
ABSTRACT
Human skin melanoma is one of the most aggressive and difficult to treat human malignancies, with an increasing incidence over the years. While the resection of the early diagnosed primary tumor remains the best clinical approach, advanced/metastatic melanoma still remains with a poor prognosis. Indeed, although enormous progress in the therapeutic treatment of human tumors has been made in recent years, patients affected by metastatic melanoma are still poorly affected by these clinical advances. Therefore, new valuable therapeutic approaches are urgently needed, to design and define effective treatments to consistently increase the overall survival rate of patients affected by this malignancy. In this review we summarize the main signaling pathways studied to kill human skin melanoma, and introduce the ferroptotic cell death as a new pathway to be explored to eradicate this tumor.
KEYWORDS:
Melanoma
Cutaneous melanoma is the most aggressive form of skin disease, occurring through melanocyte transformation and representing almost 60% of lethal skin tumors. Due to a high mortality rate in the metastatic form and a relatively high incidence among young adults, melanoma has a high socioeconomic impact [Citation1].
During the last 50 years, melanoma incidence is risen by 3–8% per year in the European population. In 1973, melanoma incidence rate, in the United States, was 6.8 per 100,000 and by 2003 to 2007, this rate has grown to 20.1 per 100,000 [Citation2]. Furthermore, the worldwide incidence of melanoma has steadily increased recent years, with an annual incidence that has risen as rapidly as 4–6% in many fair-skinned populations that predominate regions like North America, Northern Europe, Australia, and New Zealand [Citation3]. In 2020, it is estimated that there will be 100,350 new cases of melanoma in the United States and 6,850 deaths from the disease [Citation4].
Unlike other solid tumors, melanoma mostly affects young and middle-aged people, with an incidence increasing linearly after the age of 25 years until the age of 50 years and the median age at the time of diagnosis is 57 years. Melanoma is more common in men overall, approximately 1.5-times more likely than females. However, the different prevalence in both sexes must be analyzed in relation to age: its incidence rate is greater in women than men before 40 years, but after 75 years of age, the incidence is 3-times as high in men versus women [Citation5–7]. Interestingly, there also appears to be a difference in anatomical distribution by gender: melanomas are more likely to arise on the trunk in men and on the lower limbs in woman [Citation8].
It is important to note that uneven growth distribution of melanocytes results in the formation of nevi, benign clusters of melanocytes, while dysplastic nevus, which shows an increased level of cytologic and architectural atypia, is thought to be the candidate precursor for cutaneous melanoma [Citation9]. The radial growth phase (RGP) is the first recognizable malignant stage, where tumor cells do not show high proliferative capacity or metastasis. In the vertical growth phase (VGP), primary lesion, tumor cells are able to infiltrate, as an expanding mass, the dermis, and then into lymphatic and blood vessels leading to systemic dissemination. The progression to the invasive stage is driven by the accumulation of the initiating genetic alterations during the precursor stage. Finally, metastasis represents the most advanced step of tumor progression (metastatic melanoma) [Citation10].
There is a complex interaction between the exogenous, such as ultraviolet radiation (UV), and the endogenous environment, including genetic risk factors. Up to 65% of malignant melanomas are sun-related since the sunlight is a carcinogen and its UV component is the main environmental risk factor for melanoma skin cancer development [Citation11]. Nevertheless, different genetic alterations lead to the formation of melanomas. The Cyclin-Dependent Kinase Inhibitor 2A (CDKN2A) is the first gene associated with melanoma susceptibility [Citation12]. The CDKN2A gene is located in the 9p21 locus and encodes the two tumor suppressor proteins p16INK4A and p14ARF, via differential splicing and alternative reading frames. Both promote cell-cycle arrest in the G1 phase, melanocyte senescence, and apoptosis. This is a high susceptibility oncogene and has been described in 17 melanoma-prone families [Citation13]. Melanocortin 1 Receptor (MC1R) is the most important gene found to play a role in predisposition to sporadic cutaneous melanoma (CM) and it is a key regulator of skin pigmentation. MC1R allelic variants cause altered melanin synthesis associated with high risk to melanoma development [Citation14]. Moreover, different signaling pathways are involved in melanoma initiation, progression, and maintenance. An important role in melanoma development seems to be played by the phosphoinositide-3-OH Kinase (PI(3)K)/Akt pathway. Indeed, PI3K hyperactivation is commonly detected during melanoma development, due to activating mutations, which results in an intracellular increase of the second phosphatidylinositol (3,4,5)-trisphosphate (PIP3) messenger and AKT cytosolic protein activation. This hyperactive pathway promotes cell survival, proliferation, and growth of melanoma cells. Despite several gene mutations have been observed in this malignancy, oncogenic mutations in the Ras/Raf pathway are those most frequently associated with the development of melanoma [Citation15]. The RAS/RAF/MEK/ERK mitogen-activated protein kinase (MAPK) pathway is an intracellular signaling mechanism controlling cell growth, proliferation, and differentiation in response to growth factors, cytokines and hormones [Citation16]. 90% of all melanomas are characterized by activating mutations in n-ras or b-raf genes, with the most frequent mutation represented by the BRAFV600E (88–92% of cases) [Citation17]. Oncogenic BRAF mutations are responsible for the constitutive activation of the signaling pathway, resulting in melanoma proliferation, tissue invasion, evasion of immune response and resistance to apoptosis [Citation18]. However, NRAS/BRAF mutations alone are not sufficient to initiate melanomagenesis, since these mutations have also been found in benign nevi, suggesting that other factors contribute to tumor development [Citation17,Citation19]. Indeed, both autophagy and endoplasmic reticulum stress (ER stress), primarily pro-survival processes, are considered secondary events that contribute to the development of this tumor, also playing a key role in chemoresistance [Citation20]. Several reports indicated that dysregulated autophagy can promote tumor development [Citation21–Citation24], and it is now evident that the process is actively induced in wt BRAF/NRAS melanoma cells treated with pro-apoptotic drugs, resulting in apoptosis resistance and tumor growth [Citation25]. Also, the ER stress has been recognized as a key secondary event driving melanoma development through the sustained activation of the unfolded protein response (UPR) program [Citation22,Citation26,Citation27]. Moreover, oncogenic BRAF induces a chronic ER stress status directly increasing basal cell autophagy, resulting in apoptotic resistance and tumor growth [Citation28], while interfering with the UPR signaling pathway results in increased melanoma cells susceptibility to cell death [Citation23,Citation29].
Killing melanoma cells
In recent years, although enormous progress has been made in the treatment of melanoma, most patients have developed mechanisms of resistance to current therapies, indicating that effective therapy is not yet available. Indeed, early diagnosis and prevention are still the key factors in reducing the impact of the tumor. While surgical resection represents the best treatment of malignant melanoma when detected in the early stages, in the advanced stages, the tumor becomes highly aggressive, metastatic, and significantly limits the survival of patients.
Recently, the development of targeted BRAF/MEK inhibitors and immunotherapy has revolutionized treatments and certainly improved the overall survival of patients with metastatic melanoma.
BRAF and MEK inhibitors
Vemurafenib, a recently developed highly specific BRAF inhibitor, represents the drug that undoubtedly had success compared to nonspecific inhibitors, such as sorafenib. In 2011 the Food and Drug Administration (FDA) approved the treatment of metastatic melanomas with this compound while in 2013, another BRAF inhibitor, dabrafenib, was also approved [Citation30]. Unfortunately, patients quickly (6–8 months) develop resistance which is described as intrinsic or acquired resistance. The intrinsic resistance might be caused by the acquisition of new aberrations in cell cycle-regulating genes (cyclin D1), or loss of PTEN function. Importantly, other mechanisms have been described, such as the interaction of HGF (hepatocyte growth factor) with its receptor C-MET, widely expressed on cell membrane of several malignancies [Citation15]. The molecule produced by cancer cells, tumor stroma, or systemically, provides an autocrine/paracrine survival mechanism [Citation31].
On the other hand, ERK-dependent and -independent mechanisms are mainly responsible for the acquired resistance. The ERK-independent mechanism includes the activation of PI3K/AKT/mTOR pathway by PDGFRβ (platelet-derived growth factor beta) and IGF-1 R (insulin-like growth factor 1 receptor). While the ERK-dependent mechanism involves the hyperactivation of the MAPK pathway.
This is why, in addition to BRAF kinase inhibitors, highly selective MEK1/2 inhibitors, such as trametinib, selumetinib and MEK162, are also used to treat oncogenic BRAF metastatic melanomas [Citation15]. Importantly, combined MEK and BRAF inhibitors are more effective and less toxic than individual treatment and have become the standard of care for patients with BRAF-mutated melanoma [Citation32].
Immune checkpoint inhibitors
In recent years, improved knowledge of the role of the immune system in cancer control have led to the development of several immunotherapies. The immunogenicity of cancer is a process requiring several steps such as i) generation and release of neoantigens by cancer cells and capture by dendritic cells; ii) tumor antigen presentation to T cells; iii) T cell activation, and iv) direct tumor recognition and killing. The process also involves the immune checkpoints that are crucial in controlling immune responses leading to regulatory feedback mechanisms. Unfortunately, many cancers, including melanoma, exploit the process to evade eradication by the immune system [Citation33]. Therefore, immune checkpoints represent a fundamental therapeutic goal to activate/reactivate the immune system toward poorly or non-immunogenic neoplasms.
Major advances in targeting the immune evasion of melanoma have been obtained using immune checkpoint inhibitors such as the cytotoxic T-lymphocyte antigen (CTLA-4) and programmed death 1 (PD-1) [Citation34]. CTLA-4, on T lymphocytes, binds the CD80 and CD86, present on antigen-presenting cells (APCs), inhibiting T cell activation [Citation35,Citation36]. Ipilimumab, a human monoclonal anti-CTLA-4 antibody, was the first molecule to confer a significantly improved survival rate in patients with metastatic melanoma [Citation37]. Unfortunately, 60% of patients treated with anti-CTLA-4 experienced immune-related adverse events, which were severe in 10–15% of cases [Citation38]. Similarly, the binding of PD-1 with its ligands PD-L1 (CD274) and PD-L2 (CD273) delivers an inhibitory signal to T lymphocytes. PD -L1 and -L2 are predominantly localized on APC cells, but their expression is often upregulated even in tumor cells and tumor-infiltrating lymphocytes, leading to inhibition of the T cell response [Citation39]. In 2015 two PD-1 blockers monoclonal antibodies have been approved by EMA: nivolumab and pembrolizumab. Anti-PD-1 was superior to anti-CTLA-4 in the treatment of metastatic melanoma in terms of regression and patients survival, and severe side effects occurred less frequently (13.3%) compared to anti-CTLA-4 (19.9%) [Citation40]. The additive effects of CTLA-4 plus PD-1 blockers have been recently evidenced in a phase I trial [Citation41].
Cancer immunotherapies must, however, be carefully improved, since unrestrained autoimmune inflammatory responses might occur, at the potential cost of damage to normal cells.
Immunogenic Cell Death
Due to the renewed interest in the immune surveillance of cancer, an alternative and interesting new therapeutic approach has been proposed, relying on combined ability of some compounds to both stimulate the apoptotic program of cancer cells and recruitment and activation of immune system, resulting in a better eradication of the malignancy [Citation42,Citation43]. This process known as immunogenic cell death (ICD) relies on the ability of these drugs to stimulate dying cancer cells to produce/release specific signals, namely DAMPs (damage-associated molecular pattern), to efficiently stimulate the recruitment and maturation of dendritic cells (DC), that will in turn activate T lymphocytes to kill residual cancer cells [Citation44]. To date, few DAMPs have been identified [Citation45] showing the ability to be recognized by the immune system, but four of them are strictly required for an efficient induction of ICD: i) the exposure of the ER-resident calreticulin (CRT) on cell surface, (ii) the extracellular release of ATP and (iii) the non-histone chromatin-binding protein high-mobility group box 1 (HMGB1), and (iv) the production and release of type I interferons (IFNα & β) [Citation46]. In order to be effective, ICD must be induced by drugs able to cause cancer cell death and stimulate the release/exposure of DAMPs, without causing any immune cells damage [Citation42]. Only a restricted number of chemotherapeutics show these capabilities, with some of them already considered successful drugs, such as anthracyclines (doxorubicin, epirubicin and idarubicin), already used in the clinic to treat several malignancies; the anthracenedione mitoxantrone (MTX), employed in the treatment of non-Hodgkin’s lymphoma, prostate carcinoma, myeloid leukemia and breast cancer [Citation47]; oxaliplatin, a platinum derivative used in colorectal cancer treatment [Citation48]; cyclophosphamide, an alkylating agent used to treat different neoplasms and autoimmune diseases [Citation49]; the proteasomal inhibitor bortezomib, used to treat multiple myeloma and mantle cell lymphoma [Citation50]. Moreover, specific radiations and photodynamic therapy employed in the treatment of various cancers, few oncolytic viruses, elevated hydrostatic pressure and the microtubular inhibitor patupilone are also able to induce ICD [Citation46].
The activity of pro-ICD drugs on melanoma cells has also been recently explored, indicating that both MTX and DOXO (doxorubicin) were able to both stimulate the cell death program and the production/release of DAMPs [Citation51]. However, the lack of an appropriate mouse model recapitulating the human malignancy limited its further exploitation [Citation52].
Ferroptosis
Characteristics of ferroptotic cell death
In 2013, in the Stockwell laboratory a new compound, erastin, was discovered which had a selectively lethal effect on RAS mutated cancer cells, but the pattern of cell death was different from what had been described before [Citation53]. Subsequently, a RAS-selective lethal small molecule 3 (RSL3) has been identified which was able to cause the same pattern of cell death [Citation54]. Morphologically, this process mainly manifests different patterns from other modes of cell death, since it does not have the morphological characteristics of apoptosis, such as nuclear fragmentation, mitochondrial cytochrome c release, and generation of apoptotic bodies. In fact, cells are usually rounded up and detached in response to erastin; they exhibit changed mitochondrial morphology, which appear smaller with increased membrane densities and reduced cristae; there is no condensation of chromatin but, however, there is an increased level of intracellular reactive oxygen species (ROS), and cell death was completely inhibited by iron chelation or genetic inhibition of cellular iron uptake [Citation55,Citation56]. This type of cell death occurs without the involvement of most apoptotic effectors such as caspases, BAX, and BAK, and is independent of the key necroptosis factors RIPK1/RIPK3 (receptor-interacting protein kinase 1 or 3). Due to the key role played by iron, this peculiar form of cell death was named “ferroptosis”.
The first discovered pro-ferroptotic molecule erastin, inhibits the uptake of cystine, leading to glutathione (GSH) depletion and downstream inhibition of the phospholipid peroxidase glutathione peroxidase 4 (GPX4). The indirect inhibition of GPX4 through erastin, or its direct inactivation through RSL3, thus ultimately results in lipid peroxides accumulation and cell death ()[Citation57].
Figure 1. Ferroptosis. The key players of the ferroptotic cell death process have been highlighted. AKRs = Aldo-keto reductases; Cys = cysteine; FTH1 = Ferritin heavy chain 1; FTL = Ferritin light chain; GSH = Glutathione; GSSG = Oxidized Glutathione; GPX4 = Glutathione peroxidase 4; LOX = Lipoxygenases; PUFAs = Polyunsaturated fatty acids; TFR1 = Transferrin receptor 1; ERA = Erastin; Fer-1 = Ferrostatin-1; MPA = Medroxyprogesterone; RLS3 = Ras-selective lethal small molecule 3; SOR = Sorafenib.
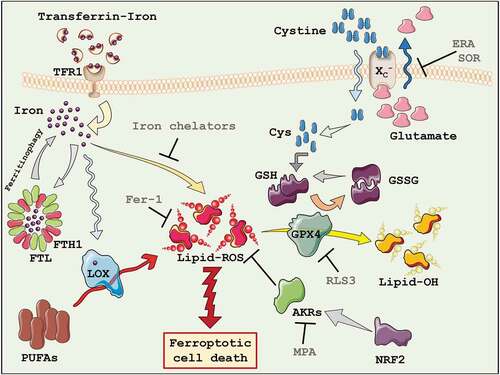
A peculiar genetic profile governs ferroptosis, compared to other forms of cell death. Iron metabolism and lipid peroxidation are an increasingly predictable marker of ferroptosis; indeed, genes regulating iron or mitochondrial fatty acid metabolism are specifically required for the induction and execution of this form of cell death. Particularly, six genes have been identified that regulate the induction of ferroptosis: gene encoding the ribosomal protein L8 (rpl8), required for translation, the iron response element-binding protein 2 (ireb2), a master regulator of iron metabolism, the ATP synthase F0 complex subunit C3 (atp5g3), the tetratricopeptide repeat domain 35 (ttc35), citrate synthase (cs), and acyl-CoA synthetase family member 2 (acsf2). In addition, the expression of the known iron uptake, metabolism, and storage genes are also involved in ferroptosis, such as transferrin receptor (tfrc) iron-sulfur cluster assembly enzyme (iscu), ferritin heavy chain 1 (fth1) and ferritin light chain (ftl) [Citation57].
Iron metabolic pathway
The iron metabolism is increasingly recognized as central mediator of ferroptosis, since iron is necessary for lipid peroxidation. In fact, supplementing the growth medium with bioavailable form of iron, but not other divalent metals, accelerates erastin-induced ferroptosis [Citation57]. Iron overload induces membrane lipid peroxidation through Fenton and Haber–Weiss reactions, in which free divalent iron catalyzes the breakdown of H2O2 to yield hydroxyl radicals and superoxide anions, two highly reactive oxygen species (ROS). Importantly, the iron-dependent accumulation of lipid peroxides (lipid-ROS), during ferroptosis execution, depends not only by inorganic chemical reactions but also by enzymatic reactions, in which iron represents a cofactor [Citation58]. Lipoxygenases (LOX), for example, are a family of non-heme iron-containing enzymes which catalyze the oxidation of polyunsaturated fatty acids (PUFAs). As a result, iron uptake, storage, and export are key contributors to sensitivity to lipid peroxidation and ferroptosis [Citation54]. The extracellular form of ferric iron (Fe3+) circulates bound to transferrin (TF) and is imported into cells through the membrane protein transferrin receptor 1 (TFR1). Then, locates in the endosome where Fe3+ is reduced to ferrous iron (Fe2+) by the ferrireductase STEAP3 (Six-transmembrane epithelial antigen of the prostate 3). Fe2+ can be release from the endosome into the labile iron pool in the cytoplasm by the divalent metal transporter 1 (DMT1). Cytoplasmatic iron can then bind ferritin, a protein complex represented by ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1), to be stored. On the contrary, intracellular iron can be mobilized and exported in the extracellular compartment trough the membrane protein ferroportin (FPN; )[Citation59].
Ferroptosis-sensitive cells with RAS mutations have improved iron uptake (increased TFR1 expression) and reduced iron storage (decreased FTL and FTH1 expression), compared with ferroptosis-resistant cells [Citation60], while iron chelators, such as Deferoxamine (DFO) and Ciclopiroxolamine (CPX), protect cells from ferroptosis. IREB2 encodes a master transcription factor that regulates iron metabolism. Its downregulation significantly increases iron metabolism-associated gene expression (e.g. TFR, FTH1 and FTL) and reduces erastin-stimulated ferroptosis [Citation57]. Notably, the autophagic degradation of ferritin, a process known as ferritinophagy, increases labile iron levels and contributes to ferroptosis. In this context, nuclear receptor coactivator 4 (NCOA4) is a cargo receptor responsible for autophagy-dependent ferritin degradation, likely because the C terminus of NCOA4 binds a conserved surface arginine (R23) on FTH1, driving the cargo to phagophores, and subsequently in autophagolysosomes [Citation61]. NCOA4-dependent ferritinophagy promotes ferroptosis through releasing free iron from ferritin, and it has been demonstrated that its levels are regulated by intracellular iron. Indeed, when intracellular levels of iron are high, NCOA4 can be ubiquitinated by HERC2 (HECT and RLD domain containing E3 ubiquitin protein ligase 2) and degraded by proteasome, suggesting a negative correlation between HERC2 and ferroptosis induction. Moreover, deficient ferritinophagy may increase the activity of IREB2 and subsequently upregulate transcription factors as a feedback mechanism [Citation62]. Recently, Beclin-1 (BECN1) has been discovered to play a non-autophagic role binding SLC7A11 (solute carrier family 7 member 11) and leading to lipid peroxidation-dependent ferroptosis through inhibiting the activity of this antiporter system. Moreover, the stability of the BECN1 mRNA increases the formation of autophagosomes and subsequently ferritinophagy-mediated ferroptosis [Citation63].
Lipid peroxidation metabolism
Lipids containing polyunsaturated fatty acids (PUFAs) with labile bis-allylic hydrogen atoms are most susceptible to lipid peroxidation and are necessary for the normal execution of ferroptosis [Citation64]. There are three known classes of lipid oxidizing enzymes: cyclooxygenases (COXs), cytochrome p450 (CYPs), and LOXs. Among those, LOXs have been demonstrated to have a pivotal role in ferroptosis, catalyzing deoxygenation of PUFAs of lipid membrane [Citation65].
Therefore, the GPX4 inactivation and ROS production contribute to alter lipid-metabolism associated gene, such as acyl-CoA synthetase long-chain family member 4 (acls4) and phosphatidylethanolamine (PE), driving the execution of ferroptosis. ACSL4 plays a key role in fatty acid metabolism, ensuring acetylated lipids to enter the metabolic pathway which represents a source of energy essential for biological processes [Citation66]. Thus, ACSL4 can catalyze the esterification of arachidonic acid (AA) into PE. Then AA-PE and derivatives are oxidized by 15-LOX to generate lipid hydroperoxides, that are proximate ferroptotic executors [Citation67]. In essence, free radicals can trigger membrane lipid peroxidation by taking an electron from PUFAs. Then, the lipid radical reacts with oxygen to produce peroxyl radical and in this form initiates a chain reaction, transforming polyunsaturated fatty acids into lipid hydroperoxides. Finally, lipid-ROS are very unstable molecules disturbing the integrity of cell membranes [Citation68]. In metastatic melanoma, the inhibition of 12/15-LOX but not 5-LOX resulted in a complete abrogation of lipid peroxides production and cell death execution under pro-ferroptotic stimulation [Citation69].
ACSL4 expression is positively correlated with the amount of erastin-induced ferroptosis in cancer cells. A previous study demonstrated that deletion of ACSL4 and LPCAT3 (lysophosphatidylcholine acyltransferase 3) prevents RSL3 induced ferroptosis in the human myeloid leukemia cell line KBM7 cells [Citation70]. Moreover, ACSL4 is overexpressed in several malignancies such as liver, kidney and colorectal cancer [Citation71].
Downstream accumulation of lipid peroxides represents a key step mediating the execution of ferroptosis. Antioxidant agents such as α-tocopherol [Citation72], butylated hydroxytoluene (BHT) [Citation73] and N-acetylcysteine (NAC) [Citation57], can block ferroptosis by inhibiting the lipid peroxidation pathway. These are potent peroxyl radical scavenger with high affinity for unpaired electrons and can break a cascade of chain reactions in the lipid peroxidation of membranes. Moreover, α-tocotrienol is also shown to regulate ferroptosis via LOX inhibition [Citation74]. Also, ferroptosis suppressor protein 1(FSP1) confers protection against ferroptosis elicited by GPX4 deletion and co-operates with GPX4 and glutathione to suppress phospholipid peroxidation and ferroptosis [Citation75,Citation76].
Other molecules such as Ferrostatins (Fer-1) and Liproxstatins (Lip-1) can also revert the ferroptotic cell death induced by erastin and RSL3 [Citation64]. Their activity depends on the aromatic amines, which specifically act as free radical-trapping antioxidants to inhibit lipid peroxidation and autoxidation. Finally, lipoxygenase inhibitors such as Baicalein, Vitamin E (scavenging hydroxyl group radicals ability) and Zileuton can depress LOXs activity to rescue cells from ferroptosis [Citation69,Citation77,Citation78]. Interestingly, dietary ingestion of omega-6 PUFAs can trigger germ-cell ferroptosis in C. elegans and human cancer cells, indicating a metabolic conserved route to trigger this non-apoptotic cell death program [Citation79].
Key regulators of ferroptosis: System XC− and GPX4
Cystine, the oxidized form of cysteine found in the extracellular space, is taken up by cells via the cystine-glutamate antiporter (system XC−). This is a heterodimeric integral cell membrane antiporter exchanging one molecule of cystine with one molecule of glutamate. It comprises the 12-pass transmembrane transporter protein SLC7A11 linked via a disulfide bridge to the single-pass transmembrane regulatory subunit SLC3A2 [Citation80]. Intracellular cystine is rapidly converted to cysteine, which is a precursor for GSH synthesis. The expression of system XC− is regulated by the nuclear factor erythroid 2-related factor 2 (NRF2), which enhances its transcription under oxidative stress [Citation81]. Furthermore, p53 can also modulate the activity of the system XC−, downregulating the expression of SLC7A11, thus resulting in a reduction in cell antioxidant capacity, accumulation of lipid-ROS and ferroptosis induction [Citation82]. Interestingly, impaired cystine intake has been implicated in several central nervous system diseases. Indeed, the import of cystine is inevitably coupled with the release of glutamate, which must be strictly controlled since high extracellular glutamate concentrations is neurotoxic [Citation83].
The inhibition of system XC− may prove useful in a number of therapeutic contexts, like tumor growth and survival and neurological dysfunctions [Citation84,Citation85]. Dixon and colleagues have demonstrated that erastin caused strong and persistent inhibition of system XC− function and this drug is more potent than the previously known best inhibitor Sulfasalazine (SAS). Also, Sorafenib triggers ferroptosis in many cancer cells types by blocking SLC7A11 activity, similar to erastin. Indeed, Sorafenib was originally reported to induce a non-apoptotic form of cell death suppressed by lipophilic antioxidants, in renal cell carcinoma and advanced hepatocellular carcinoma (HCC) [Citation86]. The key role played by the system XC− was also emphasized in colorectal cancer stem cells (CSCs) which are characterized by remarkably higher level of glutathione and SLC7A11 compared to colorectal cancer cells. Indeed, CSCs were sensitive to erastin-stimulate ferroptotic cell death compared to parental cells [Citation87].
Importantly, in addition to the intake of cysteine through the system XC−, many cells are also able to synthesize this amino acid from methionine through the trans-sulfuration pathway. Consequently, these cells are resistant to ferroptosis induced by system XC− inhibitors. Genome wide siRNA screening has revealed that the knockdown of the cysteinyl tRNA synthetase (CARS), results in the upregulation of the trans-sulfuration pathway and inhibits lipid ROS generation, thus suppressing erastin induced ferroptosis [Citation88].
Glutathione peroxidases (GPX) are a family of antioxidant enzymes responsible for reducing peroxidized molecules into lower reactive alcohols. Specifically, GPX4 is the only GPX known to catalyze the reduction of lipid hydroperoxides (Lipid-ROS), essential for life, since its knockout causes embryonic lethality in mice [Citation89,Citation90]. GPX4 converts glutathione into oxidized glutathione (GSSG) concomitantly reducing the cytotoxic lipid peroxides (L-OOH) to the corresponding alcohols (L-OH; ). Inactivation of GPX4 or depletion of GSH causes loss of cellular antioxidant capacity, leading to the accumulation of lipid peroxides, resulting in ferroptotic cell death. This explains the strong protective effect of lipophilic antioxidants, such as vitamin E, which suppress the formation of oxidized lipids upon GPX4 inhibition. RSL3 directly acts on GPX4 inhibiting its activity, thus promoting lipid-ROS accumulation, decreased GPX4 expression, and inducing ferroptosis [Citation91], while GPX4 overexpression confers resistance to ferroptosis induced by RSL3 [Citation92]. To note, GPX4 overexpression is also associated with poor prognosis in patients with diffuse large B-cell lymphoma [Citation93].
Since GSSG produced by the activity of GPX4 is subsequently reduced to GSH by glutathione reductase in the presence of NADPH, it is no surprise that the synthesis of NADPH also affects the sensitivity of cells to ferroptosis, as demonstrated by the increased sensitivity of fibrosarcoma HT1080 cells to ferroptosis, following NADPH depletion [Citation94].
Antioxidant defense in ferroptotic cell death
Maintenance of the cellular redox balance is essential for appropriate cellular functions. This balance can change in response to different stimuli, infections, chemical or physical insults. To this aim, living cells and, in particular keratinocytes that are the major cell type exposed to oxidative stress (UV irradiations), have evolved a sophisticated defense mechanism. In absence of glutathione, cells initiate a complex defense system, allowing their survival and cytoprotection. It’s known that upon chemical GSH depletion, NRF2, the antioxidant master transcription factor, can be activated. Different evidences suggest that ER stress (with unfolded protein accumulation) can activate the NRF2-KEAP1 (Kelch ECH associating protein 1) signaling pathway in a PERK–dependent manner. Once activated, PERK induces modifications of specific KEAP1 cysteine residues leading to its conformational changes and resulting in the release of NRF2 from KEAP1 [Citation95]. Then, the transcription factor can translocate from the cytosol into the nucleus where binds to the Antioxidant Responsive Elements (AREs) and enhances the expression of antioxidant factors (quinone oxidoreductase 1, NQO1), molecular chaperons, tumor suppression (such as p53) and intracellular detoxification genes (Glutathione S-transferase, GST; Aldo-keto reductase, AKR; and Heme oxygenase, HO-1) [Citation96,Citation97]. Importantly, NRF2 has a dual role in tumorigenesis since low nuclear levels are associated with tumor initiation and metastasis inhibition by eliminating carcinogenic components (ROS and DNA damage) and activating cytoprotective genes, while high nuclear levels induce chemoresistance and cancer progression. Thus, this transcription factor has protective activity only in the early stages of tumor development, but not in late tumorigenic stages [Citation98]. Consistently, the antineoplastic inhibitor Brusatol determine a rapid and transient depletion of NRF2 via post-transcriptional mechanism. In addition, PI3K inhibitors or NRF2 interfering RNA are also used to inhibit its expression and reduce the sensitivity to cytoprotective process in cancer cells. Recently, Wu and colleagues reported the strict link existing between the expression of NRF2 and GSK3β in breast cancer. They found reversed expression levels of the two factors with cell resistance to ferroptosis in low GSK3β and high NRF2 expression levels, mediated by upregulated GPX4 and downregulated Alox15. On the other hand, overexpression of GSK3β restored the cell sensitivity to erastin-stimulated ferroptosis [Citation99].
Among all genes with antioxidant activity, particularly important is the superfamily of enzymes AKRs, specially AKR1C1, AKR1C2 & AKR1C3, as described below ().
Ferroptosis and Melanoma
Cancer cell susceptibility to ferroptosis is very manifold since this is affected by several biological processes, such as loss of p53, DNA damage, metabolic state, or epithelial-mesenchymal transition (EMT), which are often dysregulated in tumors. Importantly, several studies have confirmed the key role of ferroptosis in killing cancer cells and in suppressing the growth of breast, lung, colon, central nervous system, and ovarian cancer [Citation67]. Moreover, combined chemotherapy and pro-ferroptotic drugs manifested a significant synergistic effect on their anticancer activity [Citation100]. However, although the ferroptotic process is efficiently induced in several types of tumor cells, many of them are resistant to ferroptosis execution. In this context aldo-keto reductases enzymes (AKRs) play a key role. Resistant cells to ferroptosis show low production of lipid hydroperoxides, consistent and sustained expression of AKR1C1÷ C3 enzymes, responsible for lipid peroxides reduction and, thus, cell death inhibition. Specifically, human AKR is a superfamily (AKR1 ÷ 14) of detoxifying enzymes that catalyze the conversion of aldehydes and ketones into their low-reactive primary and second alcohols, by using NAD(P)H as a cofactor [Citation101]. The AKR1C1 and AKR1C2 are involved in the metabolism of progesterone and 5α-dihydrotestosterone while AKR1C3 is associated with the formation of testosterone and prostaglandin. These enzymes display a high catalytic activity in cellular signaling involved in the regulation of endogenous substrates such as lipid aldehydes, prostaglandins and steroid hormones. For this reason, it’s not surprising that these enzymes are also implicated in pathogenesis and neurodegenerative disease. akr1c1÷ c3 genes are characterized by “ARE sequences” in their promoter regions, which are transcriptionally activated by electrophilic and antioxidant stress. These sequences allows them to bound NRF2, blocking the cancerogenic metabolism [Citation102] and tumor progression. During oxidative stress, bioactive molecules, including peroxyl radicals and peroxides, are generated; among these, aldehydes, which derive from the scission of covalent binding of PUFAs, are the major end products of lipid peroxidation [Citation103]. Downstream upregulation/activation of AKR1C1÷ C3 has been proposed as a mechanism involving the inhibition of ferroptotic cell death through the reduction of lipid peroxide levels, in metastatic melanoma cells. In fact, inhibiting the activity or gene expression of AKRs leads to the restoration of lipid ROS production and ferroptosis execution of resistant metastatic melanoma cells [Citation69]. Several studies indicated that AKRs are highly expressed in a series of tumors, such as lung, prostate [Citation104], ductal carcinoma and breast cancer [Citation105–107]. Therefore, a new potential therapeutic strategy for effectively killing cancer cells could be based on pro-ferroptosis drugs coupled with AKR1C1÷ C3 inhibitors.
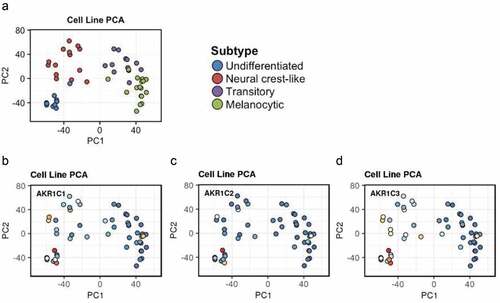
Importantly, the Graeber’s research group published data showing that primary tumor-derived melanoma cells are characterized by a wide “differentiation status” [Citation108], and identified an inverse correlation between cancer cells differentiation and susceptibility to ferroptotic cell death (a sort of “differentiation algorithm”). However, although this “differentiation algorithm” seems to be not applicable to metastasis-derived melanoma cells, potentially due to an EMT process [Citation69], interestingly the three AKRs (AKR1C1÷ C3) seems to be preferentially expressed in lower differentiated primary tumor-derived melanoma cells of the Graber’s model, potentially more metastatic cells, closely related to metastasis-derived melanoma cells ().
Figure 2. AKRs expression in primary tumor-derived melanoma. The expression of AKR1C1÷ C3 was evaluated in the “Graber cohort” of melanoma cells directly derived by primary tumors (https://systems.crump.ucla.edu/dediff/index.php). This interpolation shows that the three enzymes seem to be preferentially expressed in the less differentiated cell types, especially the AKR1C3.
Several reports also indicated that SLC7A11 basal expression seems to be correlated with poor prognosis in various malignancies, such as prostate [Citation109], breast [Citation110] and pancreatic cancer [Citation111]. Moreover, Okuno have reported that system XC− expression levels are correlated with the resistance of ovarian cancer cell line to platinum-containing compounds [Citation84]. Huang has shown that SLC7A11 can be a target for increasing the chemosensitivity of glutathione-mediated geldanamycin-resistant lung cancer cell lines [Citation112], while Angeli indicated that inhibiting SLC7A11 expression sensitizes cancer cells to ferroptosis [Citation113]. Altogether this data indicates that SLC7A11 expression might represent a valuable biomarker to predict the cancer cell susceptibility to ferroptosis, while inhibiting its expression might represent a novel therapeutic strategy to increase the efficacy of pro-ferroptotic drugs. The latter hypothesis has been also postulated by Wang which shows that pro-ferroptotic drugs induce an IFNγ-mediated SLC7A11 downregulation during cell killing, while pre-treating cells with IFNγ enhances ferroptotic cell death, through SLC7A11 downregulation [Citation114]. However it is important to underline that the basal expression of SLC7A11 could have a different predictive role, depending on the type of tumor. In fact, the basal expression of SLC7A11 seems to be inversely correlated to ferroptosis sensitivity, at least in metastasis-derived melanoma cells [Citation69].
Conclusion remarks
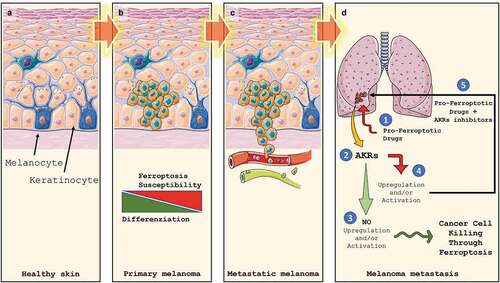
Although numerous steps forward have been made in recent years in the development of therapies capable of treating or guaranteeing adequate survival for patients affected by cancer, melanoma nevertheless represents an exception to this trend. Indeed, despite the development of specific BRAF inhibitors and the use of immune checkpoint blockers have consistently increased the overall survival rate of patients affected by metastatic melanoma, the results obtained so far are still discouraging. This is why the research in this field must go on and must be pushed forward to definitely eradicate this malignancy. At this stage of knowledge, it is time to speculate that new therapeutic strategies consisting of combined treatments capable of attacking tumor cells from different sides can be promoted by exploiting the few vulnerabilities that this tumor offers. In this scenario, ferroptosis could offer a new combination therapy opportunity with immune checkpoint blockers or BRAF/MEK inhibitors ().
Figure 3. Melanoma development, progression and Ferroptosis susceptibility. Melanocytes are cells located in the stratum basale (a) of the skin’s epidermis, the uvea, the inner ear, meninges, vaginal epithelium, heart, and bones. They are neural crest-derived cells producing melanin, a dark pigment responsible for skin color and protection from UV light. Upon transformation, melanocytes start to grow, giving rise to the primary tumor (b). At this stage, melanoma cells display a heterogeneous grade of differentiation which correlates to Ferroptosis susceptibility. In the advanced stages, the loose of intercellular junctions and the rupture of the basal lamina allow cancer cells to invade the underlying dermis, which reaching the blood and lymphatic circuits (c) will allow them to give rise to metastases (d). Ex vivo data indicate that basal expression of AKRs seems to be not predictive of Ferroptosis sensitivity while AKRs upregulation/activation (2) in pro-ferroptotic (1) treated metastatic cells does. Indeed, if no AKRs activation/upregulation is observed, metastatic melanoma cells are efficiently killed by Ferroptosis execution (3). On the other hand, AKRs upregulation/activation will result in ferroptosis inhibition and failure of treatment. However, combined pro-ferroptotic drugs with AKRs inhibitor will efficiently increase cancer cell death through effective Ferroptosis execution (5).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Tsao H, Atkins MB, Sober Arthur J. Management of cutaneous melanoma. N Engl J Med. 2004;351:998-1012.
- Markovic SN, Erickson LA, Rao RD, et al. Malignant melanoma in the 21st century, part 1: epidemiology, risk factors, screening, prevention, and diagnosis. Proceed Mayo Clin Proceed. Elsevier Ltd. 2007;82:364–380.
- All cancers; 2018. Available from: https://gco.iarc.fr/today/data/factsheets/cancers/16-Melanoma-of-skin-fact-sheet.pdf
- Table S. Tobacco use 44 sources of statistics 68 American cancer society recommendations for the early detection of cancer in average-risk asymptomatic people 71. Atlanta: American Cancer Society; 2020.
- Rigel DS. Rigel epidemiology of melanoma. Semin Cutan Med Surg. 2010 Dec;29(4):204–209. .
- Lasithiotakis KG, Petrakis IE, Garbe C. Cutaneous melanoma in the elderly: epidemiology, prognosis and treatment. Melanoma Res. 2010;20:163–170.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
- Cho E, BA R, GA. C. Risk factors for melanoma by body site. Cancer Epidemiol Biomarkers Prev. 2005;14:1241–1244.
- Kraemer KH, Greene MH. Dysplastic nevus syndrome. Familial and sporadic precursors of cutaneous melanoma. Dermatol Clin. 1985;3:225–237.
- Corazzari M, PE. L. Harnessing autophagy for melanoma benefit. Cell Biol Res Ther. 2013;2:1.
- DE B, JA R, JA S, et al. J., P. A role for sunlight in skin cancer: uVinduced p53 mutations in squamous cell carcinoma. PNAS. 1991;88:10124–10128.
- Kamb A, Gruis NA, Weaver-Feldhaus J, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436–440.
- Potrony M, Badenas C, Aguilera P, et al. Update in genetic susceptibility in melanoma. Ann Transl Med. 2015;3:210.
- Tagliabue E, Gandini S, Bellocco R, et al. MC1R variants as melanoma risk factors independent of at-risk phenotypic characteristics: a pooled analysis from the M-SKIP project. Cancer Manag Res. 2018;10:1143–1154.
- Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N Engl J Med. 2012;367:1694–1703.
- Gray-Schopfer V, Wellbrock C, Marais R. R., M. Melanoma biology and new targeted therapy. Nature. 2007;445:851–857.
- ER C-D, JJ O, OM. S. BRAFV600E: implications for carcinogenesis and molecular therapy. Mol Cancer Ther. 2011;10:385–394.
- Dhomen N, Marais R, Signaling BRAF. Targeted Therapies in Melanoma. Hematol Oncol Clin North Am. 2009;23:529–545.
- AH A-T, Li G. G., L. Genetic alterations of PTEN in human melanoma. Cell Mol Life Sci. 2012;69:1475–1491.
- Giglio P, Fimia GM, Lovat PE, et al. Fateful music from a talented orchestra with a wicked conductor: connection between oncogenic BRAF, ER stress, and autophagy in human melanoma. Mol Cell Oncol. 2015;2:e995016.
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410.
- Hersey P, Zhang XD. Adaptation to ER stress as a driver of malignancy and resistance to therapy in human melanoma. Pigment Cell Melanoma Res. 2008;21:358–367.
- Corazzari M, Lovat PE, Armstrong JL, et al. Targeting homeostatic mechanisms of endoplasmic reticulum stress to increase susceptibility of cancer cells to fenretinide-induced apoptosis: the role of stress proteins ERdj5 and ERp57. Br J Cancer. 2007;96:1062–71.
- Corazzari M, Fimia GM, Lovat P, et al. Why is autophagy important for melanoma? Molecular mechanisms and therapeutic implications. Semin Cancer Biol. 2013;23:337–43.
- JL A, Corazzari M, Martin S, et al. Oncogenic B-RAF signaling in melanoma impairs the therapeutic advantage of autophagy inhibition. Clin Cancer Res. 2011;17:2216–2226.
- DT R, SM A, Miller CN, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374.
- Corazzari M, Gagliardi M, Fimia GM, et al. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front Oncol. 2017;7:78.
- Corazzari M, Rapino F, Ciccosanti F, et al. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015;22:946–958.
- Lovat PE, Corazzari M, Armstrong JL, et al. Increasing melanoma cell death using inhibitors of protein disulfide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Res. 2008;68:5363–9.
- Niezgoda A, Niezgoda P, Czajkowski R. R., C. Novel approaches to treatment of advanced melanoma: a review on targeted therapy and immunotherapy. Biomed Res Int. 2015;2015:851387.
- TR W, Fridlyand J, Yan Y, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–509.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–1888.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10.
- Li X, Lu W, Zheng X, et al. Emerging immune checkpoints for cancer therapy. Acta Oncol. 2015;54:1706–1713.
- Weber J. Overcoming immunologic tolerance to melanoma: targeting CTLA-4 with ipilimumab (MDX-010). Oncol. 2008;13:16–25.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889–1894.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N Engl J Med. 2010;363:711–723.
- Dong H, SE S, DR S, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800.
- Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet. 2017;390:1853–1862.
- Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med. 2012;366:2517–2519.
- Galluzzi L, Vitale I, JM A, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120.
- DR G, Ferguson T, Zitvogel L. G., K. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353–363.
- AM D, Martin S, AD G. P., A. Immature, semi‐mature, and fully mature dendritic cells: toward a DC‐cancer cells interface that augments anticancer immunity.. Front Immunol. 2013;4:438.
- AD G, Galluzzi L, Apetoh L, et al. Molecular and translational Classifications of DaMPs in immunogenic cell death. Front Immunol. 2015;6:588.
- Bezu L, LC G-S, Ewitte HBK, et al. G., K. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol. 2015;6:187.
- Fucikova J, Kralikova P, Fialova A, et al. R., S. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011;71:4821–4833.
- Tesniere A, Schlemmer F, Boige V, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–491.
- Schiavoni G, Sistigu A, Valentini M, et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 2011;71:768–778.
- Spisek R, Charalambous A, Mazumder A, et al. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007;109:4839–4845.
- Giglio P, Gagliardi M, Tumino N, et al. PKR and GCN2 stress kinases promote an ER stress-independent eIF2α phosphorylation responsible for calreticulin exposure in melanoma cells. Oncoimmunology. 2018. DOI:10.1080/2162402X.2018.1466765.
- Giglio P, Gagliardi M, Bernardini R, et al. Ecto-Calreticulin is essential for an efficient immunogenic cell death stimulation in mouse melanoma. Genes Immun 2019;20:509–513.
- Dolma S, SL L, WC H, et al. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–296.
- WS Y, BR S. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–245.
- Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11.
- Galluzzi L, Vitale I, Aaronson SA, et al.. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
- Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an Iron-Dependent Form of Nonapoptotic Cell Death. Cell. 2012;149:1060–1072.
- Shah R, Shchepinov MS, Pratt DA. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent Sci. 2018;4:387–396.
- DM W. J., and K. Ferroportin-mediated iron transport: expression and regulation. Biochim Biophys Acta. 2012;1823:1426–1433.
- Gao M, Monian P, Qudri N, et al. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell. 2015;59:298–308.
- JD M, Wang X, Steven PG, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy.. Nature. 2014;509:105–109.
- JD M, VL. P, Nissim S, et al. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife. 2015;4:e10308.
- Song X, Zhu S, Chen P, et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc– activity. Curr Biol. 2018;28:2388–2399.e5.
- WS Y, KJ K, Gaschler MM, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113:E4966–75.
- JZ H, CD. F. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111:5866–5898.
- Yan S, XF Y, Liu HL, et al. Longchain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: an update. World J Gastroenterol. 2015;21:3492–3498.
- Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379.
- Birben E, UM S, Sackesen C, et al., O., K. Oxidative Stress and Antioxidant Defense. World Allergy Organ J. 2012;5:9–19.
- Gagliardi M, Cotella D, Santoro C, et al. M., C. Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis. 2019;10:902.
- SJ D, GE W, LS M, et al. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol. 2015;10:1604–1609.
- Yuan H, Li X, Zhang X, et al. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478:1338–1343.
- Burton GW, KU I. Vitamin E: application of the principles of physical organic chemistry to the exploration of its structure and function. Acc Chem Res. 1986;19:194–201.
- Lucarini M, Pedrielli P, Pedulli GF, et al. Bond Dissociation Energies of O−H Bonds in Substituted Phenols from Equilibration Studies. J Org Chem. 1996;61:9259–9263.
- Feng H, Stockwell BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLOS Biol. 2018;16:e2006203.
- Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–698.
- Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–692.
- Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90.
- QZ T, Lei P, Jackman KA, et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke.. Mol Psychiatry. 2017;22:1520–1530.
- Perez MA, Magtanong L, Dixon SJ, et al. Dietary Lipids Induce Ferroptosis in Caenorhabditis elegans and Human Cancer Cells. Dev Cell. 2020;S1534-5807(20)30498-6.
- Sato H, Tamba M, Ishii T. S., B.Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–11458.
- MC J, DD. Z. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–2191.
- Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
- Had-Aissouni L. Toward a new role for plasma membrane sodium-dependent glutamate transporters of astrocytes: maintenance of antioxidant defenses beyond extracellular glutamate clearance. Amin Acids. 2012;42:181–197.
- Okuno S, Sato H, Kuriyama-Matsumura K, et al. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br J Cancer. 2003;88:951–956.
- SC B, SL C, BR H, et al. H., S. Glutamate release by primary brain tumors induces epileptic activity. Nat Med. 2011;17:1269–1274.
- Coriat R, Nicco C, Chéreau C, et al. Sorafenib-induced hepatocellular carcinoma cell death depends on reactive oxygen species production in vitro and in vivo. Mol Cancer Ther. 2012;11:2284–2293.
- Xu X, Zhang X, Wei C, et al. Targeting SLC7A11 specifically suppresses the progression of colorectal cancer stem cells via inducing ferroptosis. Eur J Pharm Sci. 2020;152:105450.
- Hayano M, WS Y, CK C, et al. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23:270–278.
- LJ Y, Ran Q, Rao L, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496–502.
- Brigelius-Flohé R. M., M. Glutathione Peroxidases. Biochim Biophys Acta. 2013;1830:3289–3303.
- Sui X, Zhang R, Liu S, et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front Pharmacol. 2018;9:1371.
- Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell. 2014;156:317–331.
- Kinowaki Y, Kurata M, Ishibashi S, et al. Glutathione peroxidase 4 overexpression inhibits ROS-induced cell death in diffuse large B-cell lymphoma. Lab Invest. 2018;98:609–619.
- Shimada K, Hayano M, NC P, et al. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem Biol. 2016;23:225–235.
- Krajka-Kuźniak V, Paluszczak J. W., B.-D. The Nrf2-ARE signaling pathway: an update on its regulation and possible role in cancer prevention and treatment. Pharmacol Rep. 2017;69:393–402.
- Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295.
- Ma Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426.
- MB S, KT L. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564–571.
- Wu X, Liu C, Li Z, et al. Regulation of GSK3β/Nrf2 signaling pathway modulated erastin-induced ferroptosis in breast cancer. Mol Cell Biochem. 2020;10.1007/s11010-020-03821-8.
- Lu B, Chen XB, Ying MD, et al. The Role of Ferroptosis in Cancer Development and Treatment Response. Front Pharmacol. 2018;8:992.
- Pollak N, Dölle C, Ziegler M. M., Z. The power to reduce: pyridine nucleotides–small molecules with a multitude of functions. Biochem J. 2007;402:205–218.
- TM P, JE D. Human Aldo-Keto Reductases: function, Gene Regulation, and Single Nucleotide Polymorphisms. Arch Biochem Biophys. 2007;464:241–250.
- Barski OA, Bhatnagar A, Bhatnagar A. T.S.M. The Aldo-Keto Reductase Superfamily and its Role in Drug Metabolism and Detoxification. Drug Metab Rev. 2008;40:553–624.
- TM D, DW B, FL A, et al. PTHrP stimulates prostate cancer cell growth and upregulates aldo–keto reductase 1C3. Cancer Lett. 2011;306:52–59.
- HK L, Steckelbroeck S, Fung KM, et al. Characterization of a monoclonal antibody for human aldo-keto reductase AKR1C3 (type 2 3α-hydroxysteroid dehydrogenase/type 5 17β-hydroxysteroid dehydrogenase); immunohistochemical detection in breast and prostate. Steroids. 2004;69:795–801.
- AK J, Gunnarsson C, Cohen M, et al. O., S. 17β-Hydroxysteroid dehydrogenase 14 affects estradiol levels in breast cancer cells and is a prognostic marker in estrogen receptor positive breast cancer. Canc Res. 2006;66:11471–11477. .
- Sasano H, Suzuki T, Miki Y. T., M. Intracrinology of estrogens and androgens in breast carcinoma. J Steroid Biochem Mol Biol. 2008;108:181–185. .
- Tsoi J, Robert L, Paraiso K, et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell. 2018;33(890–904.e5). DOI:10.1016/j.ccell.2018.03.017.
- DW D, Gout PW, Kurita T, et al. W., P.. Sulfasalazine-Induced Cystine Starvation: potential Use for Prostate CancerTherapy. Prostate. 2007;67:162–171. .
- VS N, GM P, Gout PW, et al. Sulfasalazine-induced reduction of glutathione levels in breast cancer cells: enhancement of growth-inhibitory activity of Doxorubicin. Chemotherapy. 2007;53:210–217.
- Lo M, Ling V, YZ W, et al. The xc- cystine/glutamate antiporter: a mediator of pancreatic cancer growth with a role in drug resistance. Br J Cancer. 2008;99:464–472.
- Huang Y, Dai Z, Barbacioru C. W., S. Cystine-Glutamate Transporter SLC7A11 in Cancer Chemosensitivity and Chemoresistance. Cancer Res. 2005;65:7446–7454.
- JP FA, DV K, Conrad M. M., C. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19:405–414.
- Wang W, Green M, Choi JE, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–274.