
ABSTRACT
The proper segregation of basic elements such as the compartmentalization of the genome and the shuttling of macromolecules between the nucleus and the cytoplasm is a crucial mechanism for homeostasis maintenance in eukaryotic cells. XPO1 (Exportin 1) is the major nuclear export receptor and is required for the export of proteins and RNAs out of the nucleus. STK38 (also known as NDR1) is a Hippo pathway serine/threonine kinase with multifarious functions in normal and cancer cells. In this review, we summarize the history of the discovery of the nucleo/cytoplasmic shuttling of proteins and focus on the major actor of nuclear export: XPO1. After describing the molecular events required for XPO1-mediated nuclear export of proteins, we introduce the Hippo pathway STK38 kinase, synthetize its regulation mechanisms as well as its biological functions in both normal and cancer cells, and finally its intersection with XPO1 biology. We discuss the recently identified mechanism of XPO1 activation by phosphorylation of XPO1_S1055 by STK38 and contextualize this finding according to the biological functions previously reported for both XPO1 and STK38, including the second identity of STK38 as an autophagy regulator. Finally, we phrase this newly identified activation mechanism into the general nuclear export machinery and examine the possible outcomes of nuclear export inhibition in cancer treatment.
Eagle view of the question with a historical perspective
Nothing in biology makes sense except in the light of evolution [Citation1]. Supply logistics is a key factor in contemporary industrial manufacturing process [Citation2,Citation3]. One starts not to produce parts of a complex final product upon order of the latter: this would be a too slow process in case of necessary quick adjustments. So, the key is to stock pre-made parts that have to be brought on the assembly line in due time and step, allowing a streamlined rapid-response process.
Evolution has grasped this concept since long. In several stress situations, where quick adjustment is most welcome (starvation-induced autophagy, innate immune response via NFkB activation, etc.), parts are pre-made and accumulated in specific storage compartments: the most surprising being the nucleus that sees an extension of its function beyond being the genetic information container (see below) but helps also protein complexes manufacturing by being a “storage” place. Upon necessity, proteins stored in the nucleus are exported into the cytoplasm where they are incorporated in some final product (e.g. the initiation complex containing Beclin1) or submitted to post-translational modifications if required. This process is barely new in evolution: it takes place already in unicellular eukaryotes like baker’s yeast S. cerevisiae.
The nuclear-shuttling machinery
Back to the basics: overview and discovery of a nucleo/cytoplasmic shuttling
In eukaryotic cells, the nuclear envelope allows the proper segregation of the DNA replication and gene transcription on one side from the protein translation process on the other side. This nuclear barrier is composed of two phospholipidic membranes, where the outer one is continuous with the endoplasmic reticulum [Citation4], acting as a shield for the DNA material. The needs of higher organisms for a proper confinement of the genetic content and the translational machinery can be justified by the requirement of genetic stability, controlling with precision the access of transcriptional regulators to the chromatin. This segregation is also a way for eukaryotic cells to respond with precision to environmental signals and to control their key cellular processes.
The nuclear envelope in eukaryotic cells serves several functions: an architecture scaffold as well as a mechanical support. On the other hand, it can also be seen as a crucial center implicated in the dynamic control of key cellular processes such as cell cycle regulation and mitosis, DNA repair and integrity, apoptosis and autophagy, gene expression regulation [Citation5–13].
This nuclear physical envelope allows small molecules (<40 kDa) and ions to exchange without restriction through a simple diffusion gradient [Citation14]. Differently, the transport of macromolecules, proteins, and RNAs (>40 kDa) through this nuclear envelope is actually passively impossible and relies on an active carrier mechanism via a gatekeeper that is embedded between the inner and outer nuclear membranes [Citation4]: the nuclear pore complex (NPC) [Citation15,Citation16]. The NPC was first described in 1949, by Callan and colleagues who reported a “porous layer” when observing the nuclear envelope of amphibian oocyte nuclei using electron microscopy (EM) [Citation4,Citation17]. On the other hand, the first assumption in the possible nucleo/cytoplasmic shuttling of proteins appeared 9 years later, when Goldstein observed that radioactive proteins accumulated in the cytoplasm of non-labeled Amoeba that were grafted with radioactive nucleus [Citation18]. Finally, the first shuttling protein was identified in 1989 when Borer and colleagues discovered that nucleolin, a nuclear protein implicated in the regulation of transcription, was able to shuttle between the nucleus and the cytoplasm [Citation19].
The nuclear pore complex: structural insights and shuttling mechanism
The NPC has been extensively studied by structural methods such as cryo-EM since the past decades and has led to the detailed characterization of its molecular structure (reviewed in [Citation20]). The NPC appears as a cylindrical structure highly modular and dynamic with an octagonal symmetry [Citation21] and can be divided in three basic elements: a 40–50 nm central channel surrounded by a nuclear ring and cytoplasmic fibrils. This structure is terminated on the cytoplasmic side by 8 protein fibrils and on the nuclear part by another set of 8 protein filaments that converge and are linked by a ring-like structure called the nuclear basket [Citation22]. It is established that there is around 2000–5000 NPC per nucleus in mammalian cells [Citation23]. Despite its huge dimensions (approximately 125 000 kDa for a whole NPC), the NPC is composed of only 30 different proteins called nucleoporins (NUPs) [Citation24] that fuse the inner and outer nuclear membranes, resulting in a total of 500–1000 NUPs per entire NPC [Citation25].
As mentioned before, the NPC allows the transport of small proteins, molecules, salts, nucleotides, and other small components by passive diffusion through its aqueous channel. However, larger molecules (> 40 kDa), and the majority of proteins, rely on the assistance of energy-dependent transport receptors that recognize cognate sequences present on cargo proteins, allowing their import or export of the nucleus (reviewed in [Citation22]). For their nucleo/cytoplasmic shuttling, protein cargoes require specific transport signals, namely a nuclear localization signal (NLS) for their import into the nucleus or a nuclear export signal (NES) in contrast. These amino acid sequences bind to their proper importins or exportins and enable the release of the transport complex at the end of the shuttling [Citation22]. The known NLSs can be sorted into either classical or non-classical NLSs while the leucine-rich NES is the most characterized NES (see [Citation26] for a complete review).
The transport receptors responsible for the shuttling of the cargoes are referred to as karyopherins and can be called importins or exportins, based on their shuttling direction. Human cells count a total of 22 different karyopherins that interact directly or indirectly, trough adaptors, with unique group of cargo proteins or RNAs (reviewed in [Citation22]). The import and export cycles are relatively similar. The first step is the formation of the importin/exportin-cargo complex, where the type of transport receptor will depend on the type of NLS/NES carried by the cargo. For the nuclear import, the cargo-NLS binds to the importin, resulting into the nuclear translocation of this complex while the binding of RanGTP in the nucleus promotes the release of the cargo. Finally, the RanGTP-bound importin is then exported into the cytoplasm where the GTP hydrolysis induces the release of the importin, thus accomplishing its recycling [Citation27]. On the contrary, for the nuclear export, the cargo-NES binds to the exportin at its docking site, constituting a complex with RanGTP. This tripartite complex is then exported into the cytoplasm through the NPC where the cargo is released, the GTP hydrolyzed, and the exportin recycled into the nucleus.
Deregulation of the nuclear-shuttling machinery and implication in diseases
The nuclear import/export machinery is a highly coordinated process involving many proteins and large complexes in the vicinity of the nuclear envelope. Deregulation of this system has been associated with various diseases [Citation28,Citation29]. On one side, it has been reported that many tumor suppressor transcription factors display cytoplasmic retention, inhibiting their nuclear functions [Citation30]. On the other hand, unproperly localized onco-proteins can also lead to inadequate activity. In general, a dysregulation of this fundamental process affects other crucial cellular processes such as cell cycle, inflammatory response, apoptosis, and tumor growth [Citation22].
In addition, the involvement of a deregulated nucleo/cytoplasmic shuttling in neurodegenerative diseases has been largely reviewed in past years [Citation27,Citation31–34]. In general, diseases related to aberrant nucleo/cytoplasmic localization of proteins can be characterized either by alterations in the NLS sequence of crucial regulators, by mutations affecting directly the nuclear pore transport machinery and thus the subcellular distribution of proteins and RNAs, or also by disruption of the nucleo/cytoplasmic balance of proteins by increasing (or decreasing) the production of cytoplasmic (or nuclear) actors [Citation33].
The major actor of nuclear export: XPO1/CRM1
Characteristics and structure
Among the nuclear export receptors, XPO1 (for Exportin-1, aka CRM1 for Chromosome Maintenance 1) has been the most characterized and has been defined as the major nuclear export receptor by binding leucine-rich Nuclear Export Signals (NESs) [Citation35,Citation36]. XPO1 was first characterized in S. pombe in 1989, where mutagenesis experiments conferred nuclear chromosome domains deformations [Citation37], resulting in its now well-established function of major nuclear export receptor. The first full-length XPO1 tridimensional structures were elucidated in 2009, followed by structures of XPO1-cargo and XPO1-RanGTP-cargo complexes (see below for an introduction to the RanGTP mechanism) [Citation38], pinpointing the association of the distinct XPO1 domains with functional relevance. Human XPO1 is a 120 kDa protein composed of 1071 amino acids organized in 20 tandem HEAT repeats [Citation39]. Each HEAT repeats forms a hairpin of helices denoted A and B, defining a ring-shaped solenoid, whose outer and inner surfaces comprise A and B helices, respectively ().
Figure 1. XPO1 architecture and domains
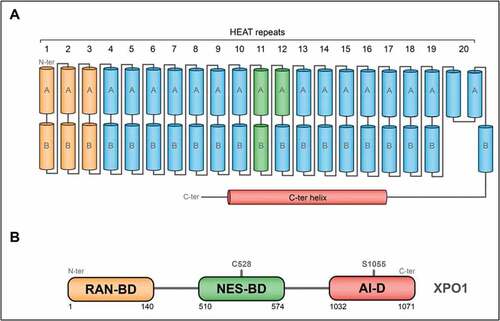
Three main domains for human XPO1 can be distinguished here. The first one, required for XPO1 export activity, has been defined as the association domain with RanGTP [Citation40,Citation41]. The second domain, comprised of the helices of HEAT repeats 11 and 12, binds the NES signal present on the cargoes by forming a hydrophobic docking site on the outer surface of XPO1 [Citation42]. Finally, the third main domain, comprised of the helix 21 and the C-terminal helix, has been reported as an auto-inhibitory region () [Citation40,Citation41]. In the ternary XPO1-cargo-RanGTP complex, this auto-inhibitory region packs next to the helix 21A, while its packs to the helices repeats 9 and 10, covering the NES-binding domain in the other conformation [Citation43]. In addition, it has been reported that deletion of the 39 last amino acid of XPO1, producing a deletion of this np-named auto-inhibitory region, enhanced drastically the NES-binding activity as well as the export activity of XPO1 [Citation44].
XPO1-mediated nuclear export
The active transport of cargoes against concentration gradients depends on an intact nuclear envelope and a nuclear pore complex barrier for retaining already transported cargoes in the desired compartment. This mechanism requires an input of metabolic energy delivered by the RanGTPase system, namely the hydrolysis of the bound GTP. The XPO1-mediated nuclear export can be divided in three main steps, the first one consisting of the formation of the trimeric XPO1-RanGTP-cargo complex. The second one consists of the translocation of this trimeric complex across the NPC followed by the last one where the dissociation of the trimeric complex leads to the release of the cargo in the cytoplasm and the recycling of both XPO1 and RanGDP into the nucleus ().
Figure 2. XPO1-dependent nuclear export
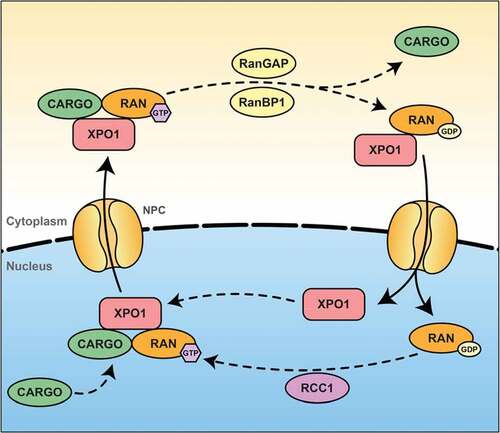
XPO1 binds cooperatively to RanGTP and the cargo, leading to the creation of a nuclear trimeric transport complex. It has been reported that the binding of NES-containing proteins with XPO1 could be regulated by phosphorylation of the cargo. For example, phosphorylation of cyclin D1 promotes its export by XPO1 [Citation45], while phosphorylation of c-Fos inhibits the nuclear export of this transcription factor [Citation46]. Interestingly, the affinity of XPO1 for the majority of its NESs is low, making the formation of this trimeric transport complex the rate-limiting step of the export transport () [Citation47]. It has been reported that RanBP3 seems to be crucial for the formation of this complex by increasing the concentration of RanGTP in the proximity of XPO1 [Citation48] and by stabilizing a conformational state that favor the binding of both RanGTP and the cargo to XPO1 [Citation49,Citation50].
The directionality of transport is mainly imposed by the Ran GTP/GDP gradient, which favors the assembly of export complexes and the disassembly of import complexes in the nucleus though the cytoplasmic nucleoporins Nup proteins. XPO1 is known to interact with several nucleoporins such as Nup358 (aka RanBP2), Nup214, and Nup88. It has been found that NES cargoes increased the affinity of XPO1 to Nup214 [Citation51], giving a first insight of this translocation mechanism. Nup358 is known to be the major component of the cytoplasmic filaments of the NPC and have a supportive role in XPO1-mediated export [Citation52].
When the trimeric RanGTP-cargo-XPO1 complex has been transported to the cytoplasmic side of the NPC through the nucleoporins, the dissociation of the cargo from this complex, as well as the recycling of both XPO1 and Ran and their transport into the nucleus occurs. The RanGAP and RanBP1 have been reported to by soluble in the cytoplasm and suggested, with Nup358, to promote the dissociation of the export complex from its terminal binding site [Citation47]. For the disassembly of the complex, RanGTP need to be hydrolyzed. To do so, RanBP1 releases RanGTP from the trimeric complex, allowing RanGAP to catalyze the hydrolysis of Ran-bound GTP to GDP [Citation53]. The mechanism responsible for the reimport of XPO1 in the nucleus remains elusive, but Nup358 seems to be involved in this reimport by transient interaction with XPO1 [Citation47].
Targeting XPO1: relevance in cancer therapy
The appropriate spatiotemporal localization of molecules in the nucleus or the cytoplasm, regulated by the bidirectional transport system channel through the NPC, is crucial for cellular homeostasis. Defect in proper localization of these molecules may alter their activities, thus disturbing the cell homeostasis and causing diseases such as cancer. It has been reported that nucleo/cytoplasmic shuttling dysregulation is involved in cancer cell survival, tumor progression, carcinogenesis, and drug resistance although it remains elusive whether this is due to a specific cargo mislocalization of which supports oncogenesis or because of a social sensitivity of cancer cells to homeostasis deregulation [Citation29]. Many tumor suppressor proteins execute their antineoplastic functions inside the nucleus, where upregulated nuclear export machinery could result in their functional inactivation [Citation54].
Due to the critical role of XPO1 in nucleo/cytoplasmic shuttling and in tumor progression (), its inhibition has emerged as a therapeutic strategy in cancer. The rational objective being increasing the nuclear concentration of tumor suppressor proteins using specific chemical inhibitors against XPO1 nuclear export activity. The first XPO1 inhibitor discovered was Leptomycin B (LMB), that covalently binds a cysteine residue (C528) in the cargo-binding domain of XPO1, preventing the cargo interaction with XPO1 [Citation55]. Where LMB demonstrated efficient antitumor activity in vitro, it displayed severe toxicity attributed to off-target effects due to its binding to several cysteine proteases, precluding its further clinical development [Citation56].
Figure 3. XPO1 alterations in cancers
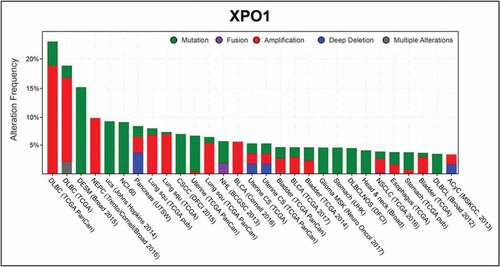
The atomic-level understanding of CRM1 function has greatly facilitated recent drug discovery and development of CRM1 inhibitors to target a variety of malignancies. Thanks to these discoveries, a novel group of small-molecule compounds, called selective inhibitors of nuclear export (SINE), have been developed. These SINEs, including KPT-115, KPT-127, KPT-185, KPT-251, KPT- 276, KPT-330 (Selinexor®), and KPT-335 (Verdinexor®), bind reversibly to the C528 residue of XPO1, with virtually no off-target effect. These SINE compounds have been shown to inhibit the nuclear export of many tumor suppressor proteins harboring key roles in genomic stability and DNA repair (TP53, TP73, and BRCA1), cell cycle progression (pRB1, CDKN1A, and CDKN1B), and apoptosis (FOXO, APC, and IκBα) in cancer cell lines and tumor biopsies [Citation57]. At this time, KPT-330 (Selinexor®) is the most represented XPO1 inhibitor in the phase I/II clinical trials registered at the ClinicalTrials.Gov database (http://clinicaltrials.gov/ct2/home).
The STK38 kinase
The Hippo pathway and STK38: an overview
The Hippo pathway was discovered more than two decades ago as a well-conserved signal transduction cascade from flies to mammals [Citation58]. The core cassette of the mammalian Hippo pathway comprises distinct signal transducers: a cascade of kinases, the STE20-like protein kinase 1/2 (MST1/2), the AGC serine/threonine protein kinases LATS1 and LATS2, the SAV1 and MOB1 scaffold proteins [Citation59,Citation60], and the transcriptional co-activators YAP and TAZ [Citation61,Citation62] (). Briefly, on its on-state, the Hippo pathway inhibits YAP/TAZ transcriptional activity through phosphorylation of YAP/TAZ on multiple residue, causing YAP/TAZ cytoplasmic retention and its latter degradation [Citation63]. Recent studies added new actors to this “canonical” core cassette: the AGC serine/threonine STK38 and STK38L (also known as NDR1 and NDR2, respectively) and the MAP4K kinase family. On one side, it has been reported that MAP4K family phosphorylate LATS1/2, activating them [Citation64]. On the other one, STK38/STK38L have been reported as directly phosphorylating YAP [Citation65].
Figure 4. The Hippo pathway and STK38
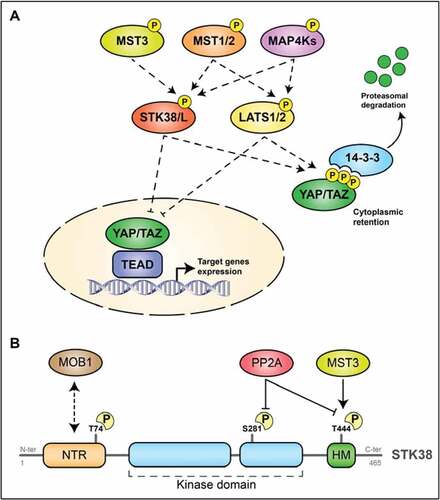
The primary structure of STK38 kinase family (STK38 and STK38L) is well conserved from yeast to human [Citation60]. In brief, STK38 is composed of three main domains. First, a N-Terminal Regulatory domain (NTR) located at its N-terminus is known to by essential for the binding of the regulatory MOB1A protein to STK38 () [Citation66], a mechanism required for STK38 activation. Interestingly, a phosphorylation site (Thr74) is located within this NTR only in humans and is required for full kinase activity [Citation67,Citation68]. The second domain, specific of the STK38/STK38L subgroup within the AGC group of protein kinases, is the catalytic/kinase domain. This domain can be divided in 12 subdomains. A second phosphorylation site (Ser281), whose phosphorylation is required for STK38 kinase activity, is embedded in this catalytic domain () [Citation60]. Finally, the last domain, located at the C-terminus of STK38, has been characterized as a hydrophobic motif and contains the last phosphorylation site (Thr444), phosphorylation of which us required for the kinase activity () [Citation69].
Regulation of STK38
In a simple mind-set, human STK38 is activated by phosphorylation and inactivated by de-phosphorylation. STK38 contains two main regulatory phosphorylation sites required for its kinase activity: one in the activation segment at Ser281 and one in the hydrophobic motif at Thr444, both well conserved in all STK38 kinase family identified [Citation70]. It has been reported that MST1, MST2 and MST3 phosphorylate both STK38 on Thr444, inducing activation of the kinase [Citation71], and that STK38 activation is also regulated by autophosphorylation on this same activation segment [Citation68]. Both phosphor-site are dephosphorylated by PP2A with subsequent inactivation of STK38 () [Citation60]. Additionally, MAP4K (mitogen-activated protein kinase kinase kinase kinase)-type kinases, which are also members of the Ste20-like kinases family, can regulate STK38/STK38L through phosphorylation on their HM [Citation64]. In this context, it has to be mentioned that RalA and MAP4K4 activate STK38 upon stress response and apoptotic signaling. Selimoglu and colleagues reported that the Ste20-like kinase MAP4K4, an effector of RalA via the exocyst complex, directly phosphorylates STK38 on its Thr444 under osmotic and oxidative stress [Citation72].
Another layer of complexity is brought by the MOB family adaptor proteins (for Mps one binder). MOB proteins bind to the conserved NTR of STK38 that precedes the catalytic domain, releasing STK38 from autoinhibition [Citation71]. It has been reported that the MOB1/STK38 complex formation seems to be essential for STK38 activation by STK38 autophosphorylation on Ser281 and HM phosphorylation at Thr444 by MST1 [Citation73]. Finally, a group of scaffolding proteins have been characterized as the third main component of the regulation of STK38 (see [Citation60] for a complete review).
STK38: biological functions
STK38, as well as STK38L, is essential for the survival of many organisms. STK38 has been associated with multifarious biological functions such as cell cycle progression, centrosome biology, apoptosis, autophagy, DNA damage signaling, and other signaling pathways, this reflecting its central and integrative role in biology. The first function attributed to STK38 is its role in centrosome duplication [Citation74] where silencing of STK38 negatively affected centrosome duplication [Citation75]. STK38 has also been linked to the progression of cell cycle through regulation of-myc and p21/Cip1 protein levels [Citation76]. Second, it has been shown that Cyclin D1 directly interacts with STK38, increasing its kinase activity required for the G1/S transition [Citation77]. In addition, STK38 regulates also the DNA damage-induced G2/M checkpoint by directly phosphorylating CDC25A at Ser76, leading to CDC25A degradation [Citation78]. Last but not least, STK38 potentiates NF-κB activation induced by TNFα whereas knockdown of STK38 inhibits such NF-κB activation [Citation79].
Recent articles have shed light on the involvement of STK38 in two other crucial cellular mechanisms: autophagy and anoïkis resistance. First, STK38 has been identified as a novel Beclin1 binding partner, a key regulator of autophagy; in its autophagic function, STK38 kinase activity is required [Citation80]. Moreover, it has been found that STK38 supports the formation of a complex composed of the exocyst component Exo84, Beclin1, and RalB, and that STK38 activity is stimulated in a MOB1-dependent manner. Finally, the contribution of STK38 to the resistance to anoïkis (apoptosis induced by the lack of cell attachment to the extracellular matrix) has been established very recently, with STK38 supporting mitophagy. This have drastic consequences on cancer cell hallmarks: it has been shown that STK38 knockdown impaired anoïkis resistance, anchorage-independent soft agar growth, and in vivo xenograft growth of Ras-transformed human cells, shedding light on the supporting oncogenic role of STK38 in Ras-dependent cancer cells [Citation81].
STK38 as the new XPO1 gatekeeper
But how STK38 intervenes in these later functions? We were tackling the STK38 kinase, being victim of our almost obsessive approach on establishing protein pathways to decipher biological functions. We had recently integrated STK38 in the Ral GTPases interactome in varied biological contexts. One was osmotic shock, where MAP4K4, an effector of RalA, via the Exocyst complex led to the activation of STK38 and subsequently to the activation of p38 Map kinase [Citation72]. Last but not least, STK38 is required and acts downstream of RalB during the autophagy process upon starvation conditions [Citation80]. Ras-induced transformation is supported by Ral GTPases through recruitment of STK38, and that STK38 contributes to a key feature of cancer cells: their survival in suspension [Citation81]. How a single kinase can perform these crucial, but clearly distinct, cellular functions? Is it through function-specific substrates? This most parsimonious and common-sense hypothesis will prove to be wrong.
We opted for a non-biased exploration by proteomics and proximity labeling assay (APEX2) [Citation82,Citation83]. We identified more than 97 proteins in nutrient starvation-induced autophagy and more than 221 ones upon anoïkis conditions [Citation6]. Overwhelmed by the number, we set up a filter and retained only the proteins identified in both conditions. We found that STK38 increased its interaction with cytoplasmic-related proteins upon nutrient starvation-induced autophagy and with nuclear ones in the context of anoïkis. Thus, STK38 is sometimes cytoplasmic, sometimes nuclear; thus, we focused our consideration on one of the proteins identified in our proteomic assay and that can be responsible for this nucleo/cytoplasmic shuttling: XPO1.
Pursuing in this direction, we found that STK38 exits the nucleus upon nutrient starvation-induced autophagy under the dependency of XPO1 and that STK38 kinase activity was required for its own nuclear exit [Citation6]. Furthermore, we found that XPO1 harbors a STK38 HxRxxS/T phosphorylation motif [Citation70] centered on the serine 1055, already reported as being phosphorylated in human cells by a yet unidentified kinase, without any related functional relevance [Citation84–87]. It is noteworthy to indicate that this serine 1055 is at the very tip of the domain that folds back toward the NES-binding domain and presumably hinders the cargo access to its mooring site [Citation43,Citation44].
An anti-phosphopeptide antibody directed toward the site once phosphorylated was raised and was a handy tool to ascertain that this site was indeed phosphorylated by STK38 [Citation6]. These observations were confirmed by the detection of this direct phosphorylation by mass spectrometry, independently of the above-mentioned antibody [Citation6]. Genetics strengthened our proposed model (): mutation of the serine 1055 by phosphomimetic residues (D or E) rendered STK38 dispensable for XPO1 nuclear export function where mutation by phosphonegative residue (A) abolished XPO1 export function and proved also that STK38 decisive action in inactivation of XPO1 is sufficient to justify its necessity for autophagy. Beyond that, STK38 is dispensable [Citation6].
Figure 5. General model of XPO1-dependent nuclear export under STK38 control
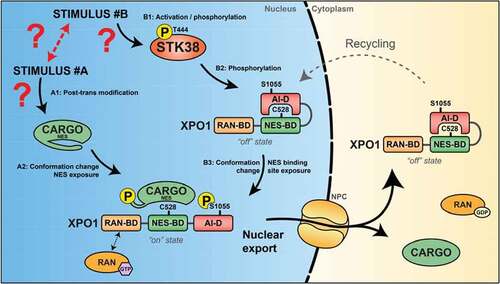
Ultimately, moving from peculiar to a more generalized model, we extended our findings by demonstrating that XPO1_S1055 phosphorylation by STK38 was regulating the subcellular localization of other cargoes. Beclin1, which shuttles physiologically from the nucleus to the cytoplasm upon nutrient starvation-induced autophagy, was found predominantly cytoplasmic even when nutrient supply was copious using phosphomimetic variants of XPO1 for serine 1055 [Citation6]. The same finding was observed for another actor: the co-transcription factor YAP1, that shuttle from the nucleus into the cytoplasm when cells get confluent. In the context of XPO1_S1055D/E, YAP1 was found in the cytoplasm, even in non-confluent cells [Citation6]. Thus, forceful nuclear export is obtained upon “constitutive” phosphomimicry at position 1055 ().
Discussion
The main outcome resulting from our work is the regulation of proteins nuclear exit by STK38 through phosphorylation, and thus activation, of XPO1 on its Ser1055. By characterizing that STK38 phosphorylates XPO1 on its serine 1055 embedded within its auto-inhibitory domain, we identified STK38 as the first activator of XPO1. A balance of RanGTP – RanGDP is maintained across the nuclear membrane, RanGTP being predominant in the nucleus. In details, Ran is efficiently loaded with GTP in the nucleus by its guanidine nucleotide exchange factor RCC1, which is tethered to chromatin through interactions with histones H2A and H2B [Citation88]. With all these information, it seems that the amount of RanGTP is not the limiting factor for the XPO1-dependent export of distinct cargoes.
The straightforward model proposed by our work is limited by its binary mode of action: NES docking to XPO1 is available or not. As a consequence of what, any NES-presenting protein should be loaded and exported. Preliminary data suggest that proteins are sorted out and not all cargoes quit the nucleus in the same time. For instance, starvation and cell confluence trigger the export selectively of Beclin1 and YAP, respectively. Since there is more than 400 possible cargoes in a given cell [Citation89], a possible selection mode can be considered along two mechanisms, both based on post-translational modifications (PTM).
For some cargoes, their effective nuclear export requires indeed their post-translational modification [Citation90]. PTM can act through revelation: in basic state the NES is rather “internalized” or hidden by a masking domain of the same protein or a partner protein (). Conversely, these modifications could mask the NES when nuclear export is to be avoided: PTM can compromise the NES-containing region by steric hindrance to interactions with the mooring domain of XPO1. Bulky PTM should be the most efficient in this effect, such as ubiquitination, and multiplication of smaller modifiers (phosphorylation, acetylation, etc …) should be equivalent. Subsequent export of the cargo would require the rejuvenation of this NES-containing domain either by active removal of the PTM (phosphatases, ubiquitin hydrolases, desacetyases, etc …) or by an in abstentia mechanism: abstinence of any PTM in neo-synthetized proteins.
As example, it is well known that p53 is exported by XPO1, this is even one of the main reasons of XPO1-dependent nuclear export inhibition in cancer: keeping p53 in the nucleus for its anti-tumorigenic activity. For this example, it has been reported that p53 nuclear export rely on its ubiquitination by MDM2, exposing the NES embedded in its C-terminal region. Other proteins rely on the masking/unmasking of their NES for their proper subcellular localization [Citation90] such as the INI1/hSNF5 integrase interactor 1 component of the SWI/SNF complex [Citation91] and the FOXO1 transcription factor that requires multiple phosphorylation on specific amino acids for the presentation of its NES [Citation92].
We start to achieve a good global understanding of the molecular mechanisms governing the XPO1-dependent nuclear export of cargoes (). However, some elements need to be characterized, such as the activator(s) of STK38 in the context of nuclear export as well as the identification of activators of all XPO1-dependent cargoes, although the final list start to take shape [Citation90]. A final element needs to be also characterized, the possible crosstalk between the activation of the two branches of the activation process (i.e. the cargo conformation change and STK38 activation).
One final remark should be assessed regarding the inhibition of XPO1 export activity in cancer treatments. Although it is unclear nuclear sequestering of which proteins will be beneficial for cancer outcome or if cancer cells are exquisitely sensitive to homeostasis imbalance. Reversely, some candidate might rise suspicion that their forced nuclear localization might be favoring cancer growth, especially YAP1.
Acknowledgments
We are in debt to our shrewd and cosmopolitan collaborators who participated to this work with creative ideas, thrilling discussions, and challenging questions/comments. We thank in particular A. Hergovitch from UCL; MK Singh from Paris; V. Aushev from Mount Sinai New-York; B. Meunier, MC. Parrini, and G. Zalcman from Institut Curie Paris; P. Codogno and C. Guerera and her collaborators from the mass spectrometry platform of INEM Paris; N. Carpi and M. Piel from IPGG Paris; and D. Daelemans and M. Jacquemyn from KU Leuven.
Disclosure statement
The authors declare that they have no conflict of interest.
References
- Dobzhansky T. Nothing in biology makes sense except in the light of evolution. Am Biol Teach. 1973;35:125–129.
- Apple’s Jobs to Obama: ‘jobs aren’t coming back’ to U.S. - News - Sarasota Herald-Tribune, Sarasota, FL.
- Supply Chain 4.0 – the next-generation digital supply chain | McKinsey.
- CALLAN HG, RANDALL JT, TOMLIN SG. An electron microscope study of the nuclear membrane. Nature. 1949;163:280.
- Huang R, Xu Y, Wan W, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57:456–466.
- Martin AP, Jacquemyn M, Lipecka J, et al. STK38 kinase acts as XPO1 gatekeeper regulating the nuclear export of autophagy proteins and other cargoes. EMBO Rep. 2019;20:e48150.
- Burke B, Stewart CL. The nuclear lamins: flexibility in function. Nat Rev Mol Cell Biol. 2013;14:13–24.
- Solovei I, Wang AS, Thanisch K, et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell. 2013;152:584–598.
- Mattout A, Pike BL, Towbin BD, et al. An EDMD mutation in C. elegans lamin blocks muscle-specific gene relocation and compromises muscle integrity. Curr Biol. 2011;21:1603–1614.
- Kind J, van Steensel B. Genome-nuclear lamina interactions and gene regulation. Curr Opin Cell Biol. 2010;22:320–325.
- Dauer WT, Worman HJ. The nuclear envelope as a signaling node in development and disease. Dev Cell. 2009;17:626–638.
- Rodriguez-Bravo V, Pippa R, Song W-M, et al. Nuclear pores promote lethal prostate cancer by increasing POM121-driven E2F1, MYC, and AR nuclear import. Cell. 2018;174:1200–1215.e20.
- Heessen S, Fornerod M. The inner nuclear envelope as a transcription factor resting place. EMBO Rep. 2007;8:914–919.
- Görlich D, Kutay U. Transport between the cell nucleus and the cytoplasm. Annu Rev Cell Dev Biol. 1999;15:607–660.
- BARNES BG, DAVIS JM. The structure of nuclear pores in mammalian tissue. J Ultrastruct Res. 1959;3:131–146.
- Fahrenkrog B, Aebi U. The nuclear pore complex: nucleocytoplasmic transport and beyond. Nat Rev Mol Cell Biol. 2003;4:757–766.
- Callan HG, Tomlin SG. Experimental studies on amphibian oocyte nuclei. I. Investigation of the structure of the nuclear membrane by means of the electron microscope. 1950;137:367–378. Proc R Soc London Ser B, Biol Sci.
- Goldstein L. Localization of nucleusspecific protein as shown by transplantation experiments in Amoeba proteus. Exp Cell Res. 1958;15:635–637.
- Borer RA, Lehner CF, Eppenberger HM, et al. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell. 1989;56:379–390.
- Beck M, Hurt E. The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol. 2016;18:73–89.
- Schwartz TU. Modularity within the architecture of the nuclear pore complex. Curr Opin Struct Biol. 2005;15:221–226.
- Cautain B, Hill R, De Pedro N, et al. Components and regulation of nuclear transport processes. Febs J. 2015;282:445–462.
- Grossman E, Medalia O, Zwerger M. Functional architecture of the nuclear pore complex. Annu Rev Biophys. 2012;41:557–584.
- Neumann N, Lundin D, Poole AM. Comparative genomic evidence for a complete nuclear pore complex in the last eukaryotic common ancestor. PLoS One. 2010;5:e13241.
- Hoelz A, Debler EW, Blobel G. The structure of the nuclear pore complex. Annu Rev Biochem. 2011;80:613–643.
- Fu X, Liang C, Li F, et al. The rules and functions of nucleocytoplasmic shuttling proteins. Int J Mol Sci. 2018;19:1445.
- Benarroch EE. Nucleocytoplasmic transport: mechanisms and involvement in neurodegenerative disease. Neurology. 2019;92:757–764.
- Mor A, White MA, Fontoura BMA. Nuclear trafficking in health and disease. Curr Opin Cell Biol. 2014;28:28–35.
- Hung MC, Link W. Protein localization in disease and therapy. J Cell Sci. 2011;124:3381–3392.
- Hill R, Cautain B, De Pedro N, et al. Targeting nucleocytoplasmic transport in cancer therapy. Oncotarget. 2014;5:11–28.
- Boehringer A, Bowser R. RNA nucleocytoplasmic transport defects in neurodegenerative diseases. In: Advances in neurobiology. Cham: Springer; 2018. p. 85–101.
- Fahrenkrog B, Harel A. Perturbations in traffic: aberrant nucleocytoplasmic transport at the heart of neurodegeneration. Cells. 2018;7:232.
- Kim HJ, Taylor JP. Lost in transportation: nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron. 2017;96:285–297.
- Veldman MB, Yang XW. Huntington’s disease: nuclear gatekeepers under attack. Neuron. 2017;94:1–4.
- Stade K, Ford CS, Guthrie C, et al. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. 1997;90:1041–1050.
- Fukuda M, Asano S, Nakamura T, et al. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;390:308–311.
- Adachi Y, Yanagida M. Higher order chromosome structure is affected by cold-sensitive mutations in a Schizosaccharomyces pombe gene crm1+ which encodes a 115-kD protein preferentially localized in the nucleus and its periphery. J Cell Biol. 1989;108:1195–1207.
- Dong X, Biswas A, Süel KE, et al. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009;458:1136–1141.
- Andrade MA, Bork P. HEAT repeats in the Huntington’s disease protein. Nat Genet. 1995;11:115–116.
- Fox AM, Ciziene D, McLaughlin SH, et al. Electrostatic interactions involving the extreme C terminus of nuclear export factor CRM1 modulate its affinity for cargo. J Biol Chem. 2011;286:29325–29335.
- Dong X, Biswas A, Chook YM. Structural basis for assembly and disassembly of the CRM1 nuclear export complex. Nat Struct Mol Biol. 2009;16:558–560.
- Güttler T, Madl T, Neumann P, et al. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17:1367–1376.
- Monecke T, Güttler T, Neumann P, et al. Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science. 2009;324:1087–1091.
- Dian C, Bernaudat F, Langer K, et al. Structure of a truncation mutant of the nuclear export factor CRM1 provides insights into the auto-inhibitory role of its C-terminal helix. Structure. 2013;21:1338–1349.
- Benzeno S, Lu F, Guo M, et al. Identification of mutations that disrupt phosphorylation-dependent nuclear export of cyclin D1. Oncogene. 2006;25:6291–6303.
- Sasaki T, Kojima H, Kishimoto R, et al. Spatiotemporal regulation of c-Fos by ERK5 and the E3 ubiquitin ligase UBR1, and its biological role. Mol Cell. 2006;24:63–75.
- Hutten S, Kehlenbach RH. CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol. 2007;17:193–201.
- Nemergut ME, Lindsay ME, Brownawell AM, et al. Ran-binding protein 3 links Crm1 to the Ran guanine nucleotide exchange factor. J Biol Chem. 2002;277:17385–17388.
- Lindsay ME, Holaska JM, Welch K, et al. Ran-binding protein 3 is a cofactor for Crm1-mediated nuclear protein export. J Cell Biol. 2001;153:1391–1402.
- Englmeier L, Fornerod M, Bischoff FR, et al. RanBP3 influences interactions between CRM1 and its nuclear protein export substrates. EMBO Rep. 2001;2:926–932.
- Hutten S, Kehlenbach RH. Nup214 is required for CRM1-dependent nuclear protein export in vivo. Mol Cell Biol. 2006;26:6772–6785.
- Walther TC, Pickersgill HS, Cordes VC, et al. The cytoplasmic filaments of the nuclear pore complex are dispensable for selective nuclear protein import. J Cell Biol. 2002;158:63–77.
- Kehlenbach RH, Dickmanns A, Kehlenbach A, et al. A role for RanBP1 in the release of CRM1 from the nuclear pore complex in a terminal step of nuclear export. J Cell Biol. 1999;145:645–657.
- Gravina GL, Senapedis W, McCauley D, et al. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J Hematol Oncol. 2014;7:85.
- Kudo N, Matsumori N, Taoka H, et al. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci U S A. 1999;96:9112–9117.
- Lapalombella R, Sun Q, Williams K, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012;120:4621–4634.
- Conforti F, Wang Y, Rodriguez JA, et al. Molecular pathways: anticancer activity by inhibition of nucleocytoplasmic shuttling. Clin Cancer Res. 2015;21:4508–4513.
- Moon S, Yeon Park S, Woo Park H. Regulation of the Hippo pathway in cancer biology. Cell Mol Life Sci. 2018;75:2303–2319.
- Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev. 2013;27:355–371.
- Hergovich A, Stegert MR, Schmitz D, et al. NDR kinases regulate essential cell processes from yeast to humans. Nat Rev Mol Cell Biol. 2006;7:253–264.
- Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015;15:73–79.
- Hong W, Guan KL. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol. 2012;23:785–793.
- Meng Z, Moroishi T, Guan K-L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016;30:1–17.
- Meng Z, Moroishi T, Mottier-Pavie V, et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat Commun. 2015;6. DOI:10.1038/ncomms9357.
- Zhang L, Tang F, Terracciano L, et al. NDR functions as a physiological YAP1 kinase in the intestinal epithelium. Curr Biol. 2015;25:296–305.
- Bichsel SJ, Tamaskovic R, Stegert MR, et al. Mechanism of activation of NDR (nuclear Dbf2-related) protein kinase by the hMOB1 protein. J Biol Chem. 2004;279:35228–35235.
- Tamaskovic R, Bichsel SJ, Rogniaux H, et al. Mechanism of Ca2+-mediated regulation of NDR protein kinase through autophosphorylation and phosphorylation by an upstream kinase. J Biol Chem. 2003;278:6710–6718.
- Stegert MR, Tamaskovic R, Bichsel SJ, et al. Regulation of NDR2 protein kinase by multi-site phosphorylation and the S100B calcium-binding protein. J Biol Chem. 2004;279:23806–23812.
- Stegert MR, Hergovich A, Tamaskovic R, et al. Regulation of NDR protein kinase by hydrophobic motif phosphorylation mediated by the mammalian Ste20-like kinase MST3. Mol Cell Biol. 2005;25:11019–11029.
- Hergovich A. The roles of NDR protein kinases in hippo signalling. Genes (Basel). 2016;7:21.
- Hergovich A. Regulation and functions of mammalian LATS/NDR kinases: looking beyond canonical Hippo signalling. Cell Biosci. 2013;3:32.
- Selimoglu R, Bettoun A, Joffre C, et al. RalA GTPase and MAP4K4 function through NDR1 activation in stress response and apoptotic signaling. HSOA J Cell Biol Cell Metab. 2014;1: 1–11.
- Cook D, Hoa LY, Gomez V, et al. Constitutively active NDR1-PIF kinase functions independent of MST1 and hMOB1 signalling. Cell Signal. 2014;26:1657–1667.
- Johnson LN, Noble MEM, Owen DJ. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85:149–158.
- Hergovich A, Lamla S, Nigg EA, et al. Centrosome-associated NDR kinase regulates centrosome duplication. Mol Cell. 2007;25:625–634.
- Cornils H, Kohler RS, Hergovich A, et al. Downstream of human NDR kinases: impacting on c-myc and p21 protein stability to control cell cycle progression. Cell Cycle. 2011;10:1897–1904.
- Du Z, Tong X, Ye X. Cyclin D1 promotes cell cycle progression through enhancing NDR1/2 kinase activity independent of cyclin-dependent kinase. J Biol Chem. 2013;288:26678–26687.
- Fukasawa T, Enomoto A, Miyagawa K. Serine-threonine kinase 38 regulates CDC25A stability and the DNA damage-induced G2/M checkpoint. Cell Signal. 2015;27:1569–1575.
- Shi DD, Shi H, Lu D, et al. NDR1/STK38 potentiates NF-kB activation by its kinase activity. Cell Biochem Funct. 2012;30:664–670.
- Joffre C, Dupont N, Hoa L, et al. The pro-apoptotic STK38 kinase is a new beclin1 partner positively regulating autophagy. Curr Biol. 2015;25:1–14.
- Bettoun A, Joffre C, Parrini MC, et al. Mitochondrial clearance by the STK38 kinase supports oncogenic Ras-induced cell transformation. Oncotarget. 2016;7(28):44142–44160. .
- Rhee H-W, Zou P, Udeshi ND, et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331.
- Hung V, Udeshi ND, Lam SS, et al. Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat Protoc. 2016;11:456–475.
- Beausoleil SA, Jedrychowski M, Schwartz D, et al. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci. 2004;101:12130–12135.
- Mertins P, Mani DR, Ruggles KV, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62.
- Mertins P, Yang F, Liu T, et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics. 2014;13:1690–1704.
- Sharma K, D’Souza RCJ, Tyanova S, et al. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014;8:1583–1594.
- Fung HYJ, Chook YM. Atomic basis of CRM1-cargo recognition, release and inhibition. Semin Cancer Biol. 2014;27:52–61.
- Kirli K, Karaca S, Dehne HJ, et al. A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning. Elife. 2015;4:1–28.
- Poon IKH, Jans DA. Regulation of nuclear transport: central role in development and transformation? Traffic. 2005;6:173–186.
- Craig E, Zhang Z-K, Davies KP, et al. A masked NES in INI1/hSNF5 mediates hCRM1-dependent nuclear export: implications for tumorigenesis. Embo J. 2002;21:31–42.
- Zhao X, Gan L, Pan H, et al. Multiple elements regulate nuclear/cytoplasmic shuttling of FOXO1: characterization of phosphorylation- and 14-3-3-dependent and -independent mechanisms. Biochem J. 2004;378:839–849.
- Neggers JE, Vercruysse T, Jacquemyn M, et al. Identifying drug-target selectivity of small-molecule CRM1/XPO1 inhibitors by CRISPR/Cas9 genome editing. Chem Biol. 2015;22:107–116.