
ABSTRACT
Basal-like triple-negative breast cancers frequently express high levels of c-Myc. This oncoprotein signals to the core cell cycle machinery by impinging on cyclin E. High levels of E-type cyclins (E1 and E2) are often seen in human triple-negative breast tumors. In the current study, we examined the requirement for E-type cyclins in the c-Myc-driven mouse model of breast cancer (MMTV-c-Myc mice). To do so, we crossed cyclin E1- (E1-/-) and E2- (E2-/-) deficient mice with MMTV-c-Myc animals, and observed the resulting cyclin E1-/-/MMTV-c-Myc and cyclin E2-/-/MMTV-c-Myc females for breast cancer incidence. We found that mice lacking cyclins E1 or E2 developed breast cancers like their cyclin Ewild-type counterparts. In contrast, further reduction of the dosage of E-cyclins in cyclin E1-/-E2+/-/MMTV-c-Myc and cyclin E1+/-E2-/-/MMTV-c-Myc animals significantly decreased the incidence of mammary carcinomas, revealing arole for E-cyclins in tumor initiation. We also observed that depletion of E-cyclins in human triple-negative breast cancer cell lines halted cell cycle progression, indicating that E-cyclins are essential for tumor cell proliferation. In contrast, we found that the catalytic partner of E-cyclins, the cyclin-dependent kinase 2 (CDK2), is dispensable for the proliferation of these cells. These results indicate that E-cyclins, but not CDK2, play essential and rate-limiting roles in driving the proliferation of c-Myc overexpressing breast cancer cells.
KEYWORDS:
Introduction
The locus encoding the transcription factor c-Myc represents one of the most frequently amplified genes across all human cancer types [Citation1]. Overexpression of the c-Myc protein and activation of the c-Myc-dependent transcriptional program represents a characteristic feature of basal-like triple-negative breast cancers [Citation2–7]. Among many different oncogenic functions of c-Myc, this protein was shown to drive cell division in part by impinging on E-type cyclins [Citation8–12]. The two E-type cyclins, cyclins E1 and E2 collectively referred to as “cyclin E”, represent the key components of the core cell cycle machinery [Citation13–16]. Upregulation of cyclins E1 and E2 is seen frequently in triple-negative, basal-like mammary carcinomas [Citation17–21]. Targeted overexpression of c-Myc in the mammary epithelium of transgenic mice results in mammary carcinomas [Citation22–24], and these tumors express high levels of E-cyclins [Citation25]. Also, mice engineered to overexpress cyclin E1 in mammary glands develop mammary hyperplasia and are prone to breast cancers [Citation26], further underscoring the importance of the c-Myc → cyclin E axis in breast tumorigenesis.
E-type cyclins are essential during mouse embryonic development but are dispensable in an adult organism [Citation27–29]. We previously demonstrated that cyclin E plays a rate-limiting role during the initiation and progression of liver cancers [Citation27]. We also demonstrated that this function of cyclin E was independent of its ability to activate the cyclin-dependent kinase 2 (CDK2) [Citation27]. In the current study, we extended these observations to the c-Myc-driven mouse breast cancer model as well as human triple-negative breast cancer cell lines expressing high levels of c-Myc.
Materials and methods
Mouse crosses
All mouse experiments were approved by the Dana Farber Cancer Institute Animal Care and Use Committee. Cyclin E1-/- and E2-/- mice were described previously [Citation28]. MMTV-c-Myc mice were purchased from Charles River Laboratories. Mice were intercrossed to generate the following genotypes: cyclin E1-/-/MMTV-c-Myc (n = 13), cyclin E2-/-/MMTV-c-Myc (n = 20), cyclin E1-/-E2+/-/MMTV-c-Myc (n = 17) and cyclin E1+/-E2-/-/MMTV-c-Myc (n = 14). Cyclin E1+/+E2+/+/MMTV-c-Myc littermates (n = 20) served as wild-type controls. Mice were checked bi-weekly, by palpation, for the presence of breast tumors. Upon sacrifice of animals, tumors were excised, measured and analyzed by histology. Tumor volumes were calculated according to the formula V = a x b2/2 (where a is tumor length and b is tumor width)
Cell culture
Human breast cancer CAL-51 cells were purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DMSZ). HCC1806, HCC1143, HCC1395, HCC1937, MDA-MB-157, MDA-MB-231, MDA-MB-436, MDA-MB-468, HCC1954, HCC38, and HCC70 cells were obtained from the American Type Culture Collection (ATCC). All cells were cultured according to the recommendation of the supplier. To inhibit asCDK2, 10 μM 3MB-PP1 (529582, Millipore) was added to the culture medium.
Immunoblotting and co-immunoprecipitation
For immunoblotting and co-immunoprecipitation (IP) experiments, cells were lysed in the lysis buffer (20 mM Tris-HCl pH 8.0, 0.1 M KCl, 5 mM MgCl2, 10% glycerol, 0.1% Tween-20, 0.1% NP40) with protease cocktail inhibitor (4693159001, Millipore). For immunoblotting, cell lysates were resolved on SDS-PAGE and transferred to PVDF membranes. For co-immunoprecipitation experiments, cell lysates were incubated with antibodies overnight. Immunoprecipitates were washed three times with the lysis buffer followed by SDS-PAGE. The following antibodies were used for immunoblotting: c-Myc (sc764, 1:1000, Santa Cruz), cyclin E1 (sc-247, 1:1000; Santa Cruz), cyclin E2 (EP454Y, 1:1,000; Millipore), CDK2 (sc-163, 1:1,000; Santa Cruz), tubulin (AC40, 1:3,000; Sigma), HSP90 (4874, 1:3,000; Cell Signaling), β-actin (A5441, 1:4,000, Sigma), GAPDH (D16H11, 1:1,000; Cell Signaling). For immunoprecipitation, the following antibodies were used: CDK1 (sc-54, Santa Cruz), CDK2 (sc-53,220, Santa Cruz), Flag (A2220, Sigma), IgG (5415S, Cell Signaling).
Viral transduction
The pLKO-puro containing shRNA against cyclin E1 (E1-sh: 5ʹ-CGACATAGAGAACTGTGTCAA-3ʹ), or cyclin E2 (E2-sh: 5ʹ-CCAGACACATACAAACTATTT-3ʹ), and pLentiCrispr-v2puro vector containing single-strand guide RNA against CDK2 (CDK2-sg: 5ʹ-CAGAAACAAGTTGACGGGAG-3ʹ) were used as previously described [Citation27]. 293 FT cells were used for the production of the virus. Briefly, lentiviral plasmids (E1-sh, E2-sh, Con-sh, Lenti-CDK2-sg, Lenti-con) were mixed with the packaging plasmid pCMV-delta8.9 and the envelope plasmid pCMV-VSV-G at the ratio of 10:9:1, and transfected into 293 FT cells using polyfect transfection reagent (301107, Qiagen). Two days later, virus-containing medium was collected, filtered, and used for infection in the presence of 10 μg/ml polybrene. After 24 h of infection, medium was changed and puromycin was added into the medium for 1 to 2 weeks.
Cell cycle analyses
Cells were cultured in a medium containing 75 μM 5-bromo-2'-deoxyuridine (BrdU) for 1 h, collected and fixed overnight in 90% ethanol. Cells were then treated with 2 N HCl containing 0.5% Triton X-100, neutralized in borate buffer pH 8.5, incubated with an anti-BrdU antibody (556028, BD Biosciences), 5 μg/ml propidium iodide and 200 μg/ml RNase A for 30 min, and analyzed by FACS. Annexin V staining was performed using FITC Annexin V Apoptosis Detection Kit (BD Pharmingen), following the manufacturer’s protocol.
Growth curves
Cells were seeded in 12 well plates (2.5 × 104 cells per well, in triplicate) and cultured in appropriate medium. Cells were harvested at the indicated time-points, fixed in 10% buffered formalin, followed by crystal violet staining. 10% acetic acid was then used to dissolve the staining and the OD was measured at 590 nm. For assays shown in ) and (d), cells were plated in 12 well plates (2.5 × 104 per well), stained with crystal violet after culturing for 7 days and 9 days, respectively, and photographed.
Generation of cells expressing asCDK2
Mouse analog-sensitive (as) Cdk2 was constructed by cloning mouse Cdk2 cDNAs carrying the F80→G mutation [Citation27] plus a C-terminal Flag-tag into pLenti-Neo vector (17392, Addgene). To generate HCC1806 cells expressing asCdk2, cells were transduced with lentiviruses expressing asCdk2 and selected with neomycin (400 μg/ml) for 2 weeks. After that, the endogenous CDK2 was knocked out using CDK2-sg CRISPR virus followed by puromycin selection (2 μg/ml).
In vitro kinase assays
Analog-sensitive CDK2 was immunoprecipitated from CDK2-knockout HCC1806 cells engineered to ectopically express Flag-tagged asCDK2. For control, immunoprecipitation with IgG was used. Kinase assays were performed in the final volume of 30 μl of the kinase buffer (50 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM DTT, 1 mM EGTA, 0.1 mM NaF), containing 10 μM ATP and 0.4 mCi [32P] γATP (Perkin Elmer) and 1 μg of recombinant histone H1 as a substrate, in the presence or absence of 10 μM 3MB-PP1. After 30 min incubation at 30°C, proteins were denatured, resolved on SDS-PAGE, transferred to nitrocellulose membranes and exposed to X-ray films.
Results
Analyses of the requirement for cyclin E in c-Myc-driven murine mammary carcinomas
To test the requirement for cyclin E in breast tumorigenesis in vivo, we took advantage of the MMTV-c-Myc transgenic mouse strain. Female mice of this strain develop mammary carcinomas due to overexpression of the c-Myc oncoprotein, which is targeted to mammary epithelium by the mouse mammary tumor virus (MMTV) promoter [Citation22–24]. We first crossed MMTV-c-Myc mice with mice lacking cyclin E1 (cyclin E1-/-) [Citation28] or cyclin E2 (cyclin E2-/-) [Citation28] and observed the resulting cyclin E1-/-/MMTV-c-Myc and cyclin E2-/-/MMTV-c-Myc females for tumor incidence. We found that cyclin E1- and E2-deficient mice developed mammary carcinomas with very similar kinetics as control cyclin E1+/+/E2+/+/MMTV-c-Myc animals (,c)). We concluded that neither cyclin E1 nor E2 is essential for the initiation of breast tumorigenesis in this experimental system.
Figure 1. Analyses of breast cancer susceptibility in cyclin E-deficient mice. (a) Comparison of breast cancer incidence between control cyclin E1+/+E2+/+/MMTV-c-Myc (WT), cyclin E1-/-/MMTV-c-Myc and cyclin E2-/-/MMTV-c-Myc females. Mice were monitored by palpation for the presence of breast tumors. (b) A similar analysis using cyclin E1+/+E2+/+/MMTV-c-Myc (WT), cyclin E1-/-E2+/-/MMTV-c-Myc and cyclin E1+/-/E2-/-/MMTV-c-Myc females (the same input for WT mice was used to generate panels a and b). ANOVA test for multiple comparisons (Friedman test) was used for calculate the p-values. ***, p < 0.001; in panel (a) differences were not significant. (c and d) The estimated total tumor burden (per mouse) in tumor-bearing animals of the indicated genotypes. Each dot corresponds to a different mouse and represents the combined tumor volume (the same input for WT mice was used to generate both panels). Tumor volumes were calculated according to the formula V = a x b2/2 (a is tumor length and b is tumor width). Horizontal lines depict mean values. There were no significant differences between the groups (Kruskal-Wallis test with Dunnett’s multiple comparison test)
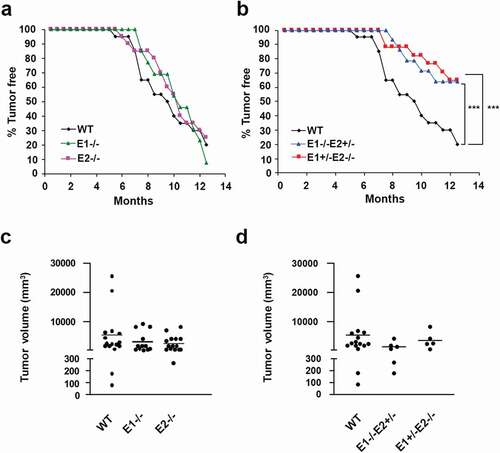
We previously observed that cyclins E1 and E2 play overlapping functions in normal mouse development [Citation28]. Therefore, we hypothesized that the same may be true during c-Myc-driven mammary tumorigenesis. To test this, we interbred MMTV-c-Myc, cyclin E1-/- and E2-/- mice. Since mice entirely lacking E-cyclins (E1-/-E2-/-) die in utero [Citation28,Citation29], we generated animals expressing only one single allele of cyclin E, namely cyclin E1-/-E2+/-/MMTV-Myc and E1+/-E2-/-/MMTV-Myc. We previously demonstrated that female mice with a reduced dosage of cyclin E genes develop normally and are viable [Citation28]. Analysis of tumor incidence revealed that the reduction of cyclin E copy number significantly diminished the incidence of breast cancers. Thus, by 13 months of age, 80% of control cyclin E1+/+E2+/+/MMTV-c-Myc mice developed mammary carcinomas, compared to only 35% in the cyclin E1-/-E2+/-/MMTV-Myc and cyclin E1+/-E2-/-/MMTV-Myc groups (,d)). These observations indicate that E-type cyclins play a rate-limiting role in the initiation of c-Myc-driven breast tumorigenesis in vivo in the MMTV-c-Myc mouse model.
Analyses of the requirement for cyclin E in human TNBC cell lines
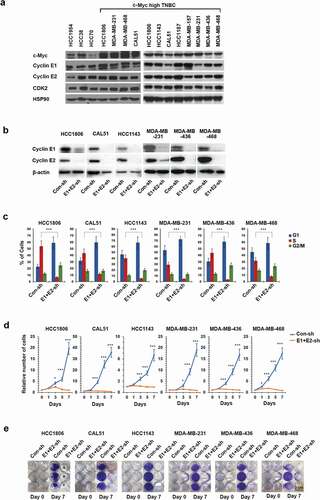
To extend these analyses to human breast cancers, we chose several human triple-negative breast cancer (TNBC) cell lines expressing relatively high levels of c-Myc ()). We verified that these cell lines also express high levels the E-cyclins ()). We next depleted both E-cyclins using previously validated anti-cyclin E1 and -E2 shRNAs [Citation27] () and Supplemental Figure S1), and examined the effect on cell cycle progression. Specifically, we pulsed cells with BrdU, stained with an anti-BrdU antibody and propidium iodide, and analyzed by flow cytometry. We observed that depletion of E-cyclins resulted in an accumulation of cells in the G1 phase of the cell cycle and a strong reduction of the fraction of cells undergoing DNA synthesis () and Supplemental Figure S2). Consistent with these findings, we determined that the depletion of E-cyclins virtually extinguished tumor cell proliferation (,e)). These results indicate that E-cyclins play an essential role in driving cell cycle progression of all six TNBC cell lines analyzed.
Figure 2. Cyclins E1 and E2 are required for proliferation of triple-negative breast cancer (TNBC) cell lines with high c-Myc expression. (a) Immunoblot analysis of c-Myc, cyclin E1, E2 and CDK2 levels in the indicated human breast cancer cell lines. HSP90 was used as a loading control. (b) Immunoblot analysis of cyclin E1 and E2 levels in the indicated six TNBC cell lines following transduction with viruses encoding anti-cyclin E1 and E2 shRNAs (E1+ E2-sh), or control shRNA (Con-sh). β-actin was used as a loading control. (c) The percentage of cells in the indicated phases of the cell cycle following depletion of E-cyclins as in panel b. Cells were pulsed with BrdU, stained with an anti-BrdU antibody and propidium iodide and analyzed by FACS. Shown are mean values, error bars indicate SD, n = 3. Unpaired two-tailed Student’s t test was used to evaluate the differences between S-phase fractions in Con-sh versus E1+ E2-sh groups. ***, p < 0.001. (d) Growth curves of the indicated human TNBC cell lines following depletion of E-cyclins as in panel b. Shown are relative numbers of cells on the indicated days; the number of plated cells (day 0) was set as 1. Error bars indicate SD, n = 3. Unpaired two-tailed Student’s t test was used for the statistical analysis of the differences between Con-sh versus E1+ E2-sh groups at the indicated time-points. *, p < 0.05; ***, p < 0.001. (e) The indicated TNBC cell lines were plated at 2.5 × 104 cells per well in 12 well plates following depletion of cyclins E1 and E2, as in panel b. Cells were stained and photographed after 7 days. Scale bar, 2 cm
Analyses of the requirement for CDK2 in human TNBC cell lines
The best-documented function of E-cyclins is the ability to activate their catalytic partner, CDK2 [Citation13]. Therefore, we verified the interaction of cyclin E1 with CDK2 in two TNBC cell lines (CAL51 and HCC1806). Specifically, we immunodepleted CDK2 using an anti-CDK2 antibody, and probed the supernatants for the presence of cyclin E1. We observed that immunodepletion of CDK2 removed essentially all cyclin E1 pool, indicating that in these cells cyclin E1 stoichiometrically binds to CDK2 ()). In contrast, very little cyclin E1 interacted in these cells with a related cell cycle kinase, CDK1 ()).
Figure 3. CDK2 is dispensable for proliferation of TNBC cell lines. (a) Immunoprecipitation (IP) using anti-CDK2 (left) or anti-CDK1 (right) antibodies, followed by immunoblotting with an anti-cyclin E1 antibody in CAL51 and HCC1806 cells. Note that immunoprecipitated CDK2 brought down large amounts of cyclin E1, and that essentially no cyclin E1 protein was left in the supernatants after CDK2 IP. In contrast, very little cyclin E1 was detected after CDK1 IP, and cyclin E1 remained in the supernatant. Whole cell extracts (WCE) were also immunoblotted. (b) Immunoblot analysis of CDK2 levels in the indicated human TNBC cell lines after CRISPR-mediated knockout of CDK2 (CDK2-sg), compared with cells transduced with control viruses (Lenti-con). Tubulin was used as a loading control. (c) Growth curves of the indicated human TNBC cell lines following knockout of CDK2 (CDK2-sg) compared with cells transduced with control viruses (Lenti-con). Shown are relative numbers of cells on the indicated days; the number of plated cells (day 0) was set as 1. Error bars indicate SD, n = 3. (d) The indicated TNBC cell lines were plated at 2.5 × 104 cells per well in 12 well plates following knockout of CDK2, as in panel b. Cells were stained and photographed after 9 days. Scale bar, 2 cm
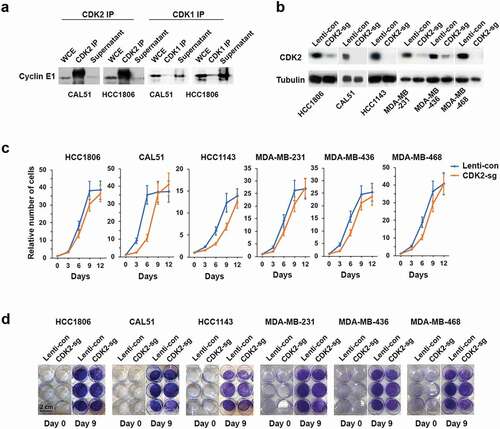
To test the requirement for CDK2 in the proliferation of TNBC cells, we used CRISPR/Cas9 to knock out CDK2 in six TNBC cell lines ()). In contrast to the depletion of E-cyclins, which arrested cell growth (,e)), knockout of CDK2 had only a very modest effect on cell proliferation (,d)). Collectively, these observations indicate that E-cyclins, but not CDK2, are essential for TNBC cell proliferation.
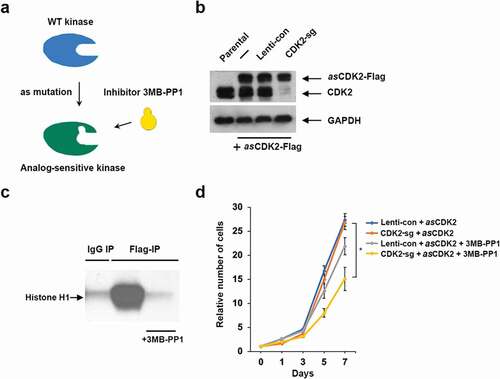
To further probe the requirement for CDK2 in driving the proliferation of TNBC cells, we knocked out CDK2 in a TNBC cell line HCC1806 and ectopically expressed the analog-sensitive (as) version of CDK2, which we have previously engineered and characterized [Citation27] (,b)). Analog-sensitive kinases contain a single amino acid substitution within the kinase ATP binding pocket, which renders this kinase inhibitable by as-specific inhibitors, such as 3MB-PP1 [Citation30,Citation31] ()). Indeed, we verified that the addition of 3MB-PP1 strongly inhibited the kinase activity of asCDK2 ()). We next cultured cells expressing asCDK2 in the presence of 3MB-PP1, and evaluated the effect on cell growth. As it was the case upon CDK2 ablation, inhibition of CDK2 kinase had only a modest effect on cell proliferation ()). These observations further support our conclusion that CDK2 is not required for the growth of TNBC cells.
Figure 4. Analyses of TNBC cells expressing analog-sensitive CDK2. (a) A diagram illustrating the principle of analog-sensitive kinases. (b) Immunoblot analysis of the levels of ectopically expressed analog-sensitive CDK2 (asCDK2-Flag) and the endogenous CDK2 (CDK2) in parental HCC1806 cells (first lane) or cells expressing asCDK2 (lanes 2–4). In lane 3, cells were transduced with a control lentivirus (Lenti-con), in lane 4 cells underwent CRISPR-mediated knockout of CDK2 (CDK2-sg). GAPDH was used as loading control. (c) asCDK2 was immunoprecipitated from CDK2-knockout HCC1806 cells expressing asCDK2 using an anti-Flag antibody, and subjected to in vitro kinase reactions using histone H1 as a substrate in the presence or absence of 3MB-PP1. As a negative control, in vitro kinase reaction was performed using immunoprecipitates obtained with IgG (IgG IP). (d) Growth curves of HCC1806 cells transfected with a control lentivirus and expressing asCDK2 in addition to the endogenous CDK2 (Lenti-con + asCDK2), or CDK2 knockout cells expressing asCDK2 (CDK2-sg + asCDK2). Cells were cultured in the presence or absence of 10 μM 3MB-PP1 (+3MB-PP1). Shown are relative numbers of cells on the indicated days; the number of plated cells (day 0) was set as 1. Error bars indicate SD, n = 3. Unpaired two-tailed Student’s t test was used for the statistical analysis, comparing CDK2-sg + asCDK2 versus CDK2-sg + asCDK2 + 3MB-PP1; *, p < 0.05
Discussion
Our earlier work revealed that E-cyclins seem to be dispensable for the physiology of adult mice, as an acute, global shutdown of cyclin E did not have any obvious adverse consequences [Citation27]. In the current study, we have shown that cyclin E is essential for the initiation of mammary carcinomas in mouse MMTV-c-myc model. We observed that mice expressing only one remaining cyclin E allele (E1-/-E2+/-, and E1+/-E2-/-) displayed reduced breast cancer incidence.
Our analyses of tumor-bearing mice indicated that the total tumor burden per mouse was not different between cyclin E-deficient versus the control group (,d)). This might indicate that once a tumor has developed, cyclin E is not required for breast cancer progression. However, a more likely scenario is that there is a differential threshold of cyclin E activity required for tumor initiation versus progression. Stated differently, we hypothesize that reduced levels of cyclin E in cyclin E1-/-E2+/- and E1+/-E2-/- mice become rate-limiting for tumor initiation, but they are sufficient to maintain tumor cell proliferation. This hypothesis can be tested using conditional cyclin E knockout mice, which will allow a complete shutdown of cyclin E in already developed tumors. We predict that the ablation of cyclin E in tumors in vivo will arrest tumor cell proliferation. Our analyses of human breast cancer cell lines are consistent with this notion. Specifically, we observed that cyclin E is essential for driving tumor cell proliferation, as depletion of cyclin E arrested the cell cycle.
Amplification of the cyclin E gene and overexpression of cyclin E protein have been documented in a large number of tumor types [Citation13,Citation32]. Cyclin E is frequently overexpressed in breast cancers, in particular in triple-negative subtype (TNBC), where it was shown to confer poor prognosis [Citation33,Citation34]. Several different mechanisms were shown to result in cyclin E overexpression in human tumors, including gene amplification as well as the loss of FBXW7, an F-box protein that is responsible for cyclin E degradation [Citation13]. Moreover, some breast cancer cell lines and tumors were shown to express truncated, proteolytically cleaved cyclin E species. These truncated cyclin E molecules bind CDK2 with higher affinity and are resistant to inhibition by KIP/CIP cell cycle inhibitors [Citation35,Citation36].
It is not entirely clear how overexpressed cyclin E contributes to tumorigenesis. Since in normal cells cyclin E regulates cell division by phosphorylating several cell cycle proteins, overexpressed cyclin E likely drives uncontrolled cell proliferation. An additional mechanism was provided by the observations that high levels of cyclin E in tumor cells lead to chromosomal instability [Citation37,Citation38]. Moreover, the hyperactive, truncated cyclin E species were detected in tumor cell cytoplasm (while full-length cyclin E is nuclear) [Citation39], suggesting that cyclin E might regulate additional cytoplasmic targets. Lastly, depletion of cyclin E in cyclin E1-amplified ovarian cancer cell lines was shown to trigger apoptosis, suggesting that cyclin E may play a pro-survival role in these cells [Citation40].
It should be noted that while the role of cyclin E in tumorigenesis is very well established, the requirement for CDK2 in this process remains unclear. In a mouse breast cancer model driven by truncated cyclin E, Cdk2 was shown to play a rate-limiting role, as genetic ablation of Cdk2 blocked tumorigenesis [Citation41]. Analyses of mouse liver cancers revealed that Cdk2 is required for tumor initiation, but it is dispensable for tumor progression and maintenance [Citation42]. Also, analyses of several human cancer cell lines revealed that CDK2 is dispensable for their proliferation [Citation43]. On the other hand, CDK2 was shown to play an essential role in the context of cyclin E1 amplified ovarian cancer cell lines as well as in melanoma cells [Citation44,Citation45].
Our analyses of TNBC cell lines revealed that while depletion of cyclin E blocked cell cycle progression, knockout of cyclin E catalytic partner, CDK2, has very little effect on cell proliferation. This observation might be hard to reconcile with the current cell cycle model indicating that cyclin E forms a stoichiometric complex with, and serves as a regulatory subunit of CDK2.
One possible explanation might be provided by the observation that in addition to CDK2, cyclin E can bind and activate a related kinase CDK1 [Citation46]. Consistent with this notion, Aleem et al. demonstrated that in cells of Cdk2-/- mice, cyclin E interacts with Cdk1 [Citation47]. Hence, the normal proliferation of CDK2-knockout TNBC cells could be explained by the compensatory formation of catalytically active cyclin E-CDK1 complexes in CDK2-knockout cells. However, our study argues against this possibility. First, we found that in TNBC cells under normal conditions (when CDK2 is present), cyclin E predominantly binds CDK2, not CDK1. Second, we engineered TNBC cells to express analog-sensitive CDK2 in place of wild-type CDK2. This scenario should not leave any room for compensation from CDK1, as cyclin E is bound to analog-sensitive CDK2 forming active complexes. Yet, we observed that inhibition of analog-sensitive CDK2 had only a very minor effect on cell proliferation. One possible caveat is that there might be a small pool of cyclin E bound to CDK1 (or to another kinase) in TNBC cells, and this pool might be sufficient to drive tumor cell proliferation even when the predominant partner of cyclin E, CDK2, has been inhibited.
An alternative explanation is that cyclin E might play a CDK2-independent, kinase-independent role in driving the proliferation of tumor cells. There are several observations to support this possibility. Thus, it was shown that cyclin E mutants that are unable to activate CDK2 can transform rat embryonic fibroblasts in collaboration with Ha-Ras [Citation48]. Another study reported that a fraction of cyclin E localizes to centrosomes and promotes S phase entry in a CDK2-independent fashion. Ectopic expression of cyclin E mutant deficient in activating CDK2 was able to accelerate S phase entry, as long as the centrosomal localization sequence was present in the mutant [Citation49]. Moreover, cyclin E was shown to promote MCM loading in cells exiting quiescence in a kinase-independent fashion [Citation50]. Our recent study of human liver cancer cell lines revealed that cyclin E-associated kinase is not required for tumor cell proliferation [Citation27]. Lastly, it was reported that in human cancer cell lines, the vast majority of cyclin E protein is present in catalytically inactive complexes [Citation51]. These observations raise a possibility that cyclin E might play a kinase-independent role in tumorigenesis. However, the molecular basis of such possible function remains unknown.
Regardless of these unresolved insights, our study indicates that targeting cyclin E expression might represent an attractive therapeutic approach in TNBC. We predict that the so-called degrader compounds [Citation52], which would selectively deplete cyclins E1 and E2, might be particularly efficacious, as they would extinguish both kinase-dependent and -independent functions of cyclin E, thereby assuring a therapeutic effect. The apparent lack of requirement for cyclin E in postnatal physiology [Citation27] raises a possibility that such degrader compounds might selectively target cyclin E-dependent cancer cells while sparing normal, non-transformed tissues.
Supplemental Material
Download Zip (2.2 MB)Disclosure statement
P.S. has been a consultant at Novartis, Genovis, Guidepoint, The Planning Shop, ORIC Pharmaceuticals, Syros and Exo Therapeutics; his laboratory receives research funding from Novartis.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905.
- Alles MC, Gardiner-Garden M, Nott DJ, et al. Meta-analysis and gene set enrichment relative to er status reveal elevated activity of MYC and E2F in the “basal” breast cancer subgroup. PLoS One. 2009;4(3):e4710.
- Chandriani S, Frengen E, Cowling VH, et al. A core MYC gene expression signature is prominent in basal-like breast cancer but only partially overlaps the core serum response. PLoS One. 2009;4(8):e6693.
- Gatza ML, Lucas JE, Barry WT, et al. A pathway-based classification of human breast cancer. Proc Natl Acad Sci U S A. 2010;107(15):6994–6999.
- Green AR, Aleskandarany MA, Agarwal D, et al. MYC functions are specific in biological subtypes of breast cancer and confers resistance to endocrine therapy in luminal tumours. Br J Cancer. 2016;114(8):917–928.
- Horiuchi D, Kusdra L, Huskey NE, et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med. 2012;209(4):679–696.
- Xu J, Chen Y, Olopade OI. MYC and breast cancer. Genes Cancer. 2010;1(6):629–640.
- Amati B, Alevizopoulos K, Vlach J. Myc and the cell cycle. Front Biosci. 1998;3:d250–68.
- Jansen-Durr P, Meichle A, Steiner P, et al. Differential modulation of cyclin gene expression by MYC. Proc Natl Acad Sci U S A. 1993;90(8):3685–3689.
- Leone G, DeGregori J, Sears R, et al. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature. 1997;387(6631):422–426.
- Perez-Roger I, Solomon DL, Sewing A, et al. Myc activation of cyclin E/Cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27Kip1 binding to newly formed complexes. Oncogene. 1997;14(20):2373–2381.
- Santoni-Rugiu E, Falck J, Mailand N, et al. Involvement of Myc activity in a G(1)/S-promoting mechanism parallel to the pRb/E2F pathway. Mol Cell Biol. 2000 May;20(10):p3497–509.
- Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24(17):2776–2786.
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–166.
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18(22):2699–2711.
- Geng Y, Whoriskey W, Park MY, et al. Rescue of cyclin D1 deficiency by knockin cyclin E. Cell. 1999;97(6):767–777.
- Chen X, Low K-H, Alexander A, et al. Cyclin E overexpression sensitizes triple-negative breast cancer to wee1 kinase inhibition. Clin Cancer Res. 2018;24(24):6594–6610.
- Gray-Bablin J, Zalvide J, Fox MP, et al. Cyclin E, a redundant cyclin in breast cancer. Proc Natl Acad Sci U S A. 1996;93(26):15215–15220.
- Keyomarsi K, Pardee AB. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc Natl Acad Sci U S A. 1993;90(3):1112–1116.
- Nie L, Wei Y, Zhang F, et al. CDK2-mediated site-specific phosphorylation of EZH2 drives and maintains triple-negative breast cancer. Nat Commun. 2019;10(1):5114.
- Zhao Z-M, Yost SE, Hutchinson KE, et al. CCNE1 amplification is associated with poor prognosis in patients with triple negative breast cancer. BMC Cancer. 2019;19(1):96.
- Sinn E, Muller W, Pattengale P, et al. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell. 1987;49(4):465–475.
- Stewart TA, Pattengale PK, Leder P. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell. 1984;38(3):627–637.
- Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411(6841):1017–1021.
- Geng Y, Yu Q, Whoriskey W, et al. Expression of cyclins E1 and E2 during mouse development and in neoplasia. Proc Natl Acad Sci U S A. 2001;98(23):13138–13143.
- Bortner DM, Rosenberg MP. Induction of mammary gland hyperplasia and carcinomas in transgenic mice expressing human cyclin E. Mol Cell Biol. 1997;17(1):453–459.
- Geng Y, Michowski W, Chick JM, et al. Kinase-independent function of E-type cyclins in liver cancer. Proc Natl Acad Sci U S A. 2018;115(5):1015–1020.
- Geng Y, Yu Q, Sicinska E, et al. Cyclin E ablation in the mouse. Cell. 2003;114(4):431–443.
- Parisi T, et al. Cyclins E1 and E2 are required for endoreplication in placental trophoblast giant cells. Embo J. 2003;22(18):4794–4803.
- Bishop AC, Shah K, Liu Y, et al. Design of allele-specific inhibitors to probe protein kinase signaling. Curr Biol. 1998;8(5):257–266.
- Bishop AC, Ubersax JA, Petsch DT, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407(6802):395–401.
- Schraml P, Bucher C, Bissig H, et al. Cyclin E overexpression and amplification in human tumours. J Pathol. 2003;200(3):375–383.
- Keyomarsi K, Tucker SL, Buchholz TA, et al. Cyclin E and survival in patients with breast cancer. N Engl J Med. 2002;347(20):1566–1575.
- Zhao Z-M, Yost SE, Hutchinson KE, et al. CCNE1 amplification is associated with poor prognosis in patients with triple-negative breast cancer. BMC Cancer. 2019;9(1):96.
- Porter DC, Zhang N, Danes C, et al. Tumor-specific proteolytic processing of cyclin E generates hyperactive lower-molecular-weight forms. Mol Cell Biol. 2001;21(18):6254–6269.
- Wingate H, Zhang N, McGarhen MJ, et al. The tumour-specific hyperactive forms of cyclin E are resistant to inhibition by p21 and p27. J Biol Chem. 2005;280(15):15148–15157.
- Spruck CH, Won K-A, Reed SI, et al. Deregulated cyclin E induces chromosome instability. Nature. 1999;401(6750):297–300.
- Teixeira LK, Wang X, Li Y, et al. Cyclin E deregulation promotes loss of specific genomic regions. Curr Biol. 2015;25(10):1327–1333.
- Hunt KK, Karakas C, Ha MJ, et al. Cytoplasmic cyclin E predicts recurrence in patients with breast cancer. Clin Cancer Res. 2017;23(12):2991–3002.
- Etemadmoghadam D, George J, Cowin PA. Amplicon-dependent CCNE1 expression is critical for clonogenic survival after cisplatin treatment and is correlated with 20q11 gain in ovarian cancer. PLoS One. 2010;5(11):e15498.
- Akli S, et al. Cdk2 is required for breast cancer mediated by the low-molecular-weight isoform of cyclin E. Cancer Res. 2011;71(9):3377–3386.
- Sonntag R, Giebeler N, Nevzorova YA, et al. Cyclin E1 and cyclin-dependent kinase 2 are critical for initiation, but not for progression of hepatocellular carcinoma. Proc Natl Acad Sci USA. 2018;115(37):9282–9287.
- Tetsu O, McCormick F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell. 2003;3:233–245.
- Au-Yeung G, Lang F, Azar WJ, et al. Selective targeting of cyclin E1-amplified high-grade serous ovarian cancer by cyclin-dependent kinase 2 and AKT inhibition. Clin Cancer Res. 2017;23(7):1862–1874.
- Du, Du J, Widlund HR, et al. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell. 2004;6:565–576.
- Koff A, Cross F, Fisher A, et al. Human cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell. 1991;66(6):1217–1228.
- Aleem E, Hirokawa H, Kaldis P. Cdc2-cyclin E complexes regulate the G1/S phase transition. Nat Cell Biol. 2005;7(8):831–836.
- Geisen C, Moroy T. The oncogenic activity of cyclin E is not confined to Cdk2 activation alone but relies on several other, distinct functions of the protein. J Biol Chem. 2002;277(42):39909–39918.
- Matsumoto Y, Maller JL. A centrosomal localization signal in cyclin E required for Cdk2-independent S phase entry. Science. 2004;306:885–888.
- Geng Y, et al. Kinase-independent function of cyclin E. Mol Cell. 2007;25(1):127–139.
- Sweeney KJ, Swarbrick A, Sutherland RL, et al. Lack of relationship between CDK activity and G1 cyclin expression in breast cancer cells. Oncogene. 1998;16(22):2865–2878.
- Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2016;16:101–114.