to adapt to the osmotic change and resuming it only when the osmotic imbalance has been corrected and homeostasis restored. Research over the last 15 years has revealed several molecular mechanisms that occur during the different phases of the cell cycle.
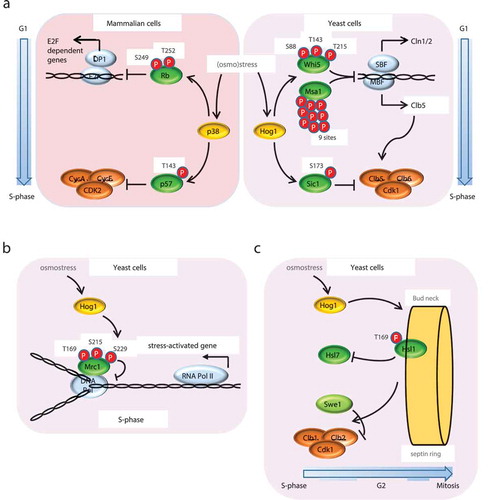
The G1-S transition in budding yeast is driven by the complex of CDK and G1 cyclins or Clns (Cln1, Cln2 and Cln3), whose expression is controlled by the Swi4/6 cell cycle box (SBF) transcription factor [Citation21,Citation22]. SBF is kept inactive by the transcriptional repressor Whi5, the yeast functional ortholog of human RB. This repression is alleviated by phosphorylation of Whi5 by CDK, which leads to the expression of several proteins, including Clns, which are essential for cell cycle progression through G1. Additional proteins are involved in G1-specific transcription; Stb1 and Nrm1 regulate the activity of the Mlu1 cell cycle box (MBF) [Citation23,Citation24], while Msa1, able to interact with Stb1 and Nrm1, is a coactivator of both SBF and MBF [Citation25]. In addition to factors involved in the expression of the CDK activators, there is an extra layer of regulation based on the CDKI (CDK Inhibitor) Sic1 [Citation26]. Sic1 inhibits the CDK-Clb5 complex. When the levels of Clb5 surpass a certain threshold both by the firing of the Clb5 promoter and by the degradation of Sic1 (determined by its phosphorylation by CDK-Clns), cells abruptly enter S phase [Citation27].
Activation of Hog1 by osmostress (or by genetic means using conditional hyperactive mutant alleles of upstream components of the MAPK pathway) yields a transient arrest in G1 via the stabilization of Sic1 and the down-regulation of G1 cyclins () [Citation28]. The stabilization of Sic1 and consequent Hog1-dependent transient G1 arrest is essential for the adaptive response to osmostress, and cells lacking Sic1 or carrying the non-phosphorylatable allele sic1T73A show reduced viability under high osmolarity [Citation29,Citation30]. Additionally, Hog1 activation represses cyclins CLN2 and CLB5 expression through Whi5 and Msa1 phosphorylation to ensure coherent passage through G1/S. The phosphorylation of these two transcriptional regulators by Hog1 is essential for inhibiting G1-cyclin expression, regulating cell morphogenesis, and ensuring maximal cell survival upon stress [Citation31]. Recently, it has been reported that the phosphatase calcineurin prolongs Hog1 activation and the extent of cell cycle arrest upon osmostress. The crosstalk between calcineurin and the MAPK contributes to the inactivation of multiple regulatory transcription factors of the cell cycle and the down-regulation of cell cycle-regulated genes [Citation32].
Figure 2. Regulation of G1, S and G2 phases by Hog1. (a) G1-S phase transition controlled by p38 and Hog1 SAPKs upon stress. Upon osmostress, p38 and Hog1 SAPKs phosphorylate the S/CDK inhibitor p57 or Sic1 respectively at a single residue. In mammalian cells (left panel), p57 phosphorylated at Thr143 has increased affinity toward the Cyclin A/Cdk2 complex, leading to G1 arrest. In budding yeast (right panel), Sic1 phosphorylation at T173 protects it from degradation by the proteasome, resulting in G1 arrest. Additionally, SAPK activation delays the G1-S phase transition by down-regulating the expression of G1 cyclins via phosphorylation of the transcription regulators Whi5 and Msa1 (yeast) and RB (mammals). (b) S phase regulation by Hog1 upon osmostress. Activated Hog1 phosphorylates the replication fork progression regulator Mrc1, delaying DNA replication and avoiding collisions between DNA Pol and the transcription machinery, which transcribes the osmo-responsive genes that are essential for cell adaptation and survival. Avoiding the concurrence of transcription and replication protects cells from DNA instability. (c) G2 regulation by Hog1 upon osmostress. In basal conditions, the CDK inhibitor Swe1 is recruited to the septin ring at the bud neck by the Hsl1–Hsl7 complex. At the neck, Swe1 is phosphorylated by Cdc5, leading to its degradation, which in turn activates Clb2–Cdc28. Activated Hog1 phosphorylates Hsl1, dissolving the complex with Hsl7, thereby preventing Swe1 migration to the neck and its degradation and, consequently, delaying the activation of the CDK and cell cycle progression through G2-M phases.
Mathematical modeling and quantitative analysis of G1 progression upon osmostress has allowed evaluation of the contribution of the different Hog1-dependent mechanisms. Whereas inhibition of CLN2 expression and Sic1 stabilization are important to prevent S phase entry in response to stress occurring close to Start, the inhibition of CLB5 expression is critical in the response to osmostress occurring at any stage of G1 [Citation33–Citation35].
Although the cell cycle machinery is more complex in mammals from a molecular perspective, the same principles described above in yeast govern cell cycle progression. CDK activity is regulated by two families of CDKIs: the INK and the Cip/Kip, which include p21CIP1, p27KIP1 and p57KIP2 [Citation36]. In response to osmostress (and other stresses), p38 delays G1 progression by directly targeting p57KIP2, by phosphorylating its Thr143, thereby increasing its affinity toward the CDK and, as a result, reducing its activity and causing a transient G1 arrest [Citation37,Citation38] (). Moreover, p38 induces p21 mRNA stabilization, without affecting its transcription or the stability of the protein. Inhibition of p38 impairs p21 accumulation and, as a result, the ability of cells to arrest in G1 in response to gamma radiation [Citation39]. p38-activated signaling leads to p27 stabilization [Citation40,Citation41]. Additionally, p38 phosphorylates the Retinoblastoma (RB; Whi5 ortholog) tumor suppressor in the N-terminal region, at Ser249 and Thr252, thereby revealing a new interaction surface between RB and E2F transcription factor. This interaction leads to an increase in RB affinity for E2F and, in turn, to a down-regulation of E2F-dependent gene expression (e.g. CycA2) and reduction of cell proliferation (). Remarkably, the p38-dependent phosphorylation of RB is dominant over the effect of CDKs, yielding RB insensitive to CDK inactivation, which is typical of cancer cells. Moreover, a p38-phosphomimetic RB mutant blocks cyclin expression, prevents cell proliferation in cancer cell lines, and leads to reduced tumor size in a mouse xenograft model. These observations thus suggest that phosphorylated RB acts as a super-repressor that prevents cancer cell proliferation [Citation17,Citation42–Citation44].
Hog1 also plays a key role in S phase by transiently delaying DNA replication in response to osmostress [Citation45]. S phase progression in yeast is driven mainly by CDK-Clb5/Clb6 activity, which phosphorylates substrates at the early and late replication origins. Cells dispose a specific S phase checkpoint pathway mediated by Rad53 to cope with genotoxic agents or stresses that endanger correct progression of DNA replication [Citation46,Citation47]. Interestingly, Hog1-dependent arrest in S phase upon osmostress is independent of the Rad53-dependent checkpoint. Hog1 interacts with and phosphorylates Mrc1, a component of the replication complex, at the N-terminal Thr169, Ser215 and Ser229 sites [Citation48], (). Mrc1 phosphorylation by Hog1 delays early and late origin firing by preventing Cdc45 loading, as well as slowing down replication-complex progression [Citation49,Citation50]. This mechanism is especially relevant because it allows cells to circumvent conflicts between DNA replication and transcription during a burst in transcription, which takes place as a crucial response for adaptation [Citation51]. The N-terminal phosphorylation of Mrc1 blocks replication and prevents transcription-associated recombination (TAR) and genomic instability during stress-induced gene expression in S phase. Interestingly, cells adapt to sudden increases in transcription caused by factors other than osmostress while replicating by the same Mrc1-dependent mechanism, although signaling and kinases other than Hog1 are involved. Thus, Mrc1 integrates multiple signals, thereby defining a general safeguard mechanism to protect genomic integrity upon transcriptional outbursts [Citation52].
Entry into mitosis is driven by the activity of the CDK-Clb2 complex, which is negatively regulated by Swe1 to ensure that cells have the required size to accomplish cell division [Citation53,Citation54]. Cells remain in G2 until Swe1 is degraded by two independent mechanisms, namely phosphorylation by CDK-Clb2 [Citation55], and degradation by the Hsl1 and Cdc5 kinases when targeted to the septin ring by Hsl7 [Citation56]. Hog1 activation stabilizes Swe1 and down-regulates the cyclin CLB2, triggering a transient arrest in G2 phase () [Citation57–Citation59]. Upon osmostress, Hog1 phosphorylates the Hsl1 kinase in Thr169, which delocalizes Hsl7 from the septin ring and impairs Swe1 recruitment to the bud neck. This prevents Swe1 degradation, leading a transient G2 arrest and, thus, a delay in progression into M phase, thereby allowing adaptation to osmostress. It should be noted that the same mechanism has been proposed in Schizosaccharomyces pombe [Citation60], together with alternative mechanisms where the SAPK sty1 regulates cdc25, the phosphatase involved in inhibiting the mitosis repressor wee1 [Citation61]. Similar to the fission yeast, activated p38 is required for a G2/M checkpoint involving Cdc25B and Cdc25C, which regulate the activity of the CDK-cyclin B1 complex during mitosis. Upon DNA damage, the downstream kinase MK2 phosphorylates Cdc25, which creates a docking site for 14-3-3 proteins that will retain Cdc25 in the cell cytoplasm, thus preventing Cdc2-CyclinB dephosphorylation and activation [Citation62–Citation64].
3. Mitosis
Mitosis ensures the accurate inheritance of genetic information. The genome, which is packed into chromosomes, is distributed between the two daughter cells during mitosis. Before mitosis, sister chromatids are tightly interlinked via the intertwining of their DNA (DNA catenation) and by specialized protein complexes called cohesins. In early mitosis, the connected sister chromatids are prepared for separation under the influence of a sophisticated regulatory system based on mitotic CDK-cyclin complexes. First, during prophase, the chromosomes are condensed into flexible rods, which are easily moved by the mitotic spindle. Entry into mitosis also leads to the separation of the two centrosomes (spindle pole bodies in yeast). In metaphase, the sister chromatids are aligned at the center of the spindle (the metaphase plate). At the onset of anaphase, the cohesin links between the sister chromatids are abruptly dissolved, and the separated sister chromatids are pulled to opposite poles of the spindle, a process called anaphase A. In anaphase B, the spindle poles move apart, completing the segregation of the sister chromatids into the two opposing halves of the dividing cell. Mitosis is completed in telophase, when the chromosomes and other nuclear components are repackaged into identical daughter nuclei and the mitotic spindle is disassembled. In vertebrate cells, the nuclear envelope breaks down in early mitosis. By contrast, yeasts do not dismantle their nuclear envelope, and the mitotic spindle forms inside the nucleus (referred to as closed mitosis).
Mitotic CDKs trigger entry into mitosis, promoting nuclear envelope breakdown, spindle assembly and organization, chromosome condensation, and Golgi fragmentation, and contribute to APC/C regulation [Citation65–Citation70]. When chromosomes are correctly attached and aligned and the bipolar tension forces are present, the spindle assembly checkpoint (SAC) is satisfied and the anaphase-promoting complex (APC/C or cyclosome) is triggered by its co-activator Cdc20, promoting the metaphase to anaphase transition. Upon activation, the APC/C-Cdc20 complex ubiquitinates several proteins, promoting their degradation by the proteasome. The most important APC/C-Cdc20 targets are securin and B-type cyclins [Citation71,Citation72].
Sister chromatid separation at the onset of anaphase is triggered upon cleavage of the Scc1 subunit of cohesin by the protease separase [Citation73]. Before anaphase, separase (Esp1 in budding yeast) is maintained inactive by the binding of securin [Citation74]. To prevent the early segregation of sister chromatids, separase must be kept inactive until the chromosomes are aligned and attached to the microtubules. At the metaphase to anaphase transition, APC/C-Cdc20 ubiquitinates securin, targeting it for proteasomal degradation and thereby activating separase. Active separase cleaves the Scc1 subunit of the cohesin complex upon its phosphorylation by Cdc5, promoting the separation of the sister chromatids [Citation73–Citation75]. The APC/C-Cdc20 complex also targets cyclins B for degradation, promoting the first wave of Cdk1 inactivation [Citation76,Citation77]. However, destruction of cyclins B by APC/C-Cdc20 is not sufficient to nullify all Cdk1 activity, which is essential for mitotic exit. Activation of the mitotic phosphatase Cdc14 is therefore essential to counteract Cdk1 activity. Cdc14 contributes to Cdk1 inactivation by promoting the accumulation of the Cdk1 inhibitor Sic1, the dephosphorylation and activation of the second APC/C co-activator Cdh1, and the dephosphorylation of Cdk1 substrates [Citation78,Citation79].
Cdc14 belongs to a family of highly conserved dual-specificity phosphatases (DUSPs) that is conserved from yeast to humans (reviewed in [Citation80,Citation81]) and tightly regulated by changes in its subcellular localization. During most of the cell cycle, Cdc14 is kept sequestered at the nucleolus by its binding to the nucleolar protein Net1 (also called Cfi) [Citation82]. The dimer Cdc14-Net1, together with Sir2 and Fob1, form the RENT (regulator of nucleolar silencing and telophase) complex, which regulates ribosomal DNA (rDNA) silencing and segregation [Citation83–Citation87]. In anaphase, Cdc14 is dissociated from the RENT complex and released from the nucleolus, allowing its translocation throughout the cell, active as a phosphatase [Citation83,Citation85]. Different localization of Cdc14 phosphatase allows the targeting of distinct substrates during anaphase progression. In addition, the net balance of Cdk1 and Cdc14 activities toward their substrates also regulates their phosphorylation status, thereby contributing to the order of substrate dephosphorylation and progression through mitosis [Citation88].
The release of Cdc14 from the nucleolus depends on two regulatory networks: the Cdc-Fourteen Early Anaphase Release (FEAR) network (reviewed in [Citation80,Citation89]), which initiates Cdc14 release, and the Mitotic Exit Network (MEN), a G-protein signaling cascade that completes Cdc14 activation and release to the cytoplasm (reviewed in [Citation90,Citation91]) (). FEAR acts in early anaphase, when Cdk1 activity is high and promotes the first wave of Cdc14 release from the nucleolus to the nucleus. Numerous proteins, including Slk19, Fob1, Spo12, Clb2, Cdc5, Zds1, PP2A-Cdc55, separase (Esp1) and Hit1, have been implicated in FEAR since their mutants exhibit delayed release of Cdc14 from the nucleolus during early anaphase [Citation80,Citation92–Citation99]. FEAR-dependent Cdc14 release requires Net1 phosphorylation at Cdk1 consensus sites [Citation95,Citation96]. In metaphase, Net1 is maintained in an under-phosphorylated state by the phosphatase PP2A-Cdc55. At anaphase onset, separase, together with Zds1/2 proteins, promotes the PP2A-Cdc55 inactivation via the Cdk1-dependent phosphorylation of the Cdc55 regulatory subunit, allowing the accumulation of phosphorylated Net1 isoforms [Citation96,Citation98,Citation100,Citation101]. Increase levels of phosphorylated Net1 by Cdk1-Clb2, with the contribution of the polo-like kinase Cdc5, stimulates Cdc14 release from the nucleolus since the phosphorylated form of Net1 has lower affinity toward Cdc14 [Citation93–Citation96,Citation102]. The early activated Cdc14 is required for anaphase progression. It leads to spindle stabilization and elongation in anaphase [Citation103,Citation104], positioning of the anaphase nucleus [Citation105], segregation of repetitive DNA regions such as rDNA and telomeres [Citation86,Citation106,Citation107], recruitment of condensin to rDNA [Citation87,Citation99], and full Cdc14 activation by a positive feedback loop activating MEN by Cdc15 dephosphorylation [92;108].
Figure 3. Molecular mechanisms for Cdc14 activation and cell cycle progression in mitosis. For mitosis to progress, substrates phosphorylated by CDK must be dephosphorylated. This action is carried out by the phosphatase Cdc14, which is sequestered in the nucleolus and kept inactive by its interaction with Net1 during all phases of the cell cycle except mitosis. There are two mechanisms devoted to releasing Cdc14, namely FEAR, which is shown on the left of the figure, and MEN, on the right. PP2A-Cdc55 phosphatase keeps Net1 dephosphorylated (and as a consequence Cdc14 sequestered) throughout the cell cycle until securin is degraded, a process that activates separase and causes chromosome separation. The inhibition of PP2A-Cdc55 along with the phosphorylation of Net1 by CDK and polo (Cdc5) allow Cdc14 release from the nucleolus to the nucleus, where it becomes active, dephosphorylating its nuclear substrates. Full release of Cdc14 to the cytoplasm requires the action of MEN, which is activated in coordination with SPB migration to the daughter cell. Fully cytoplasmic released Cdc14 triggers the end of mitosis by resetting all CDK substrates and it determines the morphogenetic mechanisms for septum formation and cell separation.
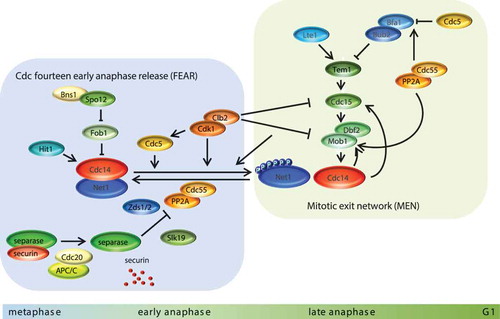
After the first wave of FEAR-Cdc14 release, when the Cdk1 mitotic kinase activity declines, the MEN kinases sustain Net1 phosphorylation and Cdc14 activation. The MEN pathway is GTPase-driven and is closely related to the mammalian Hippo pathway, which is involved in mitotic exit regulation, spindle orientation checkpoints (SPOC) and cytokinesis. The core of the MEN cascade consists of two serine/threonine kinases, Cdc15 (the Pak-like kinase) and Dbf2-Mob1 (NDR/LATS-related MEN kinases). The upstream effector of the MEN is Tem1, a small Ras-like GTPase that is localized at the centrosome (or spindle pole body, SPBs in yeast) [Citation108]. Tem1 activity is negatively regulated by the two-component GTPase-activating protein (GAP) Bfa1-Bub2 [Citation109,Citation110] and positively regulated by the Lte1 protein [Citation111–Citation113]. In metaphase, PP2A-Cdc55 keeps Bfa1 dephosphorylated, thereby contributing to the activation of Bfa1-Bub2 [Citation114]. When cells reach anaphase with a correct aligned mitotic spindle, Cdc5 phosphorylates Bfa1 and inactivates Bfa1-Bub2 GAP [Citation115,Citation116]. In addition, Cdk1-dependent PP2A-Cdc55 inhibition at early anaphase [Citation96,Citation101] also promotes the accumulation of phosphorylated Cdc5-dependent Bfa1. Active Tem1 interacts with and activates the Pak-like kinase Cdc15 [Citation117,Citation118]. In addition, the released FEAR-Cdc14 dephosphorylates Cdc15, facilitating Cdc14 activation in a positive feedback loop [Citation92,Citation119]. Once active, Cdc15 recruits the LATS-related Dbf2-Mob1 complex to the SPB and phosphorylates Dbf2 [Citation120,Citation121]. Moreover, Cdc14 and PP2A-Cdc55 dephosphorylate Mob1, thereby alleviating its Cdk1 inhibitory phosphorylation [Citation114,Citation122,Citation123]. The Dbf2-Mob1 kinase mediates Cdc14 release from the nucleus, thus maintaining Net1 phosphorylated [Citation124–Citation126] and retaining Cdc14 in the cytoplasm by phosphorylation at sites adjacent to its nuclear localization signal (NLS) [Citation125]. Cytoplasmatic Cdc14 directly promotes mitotic exit via dephosphorylation of several Cdk1 targets, the second APC/C activator Cdh1, the transcription factor Swi5, and the Cdk1 inhibitor Sic1 [Citation78,Citation127–Citation129]. Cytoplasmatic Cdc14 also regulates cytokinesis, ensuring timely septum disruption after cytokinesis (reviewed in [Citation130–Citation132]).
Mitotic exit is precisely and tightly coordinated to ensure that cell division occurs only after chromosomes are properly replicated and equally segregated between the two new daughter cells. Problems during mitotic exit can lead to genomic instability, genetic diseases and neurodegenerative disorders [Citation133,Citation134]. Various surveillance mechanisms or checkpoints delay mitotic progression to guarantee faithful inheritance of the genetic material. In budding yeast, the main mitotic checkpoints are the DNA damage checkpoint (DDC), the spindle assembly checkpoint (SAC) and the spindle position checkpoint (SPOC) (reviewed in [Citation135]). The DDC delays the metaphase to anaphase transition in response to DNA lesions to give the cell time to repair the DNA damage [Citation115,Citation136,Citation137]. In the presence of DNA damage, Pds1 is phosphorylated by the DDC-effector kinase Chk1, thereby preventing its ubiquitination and degradation by the proteasome [Citation71,Citation138]. Rad53, the other DDC-effector kinase, contributes to Pds1 stability by preventing Pds1 and Cdc20 interaction [Citation138] and halts elongation of the mitotic spindle and MEN activation by Cdc5 inhibitory phosphorylation [Citation136,Citation139].
The SAC responds to unattached kinetochores by arresting the cells in metaphase and inhibiting MEN activity (reviewed in [Citation140,Citation141]). The molecular mechanisms leading to MEN inhibition upon SAC activation remain unclear. At metaphase, SAC proteins inhibit the APC/C-Cdc20 complex in the presence of unattached microtubules [Citation142,Citation143]. In addition, Aurora B (Ipl1 in S. cerevisiae) and Shugosin (Sgo1) are also important to sense the lack of tension upon incorrect kinetochore-microtubule attachments and to promote bi-orientation of the chromosomes [Citation144–Citation147].
The SPOC inhibits MEN to prevent mitotic exit until the spindle is properly positioned. Upon spindle misalignment, the main SPOC kinase effector Kin4 regulates the activity and localization of the Bfa1-Bub2 complex, thereby preventing Bfa1 activation by Cdc5 phosphorylation [Citation148–Citation152]. Although the DDC, SAC and SPOC are induced in response to distinct stimuli, there is crosstalk between them, the polo-like kinase Cdc5 being the central hub that coordinates mitotic progression with the main cell cycle checkpoints [Citation136,Citation150,Citation153–Citation155].
4. Regulation of mitosis by the Hog1 SAPK upon stress
Mitosis is another phase in which a molecular mechanism for cell cycle control in response to osmostress has been proposed. In a MEN mutant background, enhanced activation of Cdc14 leads to accelerated mitosis upon osmostress in a Hog1-dependent manner [Citation156]. Indeed, mathematical model predictions supported the notion that cells stressed at the late G2/M phase display accelerated exit from mitosis and arrest in the next cell cycle [Citation35]. However, the same model predicted the contribution of mechanisms other than the hyperactivation of Cdc14 to mitosis regulation upon osmostress [Citation35] Along these lines, a novel molecular mechanism by which Hog1 activation regulates the M phase in response to stress has recently been described [Citation157] ().
Figure 4. Hog1 regulation of mitosis upon osmostress. Increased levels of phosphorylated Net1 by the Cdk1-Clb2 complex, with the contribution of the polo-like kinase Cdc5, promotes the release of Cdc14 from the nucleolus. Activated Hog1 phosphorylates Net1 at Thr62 and Ser385, altering its affinity for the phosphatase Cdc14 and rendering the Net1-Cdc14 complex more resistant to CDK activity. Consequently, Cdc14 is kept sequestered at the nucleolus and mitosis progression is delayed.
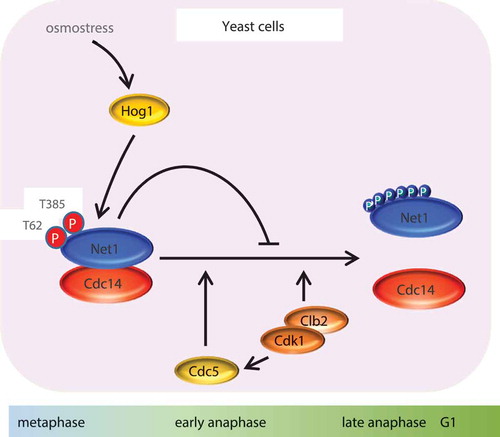
There are several lines of evidence demonstrating that cells arrest in M phase in response to osmostress. Biochemical (Clb2 destruction) and cellular (DNA content) readouts revealed that cells synchronized in early M phase by Cdc20 depletion or other methods delay the next cell cycle G1entry by around 60 min in response to 0.4 M NaCl. Moreover, genetic activation of Hog1 using a temperature-sensitive SLN1 (sln1ts) mutant allele in early metaphase showed a similar delay, thereby suggesting a direct role of the HOG pathway in this transient cell cycle arrest. Activation of Hog1 resulted in cells progressing normally through anaphase, timely elongated spindles, and apparently separated nuclei, but with a delay in spindle disassembly and physical separation [Citation157]. Accordingly, nucleolar release of Cdc14, is delayed upon Hog1 activation. Of note, Net1, which is responsible for the timely release of Cdc14, is targeted by Hog1. In vitro and in vivo experiments showed that Hog1 phosphorylates Net1 at Thr62 and Ser385, which, in fact, are distinct residues to those targeted by CDK to release Cdc14 in unperturbed cells. Net1 phosphorylation by Hog1 does not prevent Clb2-CDK phosphorylation on Net1, but stabilizes the interaction between Net1 and Cdc14, even in the presence of the Clb2-CDK complex, thereby impairing Cdc14 release. Nucleolar retention of Cdc14 upon HOG pathway activation is abolished when genomic net1T62A, S385A mutations are present. Thus, Hog1 phosphorylates Net1, altering its affinity for the phosphatase Cdc14, whose activity is essential for mitotic exit and completion of the cell cycle.
The delayed Cdc14 release from the nucleolus upon activation of Hog1 is coupled to a defect in rDNA and telomere segregation, and it eventually delays cell division. Of note, the mutant net1T62A, S385A, which cannot be phosphorylated by Hog1, displays reduced viability upon osmostress. This observation thus indicates that Hog1 contributes to maximizing cell survival upon stress by regulating mitotic exit. As a summary, the model emerging from these lines of evidence can be presented as follows; in an unperturbed cell cycle, Clb2-CDK complex phosphorylates Net1 and determines Cdc14 release, which in turn promotes the dephosphorylation of several targets to promote cell division. However, when the HOG pathway is activated at early M phase, Hog1 phosphorylates Net1, thus increasing Net1 affinity for Cdc14, and, consequently, making the complex more resistant to dissociation by CDK activity. The nucleolar retention of Cdc14 determines the transient arrest in the progression to G1. Of note, in mammalian cells, p38 is required for mitosis progression and is essential for the timely stable attachment of all kinetochores to spindle microtubules, but not for the fidelity of mitosis [Citation158,Citation159]. However, other evidence shows that loss of p38γ results in multipolar spindle formation and chromosome misalignment, which induce a transient M phase arrest [Citation160], indicating also a role in regulation of mitosis.
Concluding remarks
During the last two decades, a large body of evidence has demonstrated that cell cycle progression is delayed in a context of stress, understood as changes in the environment. In parallel with other adaptive responses at the transcriptional and metabolic level, cells stop their cell cycle and growth to provide a time window for adaptation. Only when homeostasis has been restored, does the cell cycle resume and can all the delicate processes leading to the accurate division of the genome be achieved. Several molecular mechanisms devoted to providing cells with this invaluable time for adaptation have been described in detail. Despite some idiosyncratic differences, these mechanisms are reasonably well-conserved among eukaryotes. More interestingly, these mechanisms are present in all phases of the cell cycle, in contrast to other control mechanisms that are focused on one specific phase of the cell cycle. These observations thus indicate the importance of regulation of the whole cell cycle in response to external or environmental conditions. Recently, we have added a new piece of the puzzle by describing a molecular mechanism responsible for arresting the cell cycle in its final phase, namely mitosis. This mechanism gives coherence to the remaining arresting mechanisms in G1, S and G2 phases. Additional mechanisms of cell cycle progression upon stress will appear, some of them by using strategies that are qualitatively different to targeting particular activators or inhibitors and that rather modulate more general machineries. The use of cutting-edge technologies and approaches are starting to reveal mechanisms such as the regulation exerted by the HOG pathway on the antisense RNA of the CDK Cdc28 upon osmostress [Citation161,Citation162]. Taken together, the new knowledge gained will provide a comprehensive view of cell cycle regulation by osmostress and more generally by external modulators.
Acknowledgments
We thank Dr. Silvia Tognetti and Dr. Matteo Viganò for helpful discussions and suggestions.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- de Nadal E, Posas F. Osmostress-induced gene expression–a model to understand how stress-activated protein kinases (SAPKs) regulate transcription. Febs J. 2015;282(17):3275–3285.
- Saito H, Posas F. Response to hyperosmotic stress. Genetics. 2012;192(2):289–318.
- Hohmann S. Control of high osmolarity signalling in the yeast Saccharomyces cerevisiae. FEBS Lett. 2009;583(24):4025–4029.
- Hohmann S. An integrated view on a eukaryotic osmoregulation system. Curr Genet. 2015;61(3):373–382.
- Gustin MC, Albertyn J, Alexander M, et al. MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1998;62(4):1264–1300.
- Maeda T, Wurgler-Murphy SM, Saito H. A two-component system that regulates an osmosensing MAP kinase cascade in yeast. Nature. 1994;369(6477):242–245.
- Posas F, Wurgler-Murphy SM, Maeda T, et al. Yeast HOG1 MAP kinase cascade is regulated by a multistep phosphorelay mechanism in the SLN1-YPD1-SSK1 “two-component” osmosensor. Cell. 1996;86(6):865–875.
- Posas F, Saito H. Osmotic activation of the HOG MAPK pathway via Ste11p MAPKKK: scaffold role of Pbs2p MAPKK. Science. 1997;276(5319):1702–1705.
- Posas F, Saito H. Activation of the yeast SSK2 MAP kinase kinase kinase by the SSK1 two-component response regulator. Embo J. 1998;17(5):1385–1394.
- Posas F, Witten EA, Saito H. Requirement of STE50 for osmostress-induced activation of the STE11 mitogen-activated protein kinase kinase kinase in the high-osmolarity glycerol response pathway. Mol Cell Biol. 1998;18(10):5788–5796.
- Maeda T, Takekawa M, Saito H. Activation of yeast PBS2 MAPKK by MAPKKKs or by binding of an SH3-containing osmosensor. Science. 1995;269(5223):554–558.
- Ferrigno P, Posas F, Koepp D, et al. Regulated nucleo/cytoplasmic exchange of HOG1 MAPK requires the importin beta homologs NMD5 and XPO1. Embo J. 1998;17(19):5606–5614.
- de Nadal E, Ammerer G, Posas F. Controlling gene expression in response to stress. Nat Rev Genet. 2011;12(12):833–845.
- Sheikh-Hamad D, Gustin MC. MAP kinases and the adaptive response to hypertonicity: functional preservation from yeast to mammals. Am J Physiol Renal Physiol. 2004;287(6):F1102–F1110.
- Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429(3):403–417.
- Nebreda AR, Porras A. p38 MAP kinases: beyond the stress response. Trends Biochem Sci. 2000;25(6):257–260.
- Martinez-Limon A, Joaquin M, Caballero M, et al. The p38 pathway: from biology to cancer therapy. Int J Mol Sci. 2020;21(6):1–18.
- Han J, Lee JD, Bibbs L, et al. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265(5173):808–811.
- Gupta J, Nebreda AR. Roles of p38alpha mitogen-activated protein kinase in mouse models of inflammatory diseases and cancer. Febs J. 2015;282(10):1841–1857.
- Yue J, L+¦pez JM. Understanding MAPK signaling pathways in apoptosis. Int J Mol Sci. 2020;21(7):2346.
- Costanzo M, Nishikawa JL, Tang X, et al. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell. 2004;117(7):899–913.
- de Bruin RA, McDonald WH, Kalashnikova TI, et al. Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell. 2004;117(7):887–898.
- Ho Y, Costanzo M, Moore L, et al. Regulation of Transcription at theSaccharomyces cerevisiae start transition by Stb1, a Swi6-binding protein. Mol Cell Biol. 1999;19(8):5267–5278.
- de Bruin RA, Kalashnikova TI, Chahwan C, et al. Constraining G1-specific transcription to late G1 phase: the MBF-associated corepressor Nrm1 acts via negative feedback. Mol Cell. 2006;23(4):483–496.
- Ashe M, de Bruin RA, Kalashnikova T, et al. The SBF-and MBF-associated protein Msa1 is required for proper timing of G1-specific transcription in Saccharomyces cerevisiae. J Biol Chem. 2008;283(10):6040–6049.
- Schwob E, Bohm T, Mendenhall MD, et al. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in Scerevisiae. Cell. 1994;79(2):233–244.
- Cross FR, Schroeder L, Bean JM. Phosphorylation of the Sic1 inhibitor of B-type cyclins in Saccharomyces cerevisiae is not essential but contributes to cell cycle robustness. Genetics. 2007;176(3):1541–1555.
- Feldman RM, Correll CC, Kaplan KB, et al. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell. 1997;91(2):221–230.
- Escote X, Zapater M, Clotet J, et al. Hog1 mediates cell-cycle arrest in G1 phase by the dual targeting of Sic1. Nat Cell Biol. 2004;6(10):997–1002.
- Zapater M, Clotet J, Escote X, et al. Control of cell cycle progression by the stress-activated Hog1 MAPK. Cell Cycle. 2005;4(1):6–7.
- Gonzalez-Novo A, Jimenez J, Clotet J, et al. Hog1 targets Whi5 and Msa1 transcription factors to down-regulate cyclin expression upon stress. Mol Cell Biol. 2015;35(9):1606–1618.
- Leech CM, Flynn MJ, Arsenault HE, et al. The coordinate actions of calcineurin and Hog1 mediate the stress response through multiple nodes of the cell cycle network. PLoS Genet. 2020;16(4):e1008600.
- Adrover MA, Zi Z, Duch A, et al. Time-dependent quantitative multicomponent control of the G-S network by the stress-activated protein kinase Hog1 upon osmostress. Sci Signal. 2011;4(192):ra63.
- Radmaneshfar E, Thiel M. Recovery from stress - a cell cycle perspective. JCIS. 2012;3(1–2):33–44.
- Radmaneshfar E, Kaloriti D, Gustin MC, et al. From START to FINISH: the influence of osmotic stress on the cell cycle. PLoS One. 2013;8(7):e68067.
- Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14(2):159–169.
- Joaquin M, Gubern A, Posas F. A novel G1 checkpoint mediated by the p57 CDK inhibitor and p38 SAPK promotes cell survival upon stress. Cell Cycle. 2012;11(18):3339–3340.
- Joaquin M, Gubern A, Gonzalez-Nunez D, et al. The p57 CDKi integrates stress signals into cell-cycle progression to promote cell survival upon stress. Embo J. 2012;31(13):2952–2964.
- Lafarga V, Cuadrado A, Lopez D, et al. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol Cell Biol. 2009;29(16):4341–4351.
- Cuadrado M, Gutierrez-Martinez P, Swat A, et al. p27Kip1 stabilization is essential for the maintenance of cell cycle arrest in response to DNA damage. Cancer Res. 2009;69(22):8726–8732.
- Swat A, Dolado I, Rojas JM, et al. Cell density-dependent inhibition of epidermal growth factor receptor signaling by p38alpha mitogen-activated protein kinase via Sprouty2 downregulation. Mol Cell Biol. 2009;29(12):3332–3343.
- Gubern A, Joaquin M, Marques M, et al. The N-terminal phosphorylation of RB by p38 bypasses its inactivation by CDKs and prevents proliferation in cancer cells. Mol Cell. 2016;64(1):25–36.
- Joaquin M, de Nadal E, Posas F. An RB insensitive to CDK regulation. Mol Cell Oncol. 2016;4(1):e1268242.
- Jin X, Ding D, Yan Y, et al. Phosphorylated RB promotes cancer immunity by inhibiting NF-kappaB activation and PD-L1 expression. Mol Cell. 2019;73(1):22–35.
- Yaakov G, Duch A, Garcia-Rubio M, et al. The stress-activated protein kinase Hog1 mediates S phase delay in response to osmostress. Mol Biol Cell. 2009;20(15):3572–3582.
- Lopez-Mosqueda J, Maas NL, Jonsson ZO, et al. Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature. 2010;467(7314):479–483.
- Zegerman P, Diffley JF. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature. 2010;467(7314):474–478.
- Alcasabas AA, Osborn AJ, Bachant J, et al. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Bio. 2001;3(11):958–965.
- Duch A, Felipe-Abrio I, Barroso S, et al. Coordinated control of replication and transcription by a SAPK protects genomic integrity. Nature. 2013;493(7430):116–119.
- Duch A, de Nadal E, Posas F. Dealing with transcriptional outbursts during S phase to protect genomic integrity. J Mol Biol. 2013;425(23):4745–4755.
- Duch A, de Nadal E, Posas F. The p38 and Hog1 SAPKs control cell cycle progression in response to environmental stresses. FEBS Lett. 2012;586(18):2925–2931.
- Duch A, Canal B, Barroso SI, et al. Multiple signaling kinases target Mrc1 to prevent genomic instability triggered by transcription-replication conflicts. Nat Commun. 2018;9(1):379.
- Booher RN, Deshaies RJ, Kirschner MW. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. Embo J. 1993;12(9):3417–3426.
- McMillan JN, Sia RA, Bardes ES, et al. Phosphorylation-independent inhibition of Cdc28p by the tyrosine kinase Swe1p in the morphogenesis checkpoint. Mol Cell Biol. 1999;19(9):5981–5990.
- Asano S, Park JE, Sakchaisri K, et al. Concerted mechanism of Swe1/Wee1 regulation by multiple kinases in budding yeast. Embo J. 2005;24(12):2194–2204.
- Longtine MS, Theesfeld CL, McMillan JN, et al. Septin-dependent assembly of a cell cycle-regulatory module in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20(11):4049–4061.
- Alexander MR, Tyers M, Perret M, et al. Regulation of cell cycle progression by Swe1p and Hog1p following hypertonic stress. Mol Biol Cell. 2001;12(1):53–62.
- Clotet J, Escote X, Adrover MA, et al. Phosphorylation of Hsl1 by Hog1 leads to a G2 arrest essential for cell survival at high osmolarity. Embo J. 2006;25(11):2338–2346.
- Clotet J, Posas F. Control of cell cycle in response to osmostress: lessons from yeast. Methods Enzymol. 2007;428:63–76.
- Opalko HE, Moseley JB. Dynamic regulation of Cdr1 kinase localization and phosphorylation during osmotic stress. J Biol Chem. 2017;292(45):18457–18468.
- Lopez-Aviles S, Grande M, Gonzalez M, et al. Inactivation of the Cdc25 phosphatase by the stress-activated Srk1 kinase in fission yeast. Mol Cell. 2005;17(1):49–59.
- Manke IA, Nguyen A, Lim D, et al. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol Cell. 2005;17(1):37–48.
- Reinhardt HC, Aslanian AS, Lees JA, et al. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11(2):175–189.
- Reinhardt HC, Hasskamp P, Schmedding I, et al. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol Cell. 2010;40(1):34–49.
- Miele L. The biology of cyclins and cyclin-dependent protein kinases: an introduction. Methods Mol Biol. 2004;285:3–21.
- Rahal R, Amon A. Mitotic CDKs control the metaphase-anaphase transition and trigger spindle elongation. Genes Dev. 2008;22(11):1534–1548.
- Richardson H, Lew DJ, Henze M, et al. Cyclin-B homologs in Saccharomyces cerevisiae function in S phase and in G2. Genes Dev. 1992;6(11):2021–2034.
- Fitch I, Dahmann C, Surana U, et al. Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. Mol Biol Cell. 1992;3(7):805–818.
- Deibler RW, Kirschner MW. Quantitative reconstitution of mitotic CDK1 activation in somatic cell extracts. Mol Cell. 2010;37(6):753–767.
- Chee MK, Haase SB. B-cyclin/CDKs regulate mitotic spindle assembly by phosphorylating kinesins-5 in budding yeast. PLoS Genet. 2010;6(5):e1000935.
- Cohen-Fix O, Peters JM, Kirschner MW, et al. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996;10(24):3081–3093.
- Lim HH, Goh PY, Surana U. Cdc20 is essential for the cyclosome-mediated proteolysis of both Pds1 and Clb2 during M phase in budding yeast. Curr Biol. 1998;8(4):231–234.
- Uhlmann F, Lottspeich F, Nasmyth K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature. 1999;400(6739):37–42.
- Ciosk R, Zachariae W, Michaelis C, et al. An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell. 1998;93(6):1067–1076.
- Alexandru G, Uhlmann F, Mechtler K, et al. Phosphorylation of the cohesin subunit Scc1 by Polo/Cdc5 kinase regulates sister chromatid separation in yeast. Cell. 2001;105(4):459–472.
- Yeong FM, Lim HH, Padmashree CG, et al. Exit from mitosis in budding yeast: biphasic inactivation of the Cdc28-Clb2 mitotic kinase and the role of Cdc20. Mol Cell. 2000;5(3):501–511.
- Wasch R, Cross FR. APC-dependent proteolysis of the mitotic cyclin Clb2 is essential for mitotic exit. Nature. 2002;418(6897):556–562.
- Visintin R, Craig K, Hwang ES, et al. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Molecular Cell. 1998;2(6):709–718.
- Jaspersen SL, Charles JF, Tinker-Kulberg RL, et al. A late mitotic regulatory network controlling cyclin destruction in Saccharomyces cerevisiae. Mol Biol Cell. 1998;9(10):2803–2817.
- Queralt E, Uhlmann F. Cdk-counteracting phosphatases unlock mitotic exit. Curr Opin Cell Biol. 2008;20(6):661–668.
- Mocciaro A, Schiebel E. Cdc14: a highly conserved family of phosphatases with non-conserved functions?. J Cell Sci. 2010;123(17):2867–2876.
- Traverso EE, Baskerville C, Liu Y, et al. Characterization of the Net1 cell cycle-dependent regulator of the Cdc14 phosphatase from budding yeast. J Biol Chem. 2001;276(24):21924–21931.
- Shou W, Seol JH, Shevchenko A, et al. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell. 1999;97(2):233–244.
- Straight AF, Shou W, Dowd GJ, et al. Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell. 1999;97(2):245–256.
- Visintin R, Hwang ES, Amon A. Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus. Nature. 1999;398(6730):818–823.
- Torres-Rosell J, Machin F, Jarmuz A, et al. Nucleolar segregation lags behind the rest of the genome and requires Cdc14p activation by the FEAR network. Cell Cycle. 2004;3(4):496–502.
- Clemente-Blanco A, Sen N, Mayan-Santos M, et al. Cdc14 phosphatase promotes segregation of telomeres through repression of RNA polymerase II transcription. Nat Cell Biol. 2011;13(12):1450–1456.
- Bouchoux C, Uhlmann F. A quantitative model for ordered Cdk substrate dephosphorylation during mitotic exit. Cell. 2011;147(4):803–814.
- Rock JM, Amon A. The FEAR network. Curr Biol. 2009;19(23):R1063–R1068.
- Hotz M, Barral Y. The mitotic exit network: new turns on old pathways. Trends Cell Biol. 2014;24(3):145–152.
- Baro B, Queralt E, Monje-Casas F. Regulation of mitotic exit in Saccharomyces cerevisiae. Methods Mol Biol. 2017;1505:3–17.
- Stegmeier F, Visintin R, Amon A. Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell. 2002;108(2):207–220.
- Shou W, Azzam R, Chen SL, et al. Cdc5 influences phosphorylation of Net1 and disassembly of the RENT complex. BMC Mol Biol. 2002;3:3.
- Yoshida S, Toh-e A. Budding yeast Cdc5 phosphorylates Net1 and assists Cdc14 release from the nucleolus. Biochem Biophys Res Commun. 2002;294(3):687–691.
- Azzam R, Chen SL, Shou W, et al. Phosphorylation by cyclin B-Cdk underlies release of mitotic exit activator Cdc14 from the nucleolus. Science. 2004;305(5683):516–519.
- Queralt E, Lehane C, Novak B, et al. Downregulation of PP2A(Cdc55) phosphatase by separase initiates mitotic exit in budding yeast. Cell. 2006;125(4):719–732.
- Tomson BN, Rahal R, Reiser V, et al. Regulation of Spo12 phosphorylation and its essential role in the FEAR network. Curr Biol. 2009;19(6):449–460.
- Calabria I, Baro B, Rodriguez-Rodriguez JA, et al. Zds1 regulates PP2A(Cdc55) activity and Cdc14 activation during mitotic exit through its Zds_C motif. J Cell Sci. 2012;125(12):2875–2884.
- de Los Santos-velazquez A, de Oya IG, Manzano-Lopez J, et al. Late rDNA condensation ensures timely Cdc14 release and coordination of mitotic exit signaling with nucleolar segregation. Curr Biol. 2017;27(21):3248–3263.
- Queralt E, Uhlmann F. Separase cooperates with Zds1 and Zds2 to activate Cdc14 phosphatase in early anaphase. J Cell Biol. 2008;182(5):873–883.
- Jativa S, Calabria I, Moyano-Rodriguez Y, et al. Cdc14 activation requires coordinated Cdk1-dependent phosphorylation of Net1 and PP2A-Cdc55 at anaphase onset. Cell Mol Life Sci. 2019;76(18):3601–3620.
- Rodriguez-Rodriguez JA, Moyano Y, J+ítiva S, et al. Mitotic exit function of polo-like kinase Cdc5 is dependent on sequential activation by Cdk1. Cell Rep. 2016;15(9):2050–2062.
- Higuchi T, Uhlmann F. Stabilization of microtubule dynamics at anaphase onset promotes chromosome segregation. Nature. 2005;433(7022):171–176.
- Roccuzzo M, Visintin C, Tili F, et al. FEAR-mediated activation of Cdc14 is the limiting step for spindle elongation and anaphase progression. Nat Cell Biol. 2015;17(3):251–261.
- Ross KE, Cohen-Fix O. A role for the FEAR pathway in nuclear positioning during anaphase. Dev Cell. 2004;6(5):729–735.
- Sullivan M, Higuchi T, Katis VL, et al. Cdc14 phosphatase induces rDNA condensation and resolves cohesin-independent cohesion during budding yeast anaphase. Cell. 2004;117(4):471–482.
- D’Amours D, Stegmeier F, Amon A. Cdc14 and condensin control the dissolution of cohesin-independent chromosome linkages at repeated DNA. Cell. 2004;117(4):455–469.
- Shirayama M, Matsui Y, Toh E. The yeast TEM1 gene, which encodes a GTP-binding protein, is involved in termination of M phase. Mol Cell Biol. 1994;14(11):7476–7482.
- Pereira G, Hofken T, Grindlay J, et al. The Bub2p spindle checkpoint links nuclear migration with mitotic exit. Mol Cell. 2000;6(1):1–10.
- Bardin AJ, Visintin R, Amon A. A mechanism for coupling exit from mitosis to partitioning of the nucleus. Cell. 2000;102(1):21–31.
- Falk JE, Campbell IW, Joyce K, et al. LTE1 promotes exit from mitosis by multiple mechanisms. Mol Biol Cell. 2016;27(25):3991–4001.
- Jensen S, Geymonat M, Johnson AL, et al. Spatial regulation of the guanine nucleotide exchange factor Lte1 in Saccharomyces cerevisiae. J Cell Sci. 2002;115(24):4977–4991.
- Geymonat M, Spanos A, de B G, et al. Lte1 contributes to Bfa1 localization rather than stimulating nucleotide exchange by Tem1. J Cell Biol. 2009;187(4):497–511.
- Baro B, Rodriguez-Rodriguez JA, Calabria I, et al. Dual Regulation of the mitotic exit network (MEN) by PP2A-Cdc55 phosphatase. PLoS Genet. 2013;9(12):e1003966.
- Hu F, Wang Y, Liu D, et al. Regulation of the Bub2/Bfa1 GAP complex by Cdc5 and cell cycle checkpoints. Cell. 2001;107(5):655–665.
- Geymonat M, Spanos A, Walker PA, et al. In vitro regulation of budding yeast Bfa1/Bub2 GAP activity by Cdc5. J Biol Chem. 2003;278(17):14591–14594.
- Asakawa K, Yoshida S, Otake F, et al. A novel functional domain of Cdc15 kinase is required for its interaction with Tem1 GTPase in Saccharomyces cerevisiae. Genetics. 2001;157(4):1437–1450.
- Rock JM, Amon A. Cdc15 integrates Tem1 GTPase-mediated spatial signals with Polo kinase-mediated temporal cues to activate mitotic exit. Genes Dev. 2011;25(18):1943–1954.
- Jaspersen SL, Morgan DO. Cdc14 activates cdc15 to promote mitotic exit in budding yeast. Curr Biol. 2000;10(10):615–618.
- Visintin R, Amon A, Koshland D. Regulation of the mitotic exit protein kinases Cdc15 and Dbf2. Mol Biol Cell. 2001;12(10):2961–2974.
- Rock JM, Lim D, Stach L, et al. Activation of the yeast Hippo pathway by phosphorylation-dependent assembly of signaling complexes. Science. 2013;340(6134):871–875.
- Mah AS, Jang J, Deshaies RJ. Protein kinase Cdc15 activates the Dbf2-Mob1 kinase complex. Proc Natl Acad Sci U S A. 2001;98(13):7325–7330.
- Konig C, Maekawa H, Schiebel E. Mutual regulation of cyclin-dependent kinase and the mitotic exit network. J Cell Biol. 2010;188(3):351–368.
- Mah AS, Elia AE, Devgan G, et al. Substrate specificity analysis of protein kinase complex Dbf2-Mob1 by peptide library and proteome array screening. BMC Biochem. 2005;6:22.
- Mohl DA, Huddleston MJ, Collingwood TS, et al. Dbf2-Mob1 drives relocalization of protein phosphatase Cdc14 to the cytoplasm during exit from mitosis. J Cell Biol. 2009;184(4):527–539.
- Campbell IW, Zhou X, Amon A. The mitotic exit network integrates temporal and spatial signals by distributing regulation across multiple components. Elife. 2019;8:e41139.
- Bloom J, Cristea IM, Procko AL, et al. Global analysis of Cdc14 phosphatase reveals diverse roles in mitotic processes. J Biol Chem. 2011;286(7):5434–5445.
- Kao L, Wang YT, Chen YC, et al. Global analysis of cdc14 dephosphorylation sites reveals essential regulatory role in mitosis and cytokinesis. Mol Cell Proteomics. 2014;13(2):594–605.
- Kuilman T, Maiolica A, Godfrey M, et al. Identification of Cdk targets that control cytokinesis. Embo J. 2015;34(1):81–96.
- Juanes MA, Piatti S. The final cut: cell polarity meets cytokinesis at the bud neck in Scerevisiae. Cell Mol Life Sci. 2016;73(16):3115–3136.
- Meitinger F, Palani S, Pereira G. The power of MEN in cytokinesis. Cell Cycle. 2012;11(2):219–228.
- Weiss EL. Mitotic exit and separation of mother and daughter cells. Genetics. 2012;192(4):1165–1202.
- Silk AD, Zasadil LM, Holland AJ, et al. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci U S A. 2013;110(44):E4134–E4141.
- Nicholson JM, Macedo JC, Mattingly AJ, et al. Chromosome mis-segregation and cytokinesis failure in trisomic human cells. Elife. 2015;4:e05068.
- Matellan L, Monje-Casas F. Regulation of mitotic exit by cell cycle checkpoints: lessons from Saccharomyces cerevisiae. Genes (Basel). 2020;11(2):195.
- Valerio-Santiago M, de Los Santos-velazquez AI, Monje-Casas F. Inhibition of the mitotic exit network in response to damaged telomeres. PLoS Genet. 2013;9(10):e1003859.
- Liang F, Wang Y. DNA damage checkpoints inhibit mitotic exit by two different mechanisms. Mol Cell Biol. 2007;27(14):5067–5078.
- Agarwal R, Tang Z, Yu H, et al. Two distinct pathways for inhibiting pds1 ubiquitination in response to DNA damage. J Biol Chem. 2003;278(45):45027–45033.
- Zhang T, Nirantar S, Lim HH, et al. DNA damage checkpoint maintains CDH1 in an active state to inhibit anaphase progression. Dev Cell. 2009;17(4):541–551.
- Musacchio A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol. 2015;25(20):R1002–R1018.
- Manic G, Corradi F, Sistigu A, et al. Molecular regulation of the spindle assembly checkpoint by kinases and phosphatases. Int Rev Cell Mol Biol. 2017;328:105–161.
- Rieder CL, Schultz A, Cole R, et al. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J Cell Biol. 1994;127(5):1301–1310.
- Rieder CL, Cole RW, Khodjakov A, et al. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol. 1995;130(4):941–948.
- Tanaka TU, Rachidi N, Janke C, et al. Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore-spindle pole connections. Cell. 2002;108(3):317–329.
- Pinsky BA, Kung C, Shokat KM, et al. The Ipl1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat Cell Biol. 2006;8(1):78–83.
- Indjeian VB, Stern BM, Murray AW. The centromeric protein Sgo1 is required to sense lack of tension on mitotic chromosomes. Science. 2005;307(5706):130–133.
- Marston AL. Shugoshins: tension-sensitive pericentromeric adaptors safeguarding chromosome segregation. Mol Cell Biol. 2015;35(4):634–648.
- D’Aquino KE, Monje-Casas F, Paulson J, et al. The protein kinase Kin4 inhibits exit from mitosis in response to spindle position defects. Mol Cell. 2005;19(2):223–234.
- Pereira G, Schiebel E. Kin4 kinase delays mitotic exit in response to spindle alignment defects. Mol Cell. 2005;19(2):209–221.
- Maekawa H, Priest C, Lechner J, et al. The yeast centrosome translates the positional information of the anaphase spindle into a cell cycle signal. J Cell Biol. 2007;179(3):423–436.
- Caydasi AK, Pereira G. Spindle alignment regulates the dynamic association of checkpoint proteins with yeast spindle pole bodies. Dev Cell. 2009;16(1):146–156.
- Gryaznova Y, Koca CA, Malengo G, et al. A FRET-based study reveals site-specific regulation of spindle position checkpoint proteins at yeast centrosomes. Elife. 2016;5:e14029.
- Rawal CC, Riccardo S, Pesenti C, et al. Reduced kinase activity of polo kinase Cdc5 affects chromosome stability and DNA damage response in Scerevisiae. Cell Cycle. 2016;15(21):2906–2919.
- Botchkarev VV Jr., Garabedian MV, Lemos B, et al. The budding yeast Polo-like kinase localizes to distinct populations at centrosomes during mitosis. Mol Biol Cell. 2017;28(8):1011–1020.
- Botchkarev VV Jr., Haber JE. Functions and regulation of the Polo-like kinase Cdc5 in the absence and presence of DNA damage. Curr Genet. 2018;64(1):87–96.
- Reiser V, D’Aquino KE, Ee LS, et al. The stress-activated mitogen-activated protein kinase signaling cascade promotes exit from mitosis. Mol Biol Cell. 2006;17(7):3136–3146.
- Tognetti S, Jimenez J, Vigano M, et al. Hog1 activation delays mitotic exit via phosphorylation of Net1. Proc Natl Acad Sci U S A. 2020;117(16):8924–8933.
- Cha H, Wang X, Li H, et al. A functional role for p38 MAPK in modulating mitotic transit in the absence of stress. J Biol Chem. 2007;282(31):22984–22992.
- Lee K, Kenny AE, Rieder CL. P38 mitogen-activated protein kinase activity is required during mitosis for timely satisfaction of the mitotic checkpoint but not for the fidelity of chromosome segregation. Mol Biol Cell. 2010;21(13):2150–2160.
- Kukkonen-Macchi A, Sicora O, Kaczynska K, et al. Loss of p38gamma MAPK induces pleiotropic mitotic defects and massive cell death. J Cell Sci. 2011;124(2):216–227.
- Nadal-Ribelles M, Sole C, Xu Z, et al. Control of Cdc28 CDK1 by a stress-induced lncRNA. Mol Cell. 2014;53(4):549–561.
- Sole C, Nadal-Ribelles M, de Nadal E, et al. A novel role for lncRNAs in cell cycle control during stress adaptation. Curr Genet. 2014;61:299–308.